
Abstract
There is now general agreement that osteoarthritis (OA) involves all structures in the affected joint, culminating in the degradation of the articular cartilage. It is appropriate to focus particularly on the subchondral bone because characteristic changes occur in this tissue with disease progression, either in parallel, or contributing to, the loss of cartilage volume and quality. Changes in both the articular cartilage and the subchondral bone are mediated by the cells in these two compartments, chondrocytes and cells of the osteoblast lineage, respectively, whose primary roles are to maintain the integrity and function of these tissues. In addition, altered rates of bone remodeling across the disease process are due to increased or decreased osteoclastic bone resorption. In the altered mechanical and biochemical environment of a progressively diseased joint, the cells function differently and show a different profile of gene expression, suggesting direct effects of these external influences. There is also ex vivo and in vitro evidence of chemical crosstalk between the cells in cartilage and subchondral bone, suggesting an interdependence of events in the two compartments and therefore indirect effects of, for example, altered loading of the joint. It is ultimately these cellular changes that explain the altered morphology of the cartilage and subchondral bone. With respect to crosstalk between the cells in cartilage and bone, there is evidence that small molecules can transit between these tissues. For larger molecules, such as inflammatory mediators, this is an intriguing possibility but remains to be demonstrated. The cellular changes during the progression of OA almost certainly need to be considered in a temporal and spatial manner, since it is important when and where observations are made in either human disease or animal models of OA. Until recently, comparisons have been made with the assumption, for example, that the subchondral bone is behaviorally uniform, but this is not the case in OA, where regional differences of the bone are evident using magnetic resonance imaging (MRI). Nevertheless, an appreciation of the altered cell function during the progression of OA will identify new disease modifying targets. If, indeed, the cartilage and subchondral bone behave as an interconnected functional unit, normalization of cell behavior in one compartment may have benefits in both tissues.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
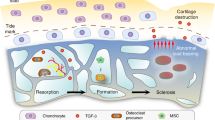
There is now general agreement that osteoarthritis (OA) involves all structures in the affected joint, culminating in the degradation of the articular cartilage. This review focuses on cartilage and the underlying (subchondral) bone, since these compartments are intimately located (Fig. 1) and characteristic changes occur in parallel in these tissues across disease progression. Changes in both the articular cartilage and the subchondral bone are mediated by the cells in these two compartments: chondrocytes in the cartilage and osteoclasts, osteoblasts and osteocytes in the bone, whose primary roles are to maintain the integrity and function of these tissues. In response to the altered mechanical and biochemical environment of a progressively diseased joint, these cells function differently and show a different profile of gene expression, suggesting direct effects of these external influences. There is also ex vivo and in vitro evidence of chemical crosstalk between the cells in cartilage and subchondral bone, suggesting an interdependence of events in the two compartments. It is ultimately these cellular changes that explain the altered morphology of the cartilage and subchondral bone. An appreciation of the altered cell function during the progression of OA is likely to identify new targets for disease modification. If, indeed, the cartilage and subchondral bone behave as a functional unit, normalization of cell behavior in one compartment may have benefits in both tissues.

Schematic representation of an articular joint and the spatial relationship between chondrocytes in the hyaline and calcified cartilage and cells in the subchondral bone and bone marrow. Also depicted is the influence of repeated mechanical load and the response of chondrocytes with respect to production of matrix modifying factors such as MMPs (curved arrows), the response of load-sensing osteocytes with respect to the production of mediators such as prostaglandins and nitric oxide [12] (curved arrows), and the possibility of bidirectional communication between the cartilage and bone by the production of soluble mediators (double headed arrow). Not depicted are the possible additional influences of osteoblasts and osteoclasts involved in remodeling the subchondral spongiosa
Chondrocytes in OA
Articular cartilage and the disease processes that lead to its degeneration in OA have been intensively studied and extensively reviewed [1]. Articular chondrocytes are the only cell type resident in articular cartilage, and comprise a mere 2 %–5 % of its volume, which otherwise consists of a hydrated extracellular matrix of collagens (predominantly type II) and proteoglycans (importantly aggrecan). The role of the chondrocytes is to maintain the integrity of the cartilage by repairing damage due to minor or acute insults to the matrix. However, in the progression of OA, in which the biomechanical and biochemical environment of the cartilage is changed, characteristic changes also occur in the behavior of the chondrocytes. As recently reviewed [2], OA chondrocytes appear to resume the default differentiation pathway that is somehow blocked in articular cartilage, which leads to proliferation, hypertrophy, and apoptosis, as seen in the growth plate [3]. This more catabolic phenotype is characterized by decreased synthesis of the extracellular matrix and increased production of degradative enzymes, such as the matrix metalloproteinases (MMPs) and the aggrecanases. The repertoire of proteins produced by the cells changes toward that of hypertrophic chondrocytes, with prominent expression of type X collagen and MMP13, in turn driven by a reprogramming of the transcriptional machinery of the cell [2].
Although the initiating factors in OA have not been identified, mechanical damage is thought to interact with the effects of aging, in a genotype-dependent manner, to trigger local inflammation in the joint. Microarray analysis of adult human articular cartilage explants found that mechanical injury significantly regulated the expression of a large number of genes, collectively consistent with a reactivation of morphogenic pathways [4]. Foremost among the genes upregulated by injury was WNT16, which was barely detectable in preserved areas of cartilage in OA joints but was highly upregulated in areas of cartilage with moderate to severe cartilage damage. Interestingly, the wingless MMTV integration (Wnt) antagonist, sclerostin, was highly expressed only in focal areas of cartilage damage in sheep and murine models of OA, as well as in end-stage human OA cartilage [5]. In the same study, the gene encoding sclerostin, SOST, was also upregulated by interleukin (IL)-1 in articular chondrocytes. The addition of recombinant sclerostin protein to explant cultures inhibited the aggrecanase-inducing effects of IL-1, suggesting an anticatabolic role for sclerostin in OA cartilage [5].
Also suggesting an important role for the Wnt pathway in OA cartilage, over-expression of the Wnt antagonist, dikkopf (DKK)-1, protected against cartilage destruction in an animal model of OA [6•]. Further, Wnt-3A, which activates the Wnt pathway, induced the expression of MMP13 and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)-4 in primary chondrocyte cultures. IL-1 and tumor necrosis factor (TNF)-α have long been known to produce a catabolic phenotype in articular chondrocytes (reviewed in [7]) and may do so via the Wnt pathway. In an analogous tissue to articular cartilage, the intervertebral disc, TNF-α increased the expression of β-catenin and MMP13, and significantly inhibited the synthesis of type II collagen and proteoglycan, an effect that could be reversed by DKK1 [8]. While inflammatory cytokines have an essential role in the repair of all tissues, chronic exposure seems to compromise repair. In cartilage, persistent inflammation increases the remodeling rate of the extracellular matrix, leading to deficient matrix and poor quality cartilage. The links between biomechanical disruption, inflammation, and protease production by chondrocytes was demonstrated in a surgical model of mouse OA, in which genes such as IL-1, IL-6, MMP3, and ADAMTS 1, 4, and 5 were all upregulated after surgery. The expression of these genes was shown to be highly mechanosensitive because it was prevented by immobilization of the joint at the time of surgery [9].
Thus, although mechanical load is necessary for healthy cartilage, and limb immobilization leads to cartilage loss [10], load induced injury or chronic overloading of the joint can lead to changes in chondrocyte behavior that have long term consequences for matrix integrity. The question posed below is whether the chondrocyte reprogramming is at least partly due to signals from the subchondral bone, or vice versa, whether chondrocyte signals can influence cells in the bone.
Osteoblasts in OA
Evidence from animal models of OA and from human bone samples obtained at surgery indicates altered bone remodeling in this disease. At the cellular level, remodeling is achieved by the actions of osteoclasts, which resorb bone, and osteoblasts, which are the bone forming cells. The activities of both of these cell types is regulated by osteocytes, embedded within the bone matrix [11, 12]. Overloading of the joints in OA seems to correspond with increased microdamage and microfracture in subchondral bone in the overloaded areas [13–15]. While it has been proposed that microdamage of bone protects articular cartilage due to enhanced energy absorption [14], it also promotes bone remodeling. Osteocytes detect damage of the mineralized bone matrix and direct its repair by initiating targeted osteoclastic resorption of the affected bone [16].
Recent evidence suggests that osteocytes produce the osteoclast differentiating cytokine, receptor activator of nuclear factor κB ligand (RANKL), in mature bone [17]. As recently discussed by Burr and Gallant [18••] the rate of bone remodeling changes across the course of the disease. Thus, increased remodeling, accompanied with increased vascularity, occurs in the subchondral bone in early OA, while late stage disease is characterized by reduced bone resorption with a bias toward bone formation. The changes in bone remodeling also vary spatially within the joint, for example medial versus lateral in the knee, but also more distally to the affected joint [19, 20]. Altered remodeling leads to altered bone structure, with increased BV/TV in cancellous bone and the formation of osteophytes [19, 21].
An increased trabecular number, decreased trabecular spacing, and reduced hardness of the bone in OA [22], due to decreased mineralization [23], characterize the subchondral bone, especially in zones underlying cartilage degeneration. Discrete zones of subchondral bone that can be imaged using magnetic resonance (MR), termed bone marrow lesions (BML), are frequently observed in both established OA and in early OA, but rarely in symptom free individuals [24, 25]. BMLs arise in regions of predicted high loading and contain abnormal bone, with areas of osteocyte death, areas of bone sclerosis with reduced mineral density [26]. Longitudinal studies have shown that BMLs occur adjacent to sites of current or future cartilage degeneration and are predictive of structural deterioration in knee OA [26–29] and future joint replacement [30].
The temporal and spatial changes in bone structure in OA are the result of altered cellular activity. The causes of this altered behavior are not yet known but are likely to be several. As stated, overloading of the joint can lead to an accumulation of damage in the bone matrix [31], which the cells attempt to resolve, and which is observed as increased remodeling in the bone. There is also some evidence for changes in perfusion of the bone, perhaps with episodes of ischemia [32], which could lead to osteocyte death followed by increased or decreased remodeling depending on whether blood supply is restored. What has been shown is that osteoblasts derived from OA bone display an altered phenotype and that gene expression in OA bone is different from that in either osteoporotic or normal bone.
For example, alkaline phosphatase and osteocalcin levels were found to be elevated in OA osteoblasts compared with normal osteoblasts, whereas osteopontin levels were similar [33]. In addition, osteoblasts from OA bone showed disturbed mineralization compared with control cells, with dramatically variable calcium: phosphate ratios compared with the value of approximately 1.6 for control osteoblasts and normal bone [34].
In addition, Couchourel et al [33] reported reduced mineralization by OA osteoblasts, accompanied by an elevated COL1A1:COL1A2 mRNA ratio, similar to the differential expression of these genes in OA bone [35]. Interestingly, OA osteoblasts produce more TGFβ1 than normal osteoblasts, and inhibiting TGFβ1 in OA osteoblasts corrected the abnormal COL1A1:COL1A2 ratio and increased cell mineralization [35]. The increased production of transforming growth factor (TGF) β1 by OA cells induced increased levels of DKK-2 and silencing of either TGFβ or DKK2 in these cells was found to normalize the OA mineralization phenotype [36]. Massicotte et al [37] reported two subgroups of osteoblasts derived from OA subchondral bone, based on production of IL-6 and prostaglandin E2, while TGFβ1 levels were increased in all osteoarthritic osteoblasts compared with normal. Kumarasinghe et al [38] performed analysis of gene expression in primary osteoblasts derived from OA and control femoral bone across differentiation. These studies showed that the dysregulated expression of TWIST1, TGFβ1, and SMAD3 mRNA observed previously in OA bone is also present in OA osteoblasts when these cells are cultured ex vivo, and proposed that at least part of the etiology of OA is due to altered intrinsic properties of the osteoblasts [34]. A recent report confirmed the high concentrations of TGFβ1 in subchondral bone in human and mouse OA, and went on to show that transgenic overexpression of TGFβ1 in the subchondral bone actually induced OA [39••]. Significantly, inhibition of TGFβ specifically in the subchondral bone improved the bone architecture in the anterior cruciate ligament transection mouse model of OA and attenuated the degeneration of articular cartilage and the percentage of chondrocytes in the cartilage expressing MMP13 and type X collagen. These positive effects of TGFβ1 inhibition were not seen with systemic approaches to inhibit TGFβ activity, since TGFβ is required for cartilage homeostasis and the systemic, but not targeted, TGFβ inhibition blocked TGFβ signaling in articular cartilage. The authors therefore concluded that high concentrations of TGFβ in the subchondral bone induced abnormal bone formation and the development of OA [39••]. The above comments are generalities because no data are available regarding the behavior of osteoblast-lineage across the course of OA or, in any systematic way, from different zones in a diseased joint.
Crosstalk Between Chondrocytes and Osteoblasts in OA
It is possible that chondrocytes and cells of the bone are responding independently to the same environmental cues, which is exhibited in an altered cell phenotype in OA. Alternatively, cellular changes in one or both compartments may influence cells in the other compartment. There is mounting evidence, both ex vivo and in vivo, that chondrocytes and osteoblasts are able to influence each other. For example, in the well-established rat model of OA, in which monosodium iodoacetate (MIA) is injected intra-articularly, it is the cartilage that is initially exposed to the drug. However, the model shows significant bone loss after only 2 weeks, followed later by increased trabecular thickness and the presence of subchondral bone sclerosis, cysts, and osteophytes, in parallel with cartilage degradation [40•]. Conversely, over-expression of the EPHB4 receptor specifically in osteoblasts and, therefore, subchondral bone, exerted a protective effect against OA in mice, induced by medial meniscus destabilization [41•]. Since the subchondral bone was also preserved in the transgenic animals, there are potentially mechanical as well as biochemical explanations for this finding, which nonetheless demonstrates the interdependence of the two tissue compartments.
There is good evidence that cartilage loss occurs in the same regions of the joint as the changes in the subchondral bone [42]. It is, therefore, perhaps not surprising that in animal models of OA, applying treatments that are thought to specifically target the subchondral bone can ameliorate or prevent disease progression. For example, in rat models of knee OA, the bisphosphonate, alendronate, suppressed both subchondral bone resorption and the development of OA in the knee joint [43]. Similarly, calcitonin reduced the levels of circulating bone turnover markers and the severity of OA lesions dog models of OA [44, 45]. Systemic injection of osteoprotegerin (OPG), to block RANKL-mediated bone remodeling in a mouse menisectomy model of OA, increased bone volume in the operated and nonoperated knee bones, as would be expected, but also dramatically protected from the meniscectomy-related OA [46]. OPG also significantly reduced ADAMTS-4 and ADAMTS-5 expression in the articular chondrocytes following meniscectomy, although not to control levels. In a similar mouse model, pamidronate dramatically preserved the bone mass and reduced the OARSI score, at the same time almost normalizing the expression of ADAMTS-4 and ADAMTS-5 in the overlying joint cartilage [47]. In a human study, strontium ranelate, which is thought to act on bone to reduce turnover, significantly reduced CTX-II, a marker of cartilage degradation, in subjects with a history of OA [48].
There is accumulating ex vivo and in vitro evidence that events in the subchondral bone have a direct effect on the overlying cartilage. Amin et al [49] reported on chondrocyte survival in bovine cartilage explants in culture, which included or excluded the underlying subchondral bone. It was found that excision of subchondral bone from articular cartilage resulted in an increase in chondrocyte death at 7 days, mainly in the superficial zone of the cartilage. However, the presence of the excised subchondral bone in the culture medium abrogated this increase in chondrocyte death, most likely due to soluble mediator(s) released from the subchondral bone. Sanchez et al [50] described a coculture system, in which osteoblasts derived, respectively, from ‘sclerotic’ or ‘nonsclerotic’ regions of human subchondral bone in OA were separated by a membrane from chondrocytes in alginate beads. Clear evidence was presented for cross-talk between the cells, with chondrocytes cultured in the presence of ‘sclerotic’ osteoblasts, but not ‘nonsclerotic’ osteoblasts, exhibiting reduced production of aggrecan and increased expression of MMP3 and MMP13. This influence was magnified when the osteoblasts were pretreated with inflammatory mediators IL-1, IL-6, or oncostatin M (OSM). These data suggest the possibility at least of disease exacerbating changes in chondrocytes proximal to phenotypically OA osteoblasts. The authors further speculate that the IL-1, IL-6, or OSM, which are overproduced by OA chondrocytes, could create a feedback loop by acting on nearby osteoblasts. Although osteocytes are the most abundant cell type in bone and are the primary mechanosensing cell type [11, 12], and interactions between chondrocytes and osteocytes are therefore likely of primary importance, very little has been reported concerning this interaction. In one such study, however, Priam et al [51••] conducted experiments in which mouse calvarial osteoblast/osteocytes were subjected to cyclic compression and mouse articular chondrocytes were exposed to the osteoblast conditioned medium. Conditioned medium from the compressed cells caused a dramatic up-regulation of MMP3 and MMP13 expression in the chondrocytes and down-regulated expression of aggrecan and type II collagen. The study identified 14-3-3ε as a soluble mediator for communication between the osteoblasts/osteocytes and chondrocytes [51••].
The above experiments demonstrate the potential for molecular crosstalk between osteoblasts/osteocytes and chondrocytes in vivo but it has long been thought that there was little possibility of communication between subchondral bone and articular cartilage. However, Imhof et al [52] described the dense subchondral vasculature in close proximity to the cartilage and the micro-channels that penetrate the subchondral mineralization zone and permit communication between the bone and the cartilage. Imhof et al [52] have further claimed that more than 50 % of the glucose, oxygen, and water requirements of cartilage are provided by perfusion from the subchondral vessels. In support of this, experimentally induced hypoxia of the femoral head led to cell death in the bony epiphysis and in the deep layer of the overlying cartilage [53]. Indeed, perfusion abnormalities have been identified in OA, in particular in zones of the subchondral bone identified by MR imaging as bone marrow ‘edema’ or bone marrow lesions [54].
As discussed above, these zones underlie regions of degraded cartilage or predict cartilage degradation [27–29]. In elegant experiments using fluorescent dyes, Pan et al [55] showed the diffusion of these small molecules between the bone marrow and the articular space. These observations suggest the possibility of direct signaling between subchondral bone and articular cartilage, at least for small molecules. The authors further suggested that the two compartments form a functional unit both mechanically and biochemically, which may play a role in the maintenance and degeneration of the joint. These results were consistent with several other observations. The ultrastructure of the interface between the subchondral bone and calcified cartilage provides numerous vascular (Haversian) canals, by which these tissues could communicate [56].
A more recent study of the human chondro-osseous junction revealed a hitherto unappreciated complexity [57]. For example, uncalcified cartilage was sometimes seen dipping through the calcified cartilage into bone and marrow spaces. The authors commented that this proximity of hyaline cartilage and marrow spaces could provide a molecular diffusion pathway, which may have nutritional, metabolic, and biomechanical roles. In addition, since this interface is profoundly affected by OA, these areas could enable trafficking of humoral mediators between these tissues (Fig. 1). Finally, a large increase in subchondral plate porosity was recently shown during disease development in a mouse model of OA [58], and it was again suggested that this may enhance mutual interaction between the bone and cartilage compartments. It remains to be formally demonstrated that cytokine-sized molecules can traverse between bone and cartilage in either healthy or diseased joints. However, larger molecules than previously thought are able to traverse osteocyte canaliculi, and this transport is increased by bone loading [59]. Similar principles may apply to transport through the porosity linking these compartments, which potentially includes the vasculature, the canalicular porosity and the pores in the subchondral bone plate and the calcified cartilage. With respect to the cartilage, diffusion of cell mediators has been well demonstrated. For example, insulin-like growth factor (IGF) has important roles in cartilage but is produced in low amounts only and is largely transported into cartilage from the circulation [60•]. This transport occurs partly by diffusion across a concentration gradient but, in addition, cyclic loading has been shown to enhance the transport of this relatively large molecule by advection [61].
Conclusions
OA is caused by factors, some known and some unknown, which result in an altered phenotype of osteoblasts and osteocytes in bone and chondrocytes in cartilage. There is much interest as to whether there is crosstalk between cells in these compartments, since there is now good evidence that these cells can signal in vitro in ways that can promote chondrocyte catabolism. There is also good evidence that small molecules can traverse between these tissue compartments in situ but this evidence is lacking for larger cell mediators. The importance of this issue is that directing treatment to either compartment may provide a circuit breaker in OA to prevent or slow the progression of this condition.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Principles of osteoarthritis - its definition, character, derivation, and modality-related recognition. Croatia: InTech; 2012.
van der Kraan PM, van den Berg WB. Chondrocyte hypertrophy and osteoarthritis: role in initiation and progression of cartilage degeneration? Osteoarthritis Cartilage. 2012;20:223–32.
Pesesse L, Sanchez C, Delcour JP, Bellahcene A, Baudouin C, Msika P, et al. Consequences of chondrocyte hypertrophy on osteoarthritic cartilage: potential effect on angiogenesis. Osteoarthritis Cartilage. 2013.
Dell'accio F, De Bari C, Eltawil NM, Vanhummelen P, Pitzalis C. Identification of the molecular response of articular cartilage to injury, by microarray screening: Wnt-16 expression and signaling after injury and in osteoarthritis. Arthritis Rheum. 2008;58:1410–21.
Chan BY, Fuller ES, Russell AK, Smith SM, Smith MM, Jackson MT, et al. Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthritis Cartilage. 2011;19:874–85.
Oh H, Chun CH, Chun JS. Dkk-1 expression in chondrocytes inhibits experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum. 2012;64:2568–78. This paper provides an example of how chondrocyte-specific gene manipulation can influence OA changes in both cartilage and subchondral bone.
Maldonado M, Nam J. The role of changes in extracellular matrix of cartilage in the presence of inflammation on the pathology of osteoarthritis. Biomed Res Int. 2013;2013:284873.
Ye S, Wang J, Yang S, Xu W, Xie M, Han K, et al. Specific inhibitory protein dkk-1 blocking Wnt/beta-catenin signaling pathway improves protectives' effect on the extracellular matrix. J Huazhong Univ Sci Technolog Med Sci. 2011;31:657–62.
Burleigh A, Chanalaris A, Gardiner MD, Driscoll C, Boruc O, Saklatvala J, et al. Joint immobilization prevents murine osteoarthritis and reveals the highly mechanosensitive nature of protease expression in vivo. Arthritis Rheum. 2012;64:2278–88.
Houard X, Goldring MB, Berenbaum F. Homeostatic mechanisms in articular cartilage and role of inflammation in osteoarthritis. Curr Rheumatol Rep. 2013;15:375.
Atkins GJ, Findlay DM. Osteocyte regulation of bone mineral: a little give and take. Osteoporos Int. 2012;23:2067–79.
Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26:229–38.
Li ZC, Dai LY, Jiang LS, Qiu S. Difference in subchondral cancellous bone between postmenopausal women with hip osteoarthritis and osteoporotic fracture: implication for fatigue microdamage, bone microarchitecture, and biomechanical properties. Arthritis Rheum. 2012;64:3955–62.
Malekipour F, Whitton C, Oetomo D, Lee PV. Shock absorbing ability of articular cartilage and subchondral bone under impact compression. J Mech Behav Biomed Mater. 2013;26:127–35.
Fazzalari NL, Kuliwaba JS, Forwood MR. Cancellous bone microdamage in the proximal femur: influence of age and osteoarthritis on damage morphology and regional distribution. Bone. 2002;31:697–702.
Mori S, Burr DB. Increased intracortical remodeling following fatigue damage. Bone. 1993;14:103–9.
O'Brien CA, Nakashima T, Takayanagi H. Osteocyte control of osteoclastogenesis. Bone. 2013;54:258–63.
Burr DB, Gallant MA. Bone remodeling in osteoarthritis. Nat Rev Rheumatol. 2012;8:665–73. Paper reviews evidence, which together makes the important point that changes in the subchondral bone during the development of OA need to be considered both as a function of time, and spatially with respect to the joint. In addition, the review provides a useful working model linking changes in the subchondral bone with those in the overlying articular cartilage.
Fazzalari N, Parkinson I. Femoral trabecular bone of osteoarthritic and normal subjects in an age and sex matched group. Osteoarthritis Cartilage. 1998;6:377–82.
Kumarasinghe DD, Hopwood B, Kuliwaba JS, Atkins GJ, Fazzalari NL. An update on primary hip osteoarthritis including altered Wnt and Tgf-beta associated gene expression from the bony component of the disease. Rheumatology. 2011;50:2166–75.
Jeffery AK. Osteophytes and the osteoarthritic femoral head. J Bone Joint Surg Br. 1975;57:314–24.
Dall'Ara E, Ohman C, Baleani M, Viceconti M. Reduced tissue hardness of trabecular bone is associated with severe osteoarthritis. J Biomech. 2011;44:1593–8.
Li B, Aspden RM. Material properties of bone from the femoral neck and calcar femorale of patients with osteoporosis or osteoarthritis. Osteoporos Int. 1997;7:450–6.
Daheshia M, Yao JQ. The bone marrow lesion in osteoarthritis. Rheumatol Int. 2011;31:143–8.
Bassiouni HM. Bone marrow lesions in the knee: the clinical conundrum. Int J Rheum Dis. 2010;13:196–202.
Hunter DJ, Gerstenfeld L, Bishop G, Davis AD, Mason ZD, Einhorn TA, et al. Bone marrow lesions from osteoarthritis knees are characterized by sclerotic bone that is less well mineralized. Arthritis Res Ther. 2009;11:R11.
Dore D, Martens A, Quinn S, Ding C, Winzenberg T, Zhai G, et al. Bone marrow lesions predict site-specific cartilage defect development and volume loss: a prospective study in older adults. Arthritis Res Ther. 2010;12:R222.
Hunter DJ, Zhang Y, Niu J, Goggins J, Amin S, LaValley MP, et al. Increase in bone marrow lesions associated with cartilage loss: a longitudinal magnetic resonance imaging study of knee osteoarthritis. Arthritis Rheum. 2006;54:1529–35.
Felson DT, McLaughlin S, Goggins J, LaValley MP, Gale ME, Totterman S, et al. Bone marrow edema and its relation to progression of knee osteoarthritis. Ann Intern Med. 2003;139:330–6.
Tanamas SK, Wluka AE, Pelletier JP, Martel-Pelletier J, Abram F, Wang Y, et al. The association between subchondral bone cysts and tibial cartilage volume and risk of joint replacement in people with knee osteoarthritis: a longitudinal study. Arthritis Res Ther. 2010;12:R58.
Taljanovic MS, Graham AR, Benjamin JB, Gmitro AF, Krupinski EA, Schwartz SA, et al. Bone marrow edema pattern in advanced hip osteoarthritis: quantitative assessment with magnetic resonance imaging and correlation with clinical examination, radiographic findings, and histopathology. Skeletal Radiol. 2008;37:423–31.
Findlay DM. Vascular pathology and osteoarthritis. Rheumatology. 2007;46:1763–8.
Couchourel D, Aubry I, Delalandre A, Lavigne M, Martel-Pelletier J, Pelletier JP, et al. Altered mineralization of human osteoarthritic osteoblasts is attributable to abnormal type 1 collagen production. Arthritis Rheum. 2009;60:1438–50.
Kumarasinghe DD, Sullivan T, Kuliwaba JS, Fazzalari NL, Atkins GJ. Evidence for the dysregulated expression of twist1, tgfβ1, and smad3 in differentiating osteoblasts from primary hip osteoarthritis patients. Osteoarthritis Cartilage. 2012;20:1357–66.
Truong L-H, Kuliwaba JS, Tsangari H, Fazzalari NL. Differential gene expression of bone anabolic factors and trabecular bone architectural changes in the proximal femoral shaft of primary hip osteoarthritis patients. Arthrit Res Ther. 2006;8:R188.
Chan TF, Couchourel D, Abed E, Delalandre A, Duval N, Lajeunesse D. Elevated dickkopf-2 levels contribute to the abnormal phenotype of human osteoarthritic osteoblasts. J Bone Miner Res. 2011;26:1399–410.
Massicotte F, Lajeunesse D, Benderdour M, Pelletier JP, Hilal G, Duval N, et al. Can altered production of interleukin-1beta, interleukin-6, transforming growth factor-beta and prostaglandin e (2) by isolated human subchondral osteoblasts identify two subgroups of osteoarthritic patients. Osteoarthritis Cartilage. 2002;10:491–500.
Kumarasinghe DD, Perilli E, Tsangari H, Truong L, Kuliwaba JS, Hopwood B, et al. Critical molecular regulators, histomorphometric indices and their correlations in the trabecular bone in primary hip osteoarthritis. Osteoarthritis Cartilage. 2010;18:1337–44.
Zhen G, Wen C, Jia X, Li Y, Crane JL, Mears SC, et al. Inhibition of TGF-beta signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med. 2013;19:704–12. Paper contains an elegant dataset demonstrating molecular links between subchondral bone and articular catilage. Specifically, TGF beta was shown to be raised in subchondral bone in human OA and animal models of OA, and overexpression of TGF beta in osteoblasts induced OA. Importantly, inhibition of TGF beta in the subchondral bone attenuated the degeneration of articular cartilage, suggesting a potential therapeutic target for OA.
Mohan G, Perilli E, Kuliwaba JS, Humphries JM, Parkinson IH, Fazzalari NL. Application of in vivo micro-computed tomography in the temporal characterization of subchondral bone architecture in a rat model of low-dose monosodium iodoacetate-induced osteoarthritis. Arthritis Res Ther. 2011;13:R210. Paper shows an experimental example of how degradative changes in the articular cartilage can affect the underlying subchonral bone. Intra-articular injection of monosodium iodoacetate (MIA) induced OA in a rat model, showing characteristic subchondral bone attrition, followed by sclerotic changes.
Valverde-Franco G, Pelletier JP, Fahmi H, Hum D, Matsuo K, Lussier B, et al. In vivo bone-specific ephb4 overexpression in mice protects both subchondral bone and cartilage during osteoarthritis. Arthritis Rheum. 2012;64:3614–25. Paper provides evidence that molecular events in the subchondral bone strongly affect the articular cartilage. It was shown that bone-specific over-expression of EphB4 in mice exerted a protective effect on all joint structures in the destabilisation of the medial meniscus (DMM) model of OA.
Neogi T, Felson D, Niu J, Lynch J, Nevitt M, Guermazi A, et al. Cartilage loss occurs in the same subregions as subchondral bone attrition: a within-knee subregion-matched approach from the multi-center osteoarthritis study. Arthritis Rheum. 2009;61:1539–44.
Hayami T, Pickarski M, Wesolowski GA, McLane J, Bone A, Destefano J, et al. The role of subchondral bone remodeling in osteoarthritis: reduction of cartilage degeneration and prevention of osteophyte formation by alendronate in the rat anterior cruciate ligament transection model. Arthritis Rheum. 2004;50:1193–206.
Behets C, Williams JM, Chappard D, Devogelaer JP, Manicourt DH. Effects of calcitonin on subchondral trabecular bone changes and on osteoarthritic cartilage lesions after acute anterior cruciate ligament deficiency. J Bone Miner Res. 2004;19:1821–26.
Manicourt DH, Altman RD, Williams JM, Devogelaer JP, Druetz-Van Egeren A, Lenz ME, et al. Treatment with calcitonin suppresses the responses of bone, cartilage, and synovium in the early stages of canine experimental osteoarthritis and significantly reduces the severity of the cartilage lesions. Arthritis Rheum. 1999;42:1159–67.
Kadri A, Ea HK, Bazille C, Hannouche D, Liote F, Cohen-Solal ME. Osteoprotegerin inhibits cartilage degradation through an effect on trabecular bone in murine experimental osteoarthritis. Arthritis Rheum. 2008;58:2379–86.
Funck-Brentano T, Lin H, Hay E, Ah Kioon MD, Schiltz C, Hannouche D, et al. Targeting bone alleviates osteoarthritis in osteopenic mice and modulates cartilage catabolism. PLoS One. 2012;7:e33543.
Alexandersen P, Karsdal MA, Byrjalsen I, Christiansen C. Strontium ranelate effect in postmenopausal women with different clinical levels of osteoarthritis. Climacteric. 2011;14:236–43.
Amin AK, Huntley JS, Simpson AH, Hall AC. Chondrocyte survival in articular cartilage: the influence of subchondral bone in a bovine model. J Bone Joint Surg Br. 2009;91:691–9.
Sanchez C, Deberg MA, Piccardi N, Msika P, Reginster JY, Henrotin YE. Osteoblasts from the sclerotic subchondral bone downregulate aggrecan but upregulate metalloproteinases expression by chondrocytes. This effect is mimicked by interleukin-6, -1beta and oncostatin m pre-treated nonsclerotic osteoblasts. Osteoarthritis Cartilage. 2005;13:979–87.
Priam S, Bougault C, Houard X, Gosset M, Salvat C, Berenbaum F, et al. Identification of soluble 14-3-3 as a novel subchondral bone mediator involved in cartilage degradation in osteoarthritis. Arthritis Rheum. 2013;65:1831–42. Paper presents data which support the concept that molecules from subchondral bone can influence articular cartilage. Compression loading of mouse osteoblasts resulted in the production of soluble mediators that induced a catabolic response in mouse chondrocytes. Among these, 14-3-3e was identified as a potential communicating molecule between these tissue compartments.
Imhof H, Sulzbacher I, Grampp S, Czerny C, Youssefzadeh S, Kainberger F. Subchondral bone and cartilage disease: a rediscovered functional unit. Invest Radiol. 2000;35:581–8.
Kim HK, Bian H, Aya-ay J, Garces A, Morgan EF, Gilbert SR. Hypoxia and hif-1alpha expression in the epiphyseal cartilage following ischemic injury to the immature femoral head. Bone. 2009;45:280–8.
Aaron RK, Dyke JP, Ciombor DM, Ballon D, Lee J, Jung E, et al. Perfusion abnormalities in subchondral bone associated with marrow edema, osteoarthritis, and avascular necrosis. Ann N Y Acad Sci. 2007;1117:124–37.
Pan J, Zhou X, Li W, Novotny JE, Doty SB, Wang L. In situ measurement of transport between subchondral bone and articular cartilage. J Orthop Res. 2009;27:1347–52.
Clark JM, Huber JD. The structure of the human subchondral plate. J Bone Joint Surg Br. 1990;72:866–73.
Lyons TJ, McClure SF, Stoddart RW, McClure J. The normal human chondro-osseous junctional region: evidence for contact of uncalcified cartilage with subchondral bone and marrow spaces. BMC Musculoskelet Disord. 2006;7:52.
Botter SM, van Osch GJ, Clockaerts S, Waarsing JH, Weinans H, van Leeuwen JP. Osteoarthritis induction leads to early and temporal subchondral plate porosity in the tibial plateau of mice: an in vivo microfocal computed tomography study. Arthritis Rheum. 2011;63:2690–9.
Wang B, Zhou X, Price C, Li W, Pan J, Wang L. Quantifying load-induced solute transport and solute-matrix interaction within the osteocyte lacunar-canalicular system. J Bone Miner Res. 2013;28:1075–86.
Zhang L, Gardiner BS, Smith DW, Pivonka P, Grodzinsky AJ. On the role of diffusible binding partners in modulating the transport and concentration of proteins in tissues. J Theor Biol. 2010;263:20–9. Paper provides an important model for the diffusion of proteins through the articular cartilage, the possibility of which is clearly important if molecules such as inflammatory mediators from the subchondral bone are to influence the overlying articular cartilage.
Zhang L, Gardiner BS, Smith DW, Pivonka P, Grodzinsky A. The effect of cyclic deformation and solute binding on solute transport in cartilage. Arch Biochem Biophys. 2007;457:47–56.
Acknowledgements
Funding from the National Health and Medical Research Council of Australia and the support of the Department of Orthopaedics and Trauma, Royal Adelaide Hospital and the University of Adelaide, Adelaide, Australia is gratefully acknowledged.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
D.M. Findlay declares that he has no conflicts of interest. G.J. Atkins declares that he has no conflicts of interest.
Human and Animal Rights and Informed Consent
All studies by the authors involving animal and/or human subjects were performed after approval by the appropriate institutional review boards. When required, written informed consent was obtained from all participants.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Findlay, D.M., Atkins, G.J. Osteoblast-Chondrocyte Interactions in Osteoarthritis. Curr Osteoporos Rep 12, 127–134 (2014). https://doi.org/10.1007/s11914-014-0192-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-014-0192-5