
Abstract
Purpose of Review
In many instances, the renin-angiotensin system (RAS) and the vasopressinergic system (VPS) are jointly activated by the same stimuli and engaged in the regulation of the same processes.
Recent Findings
Angiotensin II (Ang II) and arginine vasopressin (AVP), which are the main active compounds of the RAS and the VPS, interact at several levels. Firstly, Ang II, acting on AT1 receptors (AT1R), plays a significant role in the release of AVP from vasopressinergic neurons and AVP, stimulating V1a receptors (V1aR), regulates the release of renin in the kidney. Secondly, Ang II and AVP, acting on AT1R and V1aR, respectively, exert vasoconstriction, increase cardiac contractility, stimulate the sympathoadrenal system, and elevate blood pressure. At the same time, they act antagonistically in the regulation of blood pressure by baroreflex. Thirdly, the cooperative action of Ang II acting on AT1R and AVP stimulating both V1aR and V2 receptors in the kidney is necessary for the appropriate regulation of renal blood flow and the efficient resorption of sodium and water. Furthermore, both peptides enhance the release of aldosterone and potentiate its action in the renal tubules.
Summary
In this review, we (1) point attention to the role of the cooperative action of Ang II and AVP for the regulation of blood pressure and the water-electrolyte balance under physiological conditions, (2) present the subcellular mechanisms underlying interactions of these two peptides, and (3) provide evidence that dysregulation of the cooperative action of Ang II and AVP significantly contributes to the development of disturbances in the regulation of blood pressure and the water-electrolyte balance in cardiovascular diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Angiotensin II (Ang II) is a member of the renin-angiotensin system (RAS), which forms a cascade of highly active biological compounds regulating a variety of physiological functions. Co-activation of the RAS and the vasopressinergic system (VPS) by the same stimuli, and engagement of the same post-receptor intracellular pathways by Ang II and vasopressin (AVP), favors formation of different types of interactions, which may result in additive, synergistic, or antagonistic effects. Both Ang II and AVP directly participate in the regulation of the cardiovascular system. They also have an impact on several other processes, such as metabolism, stress, emotional disorders, and inflammation, which may exert potent secondary effects on the cardiovascular functions. The main purpose of the present review is to draw attention to the interactions of Ang II and AVP in the regulation of the water-electrolyte balance and blood pressure in healthy individuals and in patients with cardiovascular diseases. Specifically, we give a description of the regulation and function of the RAS and the VPS and we focus on the mechanisms underlying the interactions of Ang II and AVP in cardiovascular regulation and on disturbances of these interactions in cardiovascular diseases.
Renin-Angiotensin System
Prorenin and renin circulating in the blood are produced mainly in the juxtaglomerular epithelioid (JGE) cells, located in the wall of the afferent arterioles at the entrance to the glomeruli and in close vicinity to the macula densa (MD) cells, and forming together with the MD cells the juxtaglomerular apparatus (JGA) [1]. Renin is an aspartyl protease encoded by a single gene located in chromosome 1q42. During posttranslational processes, preprorenin, which consists of 401 amino acid residues, is converted to prorenin and subsequently to renin [2, 3]. Renin is a highly specific enzyme, which acts on renin substrate—angiotensinogen (Agt)—and catalyzes the first step in a cascade of RAS compounds. Acting on Agt, renin cleaves a decapeptide—angiotensin I (Ang I) which can be converted to Ang II by angiotensin-converting enzyme 1 (ACE, ACE 1) or to angiotensin-(1-7) [Ang-(1-7)] by ACE 2 and other enzymes. ACE 1 is a glycoprotein with a molecular mass of 18 kDa and two active carboxy-terminal sites, which can also metabolize bradykinin (BK), an active vasodilator and natriuretic substance, to an inactive metabolite [4, 5]. ACE 2 is a zinc metalloproteinase consisting of 805 amino acids, showing significant homology to ACE 1; however, the enzymatic activity of ACE 2 is not inhibited by ACE 1 inhibitors. ACE 2 hydrolyzes Ang I to Ang-(1-9), and Ang II to Ang-(1-7) [6].
Other angiotensin peptides [Ang-(1-9), Ang III (Ang-(2-8)), Ang IV (Ang-(3-8), Ang-(1-4), Ang-(1-5), Ang-(5-8) and Ang-(1-12)] are generated by aminopeptidases, carboxypeptidases, endopeptidases, and chymase. Prorenin, renin, and angiotensins act by means of specific renin (PRR) and angiotensin receptors, which are both widely distributed in the cardiovascular system and other organs (see below).
Local Renin-Angiotensin Systems
Renal RAS
All components of the RAS are present in the kidney [7, 8]. Renin is released from the JGA cells into the blood, but some small portion of the JGA renin is filtered through the glomeruli and reabsorbed in the proximal tubules [9]. There is also evidence for the synthesis of renin by the principal cells of the collecting ducts [10].
The synthesis and release of renin is altered by factors regulating the production of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) and by changes in the intracellular calcium and sodium content. The stimulatory cAMP-mediated effect of β-adrenergic stimulation on release of renin from the JGA cells plays a significant role during the activation of the sympathetic nervous system [11]. Prostanoids, which stimulate renin secretion via activation of prostaglandin E2 (EP2), prostaglandin E4 (EP4), and prostacyclin (IP) receptors, also act via a Gs-mediated increase in cAMP. Low level of calcium in the extracellular space and in the JGA cells exerts an intensifying effect on the release of renin, through the phenomenon known as the “calcium paradox” [12]. It has been postulated that the inhibitory effect of elevated calcium level is caused by an inhibition of renin gene transcription and destabilization of renin mRNA [13]. Nitric oxide (NO) and atrial natriuretic peptides are involved in the stimulation of renin release through a cGMP-dependent pathway [14, 15]. The release of renin is inhibited by an increase in the intraglomerular hydrostatic pressure and this effect depends on the stimulation of the adenosine A1 receptors. Experimental and clinical studies have provided evidence for the inhibition of renin secretion by a high sodium intake [16, 17]. Besides, Ang II inhibits renin release directly in a short negative feedback loop by means of the AT1 receptors (AT1R), and indirectly through a suppression of prostanoid synthesis induced by cyclooxygenase-2 (COX-2) [18•]. The role of AVP in the regulation of renin release is more complex as AVP stimulates the release by acting on the V2 receptors (V2R) in the JGA cells. However, it can also inhibit the release of renin when it acts on the V1a receptors (V1aR) in the macula densa cells [19]. In hypertensive concentrations, both Ang II and AVP exert indirect inhibitory effects, secondary to the increases in the systemic and intraglomerular blood pressure. Acting centrally, Ang II and AVP may both either stimulate or inhibit the release of renin via activation of specific groups of cardiovascular neurons in the brain (see below). Aldosterone and glucocorticoids stimulate the synthesis of renin in the JGA cells, however, both of these steroids can also exert several indirect effects resulting from their actions on sodium and the water balance and synthesis of other regulatory compounds. These actions are particularly important in salt-sensitive hypertension [20•].
Cardiovascular RAS
Renin, Agt, ACE, and Ang II receptors are all present in the myocardium. Angiotensinogen is present mainly in the cardiac atria and fibers of the conductive system. The strongest expression of ACE was found in the coronary endothelial cells and cardiac fibroblasts, and weaker expression of ACE was found in the aorta, pulmonary arteries, valves, endocardium, and epicardium [21,22,23]. Angiotensin I and Ang II are synthesized de novo in the heart and their synthesis is regulated by glucocorticoids, estrogen, and thyroid hormones [24]. In the rat, endothelial cell ACE gene expression is stimulated by aldosterone [25]. Stimulation of beta receptors upregulates renin and Agt expression in the heart and mechanical stretching of the ventricular myocytes increases the release of Ang II [21, 26].
Brain RAS
All components of the RAS have been detected in the brain, in particular, in the brain cardiovascular regions, including the paraventricular nucleus, the medullary cardiovascular regions, and the circumventricular organs. There is strong evidence that the intrabrain RAS plays a significant regulatory role in cardiovascular regulation and is significantly affected in cardiovascular diseases [27,28,29,30]. For example, it has been shown that the intracerebroventricular (ICV) administration of renin antisense into the brain reduces blood pressure in spontaneously hypertensive rats (SHR) (see also below) [31, 32]. It has also been shown that hyperosmotic stimulation increases the release of Ang II in the paraventricular nucleus [33] which suggests that it may play a role under physiological conditions.
Adrenal Gland RAS
The components of the RAS have been identified in the adrenal medullary chromaffin cells [34]. Significant renin activity was detected in the zona glomerulosa of the adrenal cortex and smaller expressions were present in the zona fasciculata and reticularis [35]. Sodium restriction or a high potassium diet increased renin concentration and aldosterone synthesis in the adrenal gland [36]. It was also shown that stimulation of aldosterone secretion by potassium or adrenocorticotropic hormone (ACTH) involves activation of ACE and AT1R [37].
Cellular Effects of Angiotensins
AT1 Receptors
The human angiotensin AT1 receptor gene (at1r) has been mapped to chromosome 3 [38]. The AT1R protein is composed of 359 amino acids and belongs to G protein-coupled receptors (GPCR) [39, 40]. After interaction with Ang II, the AT1Rs are internalized but approximately 25% of the internalized receptors recycle back to plasma membrane. The remaining AT1R serve their intracellular functions or are degraded in lysosomes [41]. The ACE→Ang II→AT1R axis forms the vasoconstrictor/proliferation/profibrotic wing of the RAS. Angiotensin AT1Rs are widely distributed in the heart and vessels, kidney, brain, lungs, endocrine glands, and several other organs [42, 43]. Stimulation of AT1Rs by Ang II involves activation of a broad scope of intracellular pathways, which may have different roles in specific groups of cells [44]. With respect to the regulation of the cardiovascular system and Ang II and AVP interactions, the most essential is the activation of phospholipase C (PLC) and phospholipase D (PLD) with subsequent formation of inositol triphosphate (IP3) and diacylglycerol (DAG). Increased production of IP3 results in the release of calcium from intracellular stores and subsequent activation of Ca2+-dependent myosin light-chain kinase (MLCK) [45]. Activation of PLD causes hydrolysis of phosphatidylcholine and production of phosphatidic acid, which can be transformed into diacylglycerol, an activator of conventional and novel classes of protein kinase of type C (PKC) [46•].
Activation of AT1R also results in the stimulation of phospholipase A2 (PLA2) and the production of arachidonic acid. Metabolism of arachidonic acid by cyclooxygenases, lipoxygenases and cytochrome P450 enzymes results in the formation of eicosanoids which regulate the contractility of the vascular smooth muscle cells (VSMC) [47]. The main signaling pathways activated by AT1R stimulation involve nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, metalloproteinases (MMP), and the peroxisome proliferator-activated receptors (PPAR). The stimulation of AT1R results in the activation of NADPH oxidase and the subsequent generation of reactive oxygen species (ROS). This promotes the formation of several molecules engaged in inflammatory and profibrotic processes. Among them are mitogen-activated protein kinases [p38 MAPK, c-Jun N-terminal kinases (JNK), extracellular signal-regulated protein kinases 1 and 2 (ERK1/2), extracellular-signal-regulated kinase 5 (ERK5)] and transcription factors [nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), activator protein 1 (AP-1), hypoxia-inducible factor 1-alpha (HIF-1α)]. There is also evidence that the stimulation of AT1R is associated with increased activity of Na+,K+-ATPase (NKA). The activation of AT1R also results in the downregulation of PPAR, which acts as an anti-inflammatory link [46•, 48, 49]. The activation of ERK1/2, p38MAPK, and c-Jun kinase pathways mediates the migratory and pro-proliferative actions of AT1R whereas an increase of interstitial matrix MMP activity and reduced production of tissue inhibitor of MMP-1 contribute to increased collagen accumulation. To some extent, the activation of ERK1/2 and p38 depends on the transactivation of epidermal growth factor receptor (EGFR) and the stimulation of proteins of the Src family of tyrosine kinase by a mechanism involving MMP [50]. The process of transactivation critically depends upon agonist binding by β-arrestin2 [51]. AT1R can also be activated by mechanical stretching through the mechanism independent of Ang II [52, 53•].
Homologous regulation of AT1 receptors by Ang II appears to depend on the type of cells. In a culture of vascular smooth muscle cells, Ang II downregulated AT1R mRNA and protein expression [54], whereas Ang II increased the expression of AT1R in neurons and the effect was associated with elevated expressions of NF-κB and Ets-like protein (Elk-1) [55••]. In renal mesangial cells, stimulation of AT1R by Ang II stimulated stress activated protein kinase (SAPK) through the mechanism of engaging stimulation of pertussis toxin-sensitive and pertussis toxin-insensitive G proteins and activation of tyrosine kinase [56]. Significant upregulation of AT1R mRNA was found in the rat subfornical organs (SFO) and anterior pituitary after sustained (5 days) dehydration [57]. In healthy human subjects, prolonged (8 days) blockade of AT1R increased PRA and Ang II concentrations, elevated urinary sodium excretion, decreased plasma aldosterone concentration, and reduced filtration fraction (FF) [58].
There is substantial evidence that AT1Rs are overexpressed in cardiovascular diseases. Expression of AT1R is elevated in SHR arterial vessels, in the brain and hearts of rats with 2K-1C hypertension, and in various brain regions [paraventricular nucleus (PVN), rostral ventrolateral medulla (RVLM), SFO] of hypertensive or myocardially infarcted rats and of rats exposed to oxidative stress or drastic acute and mild chronic stress [29, 59,60,61,62,63,64,65]. There is evidence that AT1Rs are upregulated by aldosterone, endothelin, and tumor necrosis factor alpha (TNF-α) [61, 66] and downregulated by Ang-(1-7), NO, high-density lipoprotein (HDL) cholesterol, and stimulation of AT2R [67,68,69]. In neuronal cultures prepared from the brains of SHR and Wistar Kyoto (WKY) rats, the expression of AT1R was significantly greater in SHR than in WKY and in both strains it was upregulated by elevated sodium concentration [70]. Several studies have provided evidence that the upregulation of AT1R or other compounds of the RAS may have functional consequences in cardiovascular pathology. For example, overactivation of AT1R in the RVLM plays an important role in the overstimulation of the sympathetic activity in stroke prone SHR [71•]. A significant reduction of blood pressure and a decreased release of Ang II in the hypothalamus was found in chronic 2K-1C hypertensive rats after antisense inhibition of the synthesis of Agt in the brain [72], and deficiency in Agt expression in the brain of Agt(−) transgenic rats was associated with reduced sympathoexcitation and pressor responses to ouabain [73]. Moreover, intracardiac pretreatment with AT1R antisense prevented the development of hypertension and cardiac hypertrophy and reduced cardiovascular contractility in SHR exposed to chronic infusion of Ang II [74]. Oral administration of AT1R antagonists reduced hypertension and cerebral edema in stroke-prone SHR, and oral administration of captopril decreased the resting blood pressure and cardiovascular responses to stress in myocardially infarcted rats [75•, 76, 77]. Furthermore, the Macaca fascicularis monkey maintained on a high cholesterol diet responded to prolonged administration of losartan with a significant reduction of the size of atherosclerotic fatty streaks in the aorta, coronary arteries, and carotid artery [78]. Similarly, blockade of AT1Rs with telmisartan reduced the size of the atherosclerotic damage and superoxide production in apolipoprotein E (ApoE)-deficient mice [79].
AT2 Receptors
The gene for AT2 receptor (AT2R) is located on the X chromosome [80]. The AT2R protein belongs to a family of G protein-coupled proteins and has a low homology of amino acid sequence (~ 34%) with AT1R [81]. Stimulation of AT2R activates phosphotyrosine phosphatases, especially serine/threonine phosphatase 2A, protein kinase phosphatase, and SHP-1 tyrosine phosphatase. This is associated with an inactivation of MAPK (specifically p42 and p44 MAPK) and ERK [82]. The most prominent expression of AT2R was found in the kidney, heart, blood vessels, and brain, especially in the soma and dendrites of the PVN [83•]. During cold-restraint stress, expression of AT2R in the brain increases after blockade of AT1R [84]. In the kidney, the AT2Rs are present in the glomerular epithelial cells, cortical tubules, and interstitial cells [85]. In the heart, AT2Rs are present in the atrial and ventricular myocardium and in the vascular smooth muscle cells of the coronary arteries [86]. Expression of AT2R is upregulated by sodium depletion and by insulin and insulin-like growth factor 1 (IGF-1). It is inhibited by Ang II and growth factors such as platelet-derived growth factor (PDGF) and epidermal growth factor (EGF) [85, 87, 88].
In the cardiovascular system, activation of AT2R exerts opposite effects to those following stimulation of AT1R. After blockade of AT1R, administration of Ang II may result in hypotension, partly mediated by an increased production of BK, NO, and cGMP [89]. In addition, stimulation of AT2R exerts antiproliferative and proapoptotic effects on smooth muscle cells, and reduces expression of AT1R and transforming growth factor beta (TGF-β) receptors [90]. In the heart, stimulation of the AT2Rs inhibits growth and remodeling and induces coronary vasodilation [91]. The role of AT2R increases under pathological conditions. They play a buffering role by preventing cardiac hypertrophy and fibrosis during the administration of Ang II and participate in cardiac remodeling during post myocardial infarction. It is also postulated that they act nephroprotectively in chronic kidney diseases [92].
Angiotensin-(1-7) Receptors
Ang-(1-7) is the main non-classical RAS peptide, which exerts actions via the G protein-coupled receptor Mas [93]. In many aspects, stimulation of Mas receptors opposes the negative effects of stimulation of AT1R and has similar functional consequences as the stimulation of AT2R. It has been found that stimulation of the ACE2→Ang-(1-7)→Mas receptor axis exerts significant vasodepressor and antihypertensive actions. These actions can be unmasked after the inhibition of ACE or AT1R. Ang-(1-7) can also act indirectly through the stimulation of the release of prostaglandins (PG) and NO and via interaction with bradykinin [94, 95]. Ang-(1-7) inhibits the growth of cardiomyocytes and smooth muscle cells, reduces the activity of MAP kinase, inhibits the generation of ROS, reduces Ang II-induced stimulation of ERK1/2 and Rho kinases, causes the downregulation of AT1Rs, and attenuates the unfavorable structural and functional consequences of post-infarct heart failure and cerebral ischemia [67, 93, 96,97,98]. In the kidney, Ang-(1-7) counteracts the stimulatory effect of Ang II on Na+,K+-ATPase and promotes water transport in the rat inner medullary collecting duct. The latter effect can be abolished both by Mas and V2R antagonists, which suggests involvement of AVP in this process [99, 100]. Stimulation of Mas receptors reduces symptoms of experimentally evoked glomerulosclerosis, namely, decreases expression of TGF-β, plasminogen activator inhibitor-1 (PAI-1), fibronectin (FN), and collagen I [101].
Current evidence indicates that Ang-(1-7) acting in the brain may exert both depressor and pressor effects depending on the locus of action. Presumably, some of the pressor effects are mediated by AT1 receptors or by AVP, which is released by Ang-(1-7) [102,103,104]. It was found that administration of Ang-(1-7), either ICV or into the nucleus of the solitary tract (NTS), induces hypotensive and bradycardic effects and facilitates baroreflex bradycardia, whereas the intrabrain administration of Ang-(1-7) antibody in transgenic hypertensive rats induces tachycardia and hypertension [105, 106•]. Moreover, selective overexpression of ACE 2 in the brain prevented development of neurogenic hypertension in mice through improvement of spontaneous baroreflex sensitivity and parasympathetic activity [107•]. In contrast, blockade of Mas receptors in the PVN resulted in a reduction of the renal sympathetic nerve activity (RSNA), which may suggest involvement of Ang-(1-7) in tonic maintenance of RSNA by the PVN [108]. Administration of Ang-(1-7) antagonist accelerated and intensified development of hypertension in the two-kidney, one-clip Goldblatt model [109•] and Mas knockout mice manifested reduced diuresis and natriuresis, and this was associated with glomerular hyperfiltration and increase of collagen III and fibronectin content in the mesangium and interstitium. They also showed upregulation of AT1R and TGF-β mRNA expression [110]. Altogether, the above results indicate that Ang-(1-7) may protect the cardiovascular system to some extent from destructive changes induced by Ang II.
Other Angiotensin Receptors
Blood pressure and renal functions are also regulated by Ang III (Ang 4-8) and Ang IV (Ang 3-8) [111]. Angiotensin III is formed from Ang II via aminopeptidase A and acts via stimulation of the AT1 receptors [111]. Angiotensin IV has its own receptor—the transmembrane enzyme, insulin-regulated aminopeptidase (IRAP, AT4R), which is present in the brain, heart, blood vessels, and kidney. Some of the actions of Ang IV can be mediated by AT1R [112]. Expression of AT4R in the carotid body is significantly elevated by hypoxia [113].
Role of the Renin-Angiotensin System in Cardiovascular Regulation
The renin-angiotensin system regulates blood pressure through actions exerted either directly on the cardiovascular system or indirectly by means of the autonomic nervous system and vasoactive endocrine factors affecting blood circulation and the water-electrolyte balance.
Direct Vascular Effects of Angiotensins
Several studies have shown that systemic and locally released Ang II plays a key role in the regulation of vascular tone and that overactivation of the RAS causes excessive vasoconstriction and vascular remodeling, fibrosis, and atherosclerosis of the vascular wall resulting from endothelial dysfunction, as well as migratory, growth promoting, proliferative, apoptotic, and inflammatory processes [49, 114]. The vasoconstriction is caused by activation of AT1R conjugated to Gq/11 and G12/13 classes of G proteins [115, 116]. The vasoconstrictory effect of Ang II can only be partly reduced by the intracellular effect of Ang II on the potassium current (Kv), which is associated with hyperpolarization of smooth muscle cells [117]. In the vessels, heart, and kidney, Ang II promotes proinflammatory processes through induction of NF-κB and upregulation of cytokines and chemokines [interleukin 6 (IL-6), TNF-α, monocyte chemoattractant protein 1 (MCP-1)] which is followed by an increased expression of P-selectin, E-selectin, intercellular cell adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) [118, 119]. Increased shear stress, which enhances ACE and basic fibroblast growth factor (bFGF) expression in the vascular smooth muscle cells, may additionally promote the negative effects of RAS overactivation [120].
Cardiovascular diseases are associated with inappropriate activation of the RAS in the vascular wall. For example, SHRs show greater expression of AT1R and lower expression of AT2R in mesangial vascular cells than WKY rats. Furthermore, ACE 2 gene transfer into SHR results in reduced perivascular fibrosis and blockade of AT1R inhibits Ang II-induced NF-κB activation and VCAM-1 accumulation in smooth muscle cells [119, 121].
Direct Cardiac Effects of Angiotensins
Angiotensin II, its precursors and cleavages are produced in the heart and distributed heterogeneously in the cardiac walls, endocardium, epicardium, conductive system, valves, aorta, pulmonary arteries, and coronary vessels [21, 23, 122]. Expression of elements of the RAS is higher in the fetal developing heart and in cardiac pathology than in the healthy heart of adults [23, 122]. Cardiac AT1Rs are coupled with Gαq and use ERK1/2 and phosphatidylinositol 3-kinase (PI3K) pathways and protein kinase B (Akt) phosphorylation as the main transduction cascades in Ang II-mediated cell proliferation. By stimulating AT1R, Ang II may also interact with Gαi, and may regulate adenylyl cyclase and the unconventional arrestin beta 2 pathway as well as heterotrimeric signaling processes [123]. Systemic administration of Ang II evokes positive chronotropic and inotropic effects, modifies cardiac metabolism, and induces vasoconstriction of coronary blood vessels [124]. Exposure of cardiac fibroblasts to elevated Ang II concentration or overexpression of AT1R increases expression of focal adhesion kinases (FAK) and integrins in cultured neonatal cells, and elevates expression of c-fos, EGFR1, TGF-β, and extracellular matrix proteins in the cardiomyocytes of adults [125•, 126]. The above effects can be abolished by PKC inhibitors or EGF receptor kinase antagonists [127]. Renin and other components of the RAS play a role in the regulation of cell volume and chemical communication in the heart, for example, renin in cooperation with Agt impairs permeability of gap junctions [128•]. Moreover, it has been shown that Ang II, acting in the heart, increases the generation of hydroxyl radicals and impairs cell-to-cell communication, impulse propagation, and conduction velocity [129•, 130•]. Mechanical stretch stimulates secretion of Ang II from myocytes and increases Agt gene expression in cardiac myocytes and fibroblasts [21]. It is suggested that the cardiac AT1R functions as a mechanosensor and that the pressure overload may induce cardiac remodeling through mechanic activation of AT1R, independently of Ang II [53•]. Rats with hypertension induced by coarctation of the abdominal aorta show significant overexpression of AT1R mRNA and TGF-β mRNA, which are associated with cardiac hypertrophy. The latter two effects could be abolished by blockade of AT1Rs [131]. Studies on rats or mice with deletion or overexpression of various components of the RAS and on SHR provide additional arguments for the negative effects of the overexpression of Ang II in hypertension and post-infarct heart failure [132, 133]. In the samples of the left ventricle obtained from patients with post-infarct heart failure, the expression of AT1R was markedly elevated in patients with acute myocardial infarction and reduced in those with an old infarction or dilated cardiomyopathy. Expression of AT2R in the failing hearts was upregulated and this effect was particularly well seen in the fibrous regions of the myocardium [134].
Central Cardiovascular Effects of Angiotensins
The first evidence that Ang II induces a central pressor effect appeared over 50 years ago [135] and was confirmed in multiple studies [136, 137•, 138]. The pressor response may be evoked by intracerebroventricular (ICV) administration of Ang II or Ang III and by local applications of Ang II into the SFO, PVN, lateral septal area (LSA), bed nucleus of the stria terminalis (BNST), RVLM, and NTS [139•, 140, 141, 142••]. The central pressor action of Ang II is mediated by the sympathoadrenal system and by AVP acting on V1 receptors [137•, 142••, 143, 144, 145•]. At the cellular level, the central pressor effect of Ang II is mediated by AT1R and is associated with activation of the Rho/Rho-kinase pathway, increased production of O*-generated by NADPH oxidase, enhanced stimulation of P38 MAPK and ERK1/2, and decreased SK current in the PVN neurons projecting to the RVLM [146,147,148]. Local application of Ang II decreases the frequency of GABAergic miniature postsynaptic inhibitory currents in the PVN and it is likely that Ang II excites PVN neurons projecting to the brain stem and spinal cord via an inhibition of GABAergic input to the PVN. As the inhibitory effect of Ang II could be abolished by pertussis toxin and superoxide dismutase, it has been postulated that the inhibition is mediated by the pertussis toxin-sensitive G(i/o) protein and superoxide anions [149]. Acting in the PVN, Ang II potentiates also the cardiac sympathetic afferent reflex [150]. It is likely that sympathoexcitation induced by stimulation of AT1R may be partly reduced by parallel stimulation of AT2R as it has been shown that ICV infusion of Compound 21, a non-peptide AT2R agonist, decreases norepinephrine secretion and blood pressure via the NO signaling pathway in the PVN [151]. Moreover, ICV Ang II injections in AT2R gene knockout mice resulted in higher blood pressure elevations than in wild-type mice [152]. Resetting of AT1R and AT2R activation has been demonstrated in rats with chronic heart failure (CHF) as they manifest significantly higher pressor, tachycardic and sympathoexcitatory responses to stimulation of AT1R in the RVLM. They also show markedly reduced hypotensive, bradycardic, and sympathoinhibitory responses to activation of RVLM AT2R than the sham-operated individuals [153].
Regulation of the Autonomic Nervous System and Cardiovascular Reflexes by Angiotensins
Numerous studies have provided evidence that Ang II is an essential modulator of the transmission of signals in the sympathetic and parasympathetic portions of the autonomic nervous system (ANS) and that local changes of Ang II concentration in the ganglia may play an essential role in the final adjustment of the ANS activity to specific physiological/pathological conditions. Dysregulation of the cardiovascular system by the ANS with enhanced activity of the sympathetic nervous system (SNS) and reduced parasympathetic nervous system (PNS) tone is among the characteristic symptoms of hypertension and cardiac failure [154, 155].
At the supraspinal level, Ang II stimulates the presympathetic neurons of the PVN and pacemaker cells of the RVLM. It also depolarizes the preoptic neurons and increases resting K+ conductance in the bulbospinal neurons of the C1 area. There is a positive feedback loop between Ang II and SNS, i.e., Ang II stimulates the presympathetic neurons of the PVN and RVLM and facilitates sympathoadrenal transmission [156], whereas sympathetic stimulation enhances the release of renin in the kidney [157]. Circulating Ang II may increase presympathetic stimulation in the central nervous system (CNS) through binding to AT1R in certain autonomic regions, which have no blood-brain barrier, such as the area postrema (AP) and SFO [158, 159].
Numerous studies have provided evidence for the involvement of Ang II and AT1R in the regulation of the peripheral sympathetic system [160]. Angiotensin-immunoreactive neurons are present in the sympathetic ganglia and Ang II facilitates the transmission of signals from the preganglionic to the postganglionic neurons, enhances the release of norepinephrine (NE) during ganglionic excitation, and promotes the release of NE from the sympathetic termini [161, 162]. Abundant specific AT1 receptor binding sites were found at the somatodendritic membrane of the superior cervical ganglion neurons [163]. In the aortic, renal, and celiac ganglia, Ang II was able to trigger an increase in [Ca2+]i through an effect dependent on the inflow of extracellular calcium via specific N-type and L-type calcium channels and activation of PKC [160, 164, 165]. Acting on AT1R in the superior cervical ganglion, Ang II modulates also M type K+ channels and intensifies the IKv current through an ATP-dependent effect, which is mediated by PKC [165, 166].
Ang II receptor binding sites have been found along the vagal components subserving the baroreceptor reflex, including the nodose ganglion, NTS, and the dorsal motor nucleus of the vagus (DVMNc) [167]. Impaired efferent vagal tone was found in rats with myocardial infarction and the impairment could be attenuated by the administration of losartan [168]. Reduced parasympathetic tone is also found in human patients with chronic heart failure and can be improved by ACE inhibitor therapy [169, 170]. AT1 receptors are expressed at multiple sites of the baroreceptor reflex pathway including the sensory afferent neurons and their terminals in the NTS [171•]. It has been shown that denervation of baroreceptors significantly increases pressor responses to the central application of Ang II, which argues for an essential role of baroreceptors in buffering the central pressor effect of this peptide [172]. However, during administration of Ang II, the buffering role of baroreceptors is not as effective as in the case of other vasoconstrictory substances and this is related to the resetting of the baroreflex by Ang II. Namely, during administration of Ang II, the baroreflex relationship between blood pressure and heart rate was shifted to a higher pressure level [173]. Ang II tonically modulates cardiovascular regulation through an effect on neurons forming the baroreflex circuitry in the NTS, RVLM, and caudal ventrolateral medulla (CVLM) [174•, 175,176,177]. Enhanced stimulation of AT1R in the CVLM is presumably responsible for the inhibition of the cardiac vagal baroreflex gain during neurogenic stress [178], whereas AT1R located in the NTS may participate in the transmission of exaggerated excitation from the cardiac sympathetic afferents and in the potentiation of the chemoreceptor reflex during hypoxia [179]. There is also evidence that overactivation of central AT1R accounts for the desensitization of the baroreflex function, elevated sympathoexcitation, and decreased sympathetic inhibition after myocardial infarction in rats and rabbits [180, 181].
In contrast, stimulation of AT2R potentiates baroreceptor reflex sensitivity, though this effect is normally masked by the stimulation of AT1R [182]. Inhibition of ACE 2 in the NTS significantly reduces the reflex heart rate control [183], whereas microinjections of Ang-(1-7) and the transfer of the ACE 2 gene into the NTS of the SHR, as well as microinjections of Ang-(1-7) into the CVLM of rats with renovascular hypertension significantly improve the baroreflex control of the heart rate [184, 185]. On the other hand, administration of Ang-(1-7) into the RVLM of rats with renovascular hypertension appears to exert the opposite effect, as it has been found that it enhances the cardiac sympathetic afferent reflex and increases the sympathetic outflow and blood pressure via Mas receptor activation and stimulation of the cAMP-protein kinase A (PKA) pathway [186].
Interestingly, in the majority of sensory neurons of the nodose ganglion, Ang II inhibited [Ca 2+]i in conotoxin-sensitive calcium channels. However, the facilitation of the calcium current by Ang II, involving activation of a reserve pool of dihydropyridine sensitive channels, could be also observed after blockade of conotoxin-sensitive channels [187].
Interaction of Ang II with Vasopressin and Aldosterone
The presence of the (pro)renin receptor, AT1R and AT2R mRNAs in the PVN and supraoptic nucleus (SON) and their co-localization with AVP in the magnocellular neurons strongly suggests that the RAS plays a significant role in the regulation of the brain vasopressinergic system [188]. Indeed, several studies have shown that ICV infusion or local application of various constituents of the RAS into the PVN and SON elicits the release of AVP [105, 189, 190]. There is also evidence that overactivation of the RAS may play a significant role in the increased release of AVP in cardiovascular pathology. For example, blockade of AT1R or ACE was found to enhance central and peripheral release of AVP in experimental left ventricular hypertrophy induced by aortic banding. Moreover, experiments on transgenic rats (TG) carrying the mouse renin gene showed a significantly greater content of AVP in the dorsal lower brain stem and an elevated intrabrain release of AVP in response to Ang II application [105]. Angiotensin II plays a significant role in the regulation of the synthesis and release of aldosterone from the adrenal gland [191]. Moreover, there is cross-talk between AT1Rs and mineralocorticoid receptors (MR) in the cardiovascular system so that the stimulation of AT1R increases the effectiveness of the action of aldosterone [192]. Mineralocorticoids increase the expression of Ang II receptors in the arterial smooth muscle cells whereas blockade of AT1R reduced the pro-oxidant effect of aldosterone on the arterial wall [193, 194]. In VSMC, aldosterone potentiated Ang II-induced ERK-1/2 and JNK phosphorylation and increased NADPH oxidase activity. The latter effect was particularly prominent in the mouse model of atherosclerosis [195].
The Renin-Angiotensin System and the Water-Electrolyte Balance
There is substantial evidence that various components of the RAS, and especially Ang II, considerably contribute to the regulation of fluid balance through parallel stimulation of sodium and water intake and sodium and water retention. Central or peripheral infusions of renin or angiotensins stimulate sodium ingestion [196, 197]. The circulating Ang II stimulates salt appetite by an action exerted at the forebrain circumventricular organs (CVO) [198]. The stimulation is mediated by AT1R and AT2R and is potentiated by aldosterone [197, 199].
Renin, Ang II, and Ang III are potent dipsogens and avid stimulation of thirst belongs to the most spectacular effects of these peptides. Several studies indicate that the dipsogenic action of Ang II engages AT1R in the median preoptic nucleus and lateral septal area [200,201,202,203]. The dipsogenic effect is adjusted to the actual blood volume and hemodynamic conditions and can be partly damped by the parallel inhibition of thirst by an enhanced input from baroreceptors and cardiopulmonary receptors. These mechanisms are partly abolished in hypertension and heart failure [197, 202, 204].
Angiotensin AT1Rs are widely distributed in the kidney [205]. As discussed in several studies, stimulation of the RAS causes retention of sodium and water and this results both from the direct renal effects of Ang II and from the stimulation of aldosterone secretion [206,207,208]. Acting directly in the kidney via AT1R, Ang II constricts the smooth muscle cells of afferent and efferent arterioles and increases sodium absorption in the proximal tubule. This is associated with a reduction of the renal cortical and papillary blood flows, impairment of tubuloglomerular feedback sensitivity, and resetting of the pressure natriuresis relationship [44, 209]. Removal of one or several of the genes of the RAS results in significant disturbances of the renal functions, manifested by polyuria and inefficient urine concentration, and in abnormalities of the renal structure [210, 211]. Overactivation of the RAS associated with increased activity of aldosterone, vasopressin, and TGF-β are considered to be key pathogenic factors in chronic kidney diseases [206, 212•].
Vasopressin
Organization of the Vasopressinergic System
Arginine vasopressin, the principal vasopressin peptide in mammals, is synthesized mainly in the neuroendocrine neurons of the supraoptic, paraventricular, and suprachiasmatic nuclei of the hypothalamus. Vasopressin expressing cells have also been identified in the BNST, medial amygdaloid nucleus, dorsomedial hypothalamic nucleus (DMH), nucleus of the diagonal band of Broca (DBB), and the olfactory bulb [213,214,215]. The magnocellular neurons (MCN) of the SON and the PVN send axons to the capillary vessels of the posterior pituitary where they release their products into the systemic circulation. Circulating AVP is involved in the regulation of the water-electrolyte balance, blood pressure, and metabolism. Some of the neurons send axons to the median eminence and release AVP into the hypophyseal portal system and form a link regulating the secretion of ACTH and other pituitary hormones. The parvocellular neurons of the PVN, DBB, BNST, and suprachiasmatic nucleus (SCN) send axons to multiple regions of the CNS located at virtually all levels of the brain, including the brain cortex, brain stem, CVO, and the spinal cord [216, 217, 218•, 219]. The CNS vasopressinergic neurons participate in the regulation of cardiorespiratory functions, the water-electrolyte balance, circadian rhythmicity, food intake, metabolism, pain, neurogenic stress, emotions, social behavior, and immunological processes. There is evidence for a dendritic release of AVP and its role in the regulation of local neuronal activity [220]. Some evidence indicates that synthesis of AVP occurs in the heart, adrenal medulla, and pancreas [221,222,223].
Regulation of the Release of Vasopressin. Role of Angiotensins and Mineralocorticoids
The release of vasopressin is significantly influenced by changes in plasma osmolality, blood volume, and blood pressure. At the cellular level, osmotic stimulation of AVP release involves the activation of transient receptor potential cation channel subfamily V member (TRPV1 and TRPV4) channels [224, 225]. Hypothalamic AVP synthesizing neurons receive inputs from multiple neurotransmitting and neuromodulating neurons releasing catecholamines, serotonin, acetylcholine, histamine, angiotensins, neuropeptide Y, orexins, substance P, glutamate, and ATP. Several cardiovascular compounds, including Ang II, have receptors in the PVN and SON and stimulate the release of AVP [212•, 226,227,228]. The inhibition of AVP release occurs after administration of GABA, NO, natriuretic peptides, apelin, and relaxin-3 [229,230,231,232]. As discussed above, Ang II is a potent stimulator of AVP release. In the context of angiotensinergic-vasopressinergic interactions, special attention should be given to the role of GABA, serotonin, and steroid hormones. In most studies performed on normotensive animals, stimulation of the GABAA receptors resulted in the inhibition of vasopressinergic neurons and was mediated by Cl− influx via Na+,K+,2Cl− cotransporter (NKCC1) channels [233]; however, an excitatory effect of GABAA on AVP secreting neurons was also observed [234] and it has been suggested that the inhibitory input of GABA innervation may be replaced by an excitatory drive under some pathological circumstances, for example, in DOCA-salt hypertension [235]. At present, it seems that the release of AVP can be adjusted to specific conditions by means of different neuroactive modulators. For example, different types of serotonin (5-HT2A, 5-HT2C, 5-HT3, and 5-HT4), Ang II, and cytokine receptors are engaged in vasopressin release during various types of stress [30, 212•, 236].
The role of steroid hormones in the regulation of AVP secretion has been explored in detail. Specific 11β-HSD2 immunoreactivity and mineralocorticoid receptors were identified in the cytoplasm of the SON and PVN neurons [237]. Colocalization of MR with the membrane conductance regulating epithelial sodium channels (ENaC) was found in the MCN neurons immunoreactive for AVP. Interestingly, both normotensive and SHR rats respond with the upregulation of AVP mRNA and V1a receptors to the administration of mineralocorticoids, however, SHR rats show greater expression of AVP, V1aR, and MR than WKY rats [238•]. Moreover, abundant expression of MR and α, β, and γ subunits of ENaC are present in the cardiovascular brain regions innervated by vasopressinergic neurons [239, 240].
Cellular Effects of Vasopressin
Arginine vasopressin acts via three primary types of receptors (V1aR, V1bR, and V2R) which belong to the family of monomeric G protein receptors (GPCR) [241, 242].
V1 Receptors
Vasopressin V1aR have been detected in the brain, heart, vessels, lungs, kidney, liver, pituitary gland, adrenals, uterus, and in other peripheral organs. In the nervous system, V1aR mRNA and protein were identified at virtually all levels of the brain and spinal cord [212•, 243•, 244•, 245•]. There is also functional evidence for the presence of V1aR in the glial cells of the brain and dorsal ganglia. These cells responded to the application of AVP with an increase of intracellular Ca2+ concentration, which could be abolished by V1aR and PLC antagonists [246]. In the heart, V1aR were detected in the cardiac muscle and in the coronary vessels. In the kidney, they were found in vessels (interlobular arteries, efferent arterioles, vasa recta, glomerular mesangium) and in the JGA, the macula densa, thick ascending limb of the loop of Henle, the distal convoluted tubule, and the principal and intercalated cells of the collecting ducts [247, 248, 249•].
Distribution of V1bR is less widespread than of V1aR; however, they have been detected in the brain, the pituitary gland, and the pancreas [212•, 250•, 251–252].
Stimulation of V1R activates intracellular pathways engaging Gq/11/PLC/IP3/PKC/MEK1/2/MAPKK, PI3-kinase, calcium-calmodulin kinase, and adenylyl cyclase of type III. The receptors are coupled to phospholipase C via a G protein of the Gq/11 type and their activation results in generation of inositol 1,4,5-triphosphate (IP3). In smooth muscle cells, IP3 interacts with IP3 receptors (IP3R) of the sarcoplasmic reticulum (SR), and the subsequent release of calcium results in stimulation of the ryanodine receptors (RyR). In cells expressing adenylyl cyclase of type III, stimulation of V1R may also potentiate cAMP accumulation, induced primarily by exposure to isoproterenol or prostaglandin I2 (PGI2) [253•]. More recently, it has been shown that AVP significantly modulates the ionic transport of vascular smooth muscle cells. In picomolar concentrations, it elicits calcium transients that are associated with activation of protein kinase C (PKC) and the inflow of calcium through voltage-sensitive L-type channels (VSCC) [254•, 255•, 256•]. In A7r5 vascular smooth muscle cells, application of 100 pm of AVP increases the frequency of calcium spikes through an effect associated with proline-rich tyrosine kinase 2 (PYK2), tyrosine kinase phosphorylation and activation of PKC. Vasopressin also regulates the potassium current engaging the delayed rectifier K+ channels, and this effect depends on phosphorylation of the Kv1.2 channel protein and inhibition of the voltage-dependent potassium current (IKv), which is associated with gradual membrane depolarization and the generation of action potentials [256•]. In A-10 rat aortic smooth muscle cells, AVP stimulates sodium-dependent phosphate transport through a mechanism involving activation of V1R and multiple V1R-dependent signaling pathways, such as PKC, PI3-kinase, S6 kinase, and Jun kinase [257•].
In some forms of hypertension, there is an overactivation of the vasopressinergic system. A higher number of AVP mRNA expressing cells and V1aR-immunopositive cells has been found in the PVN of the SHR than in the PVN of WKY rats. In the SHR, both these parameters were significantly elevated after the administration of DOCA, whereas WKY rats responded only with an increase in the number of V1aR-positive cells [238•]. On the other hand, a decreased expression of V1aR mRNA in the brain medulla and an increased expression of V1bR mRNA in the mesencephalon-pontine region were found in hypertensive renin transgenic TGR(mRen2)27 rats in comparison with the parent normotensive SD strain. The TGR(mRen2)27 rats manifested also a higher expression of V1aR in the renal medulla than SD rats [244•, 250•]. Significant changes in expression of V1aR in the brain and kidneys were also found in rats with renovascular hypertension, in post-infarct heart failure, and in rats exposed to chronic stress [258•, 259••].
V2 Receptors
Vasopressin V2R are expressed mainly in the kidney where they are located on the basolateral membrane of the thick ascending limb (TAL), the distal convoluted tubule, and the collecting ducts [249•, 260•]. The stimulation of V2R causes activation of adenylyl cyclase and formation of cAMP. This is followed by an activation of protein kinase A, and a subsequent phosphorylation of aquaporin 2 (AQP2). The phosphorylated AQP2 is subsequently incorporated into the luminal membrane of the cell and serves as a water channel. Vasopressin also regulates the transcription of the AQP2 gene [261•, 262•, 263•].
Dimers and Dual Receptors
Studies on cell cultures have shown that AVP receptors can form various variants of homo- (V1aR/V1aR, V1bR/V1bR, V2R/V2R) and heterodimers (OTR/V1R, V1aR/V2R, V2R/OTR, V1bR/CRH). It is likely that dimerization plays a role in the process of intracellular internalization of the receptors and may determine the effectiveness of the cellular action of AVP. Thus far, there is no evidence for the dimerization of vasopressin and angiotensin receptors. This issue was approached in the study of Szalai et al. [264] who were unable to prove the formation of heterodimers of vasopressin V2R with angiotensin AT1R or AT2R.
A dual Ang II/AVP receptor coupled to adenylate cyclase has also been characterized and its immunocytochemical distribution in the kidney has been determined. The immunopharmacological characteristics of this receptor indicated that it may have functional properties of the AT1R and V2R. It has been suggested that it may play an essential role in sodium and fluid resorption in the inner medullary collecting ducts [265•].
Role of Vasopressin in Cardiovascular Regulation
It has long been known that moderate elevation of AVP in the systemic circulation increases arterial blood pressure, central venous pressure, and central blood volume, and resets the reflex regulation of blood pressure by baroreceptors [266•, 267•]. Similarly as Ang II, AVP participates in the regulation of blood pressure via multiple actions exerted either directly on vascular smooth muscle cells and cardiac myocytes or indirectly through the regulation of the water-electrolyte balance and blood volume. Besides, AVP stimulates the pre-autonomic neurons in the brain as well as modulates the synaptic transmission in the sympathetic ganglia [268•].
Direct Vascular Effects of Vasopressin
Vasopressin is one of the most potent vasoconstrictors, although responsiveness of various vascular beds to its contractile action significantly differs. The most sensitive vessels to vasopressin are those of the skin, muscle, and splanchnic circulation [269•, 270•]. The vasoconstrictory effect of AVP is mediated by V1aR. Vasopressin elicits contractile and growth-promoting effects in vascular smooth muscle cells, increases smooth muscle actin and SM22 promoter activities, and stimulates the JNK and p38 MAPK pathways [271•]. Moreover, experiments on ring segments of gastroepiploic arteries obtained from human patients have shown that AVP is able to restore the contractile response to Ang II after tachyphylaxis [270•]. The vasoconstrictory potency of AVP may be significantly altered under pathological conditions [272•].
The interaction of Ang II, AVP, and aldosterone in the regulation of cellular sodium content in the rat tail arteries was approached by Friedman [273•], who showed that exposures to aldosterone, Ang II, and AVP were associated with a significant reduction of free cell Na+ and that the effects of Ang II and AVP were additive with the effect of aldosterone.
Direct Cardiac Effects of Vasopressin
Vasopressin stimulates IP3 accumulation in the atrial and ventricular cardiomyocytes [274•]. Current evidence indicates that V1aR is the main type of AVP receptors in the adult heart although it appears that, in newborns, V2R are also present and may play an essential role in differentiation of the embryonic stem cells into contracting cells which display cardiac specific transcription factor GATA-4 and ventricle-specific myosin light chain [275•]. Studies in vivo have shown that transfer of the V2R gene into the myocardium increases cardiac contractility [276•]. It has been suggested that by affecting cardiac contractility, AVP may contribute to adjustment of newborns to the hypoxic conditions during posthypoxic reoxygenation [277•].
There is evidence that excessive stimulation of V1aR promotes pathological changes in the heart. It has been shown that it causes cardiac hypertrophy and fibrosis through increased synthesis of collagen and TGF-β1, and enhanced proliferation and differentiation of fibroblasts. Exposure of the cultured neonatal cardiomyocytes for 24 h to AVP resulted in an increase of the cell surface areas, elevation of expression of the atrial natriuretic peptide (ANP), and activation of ERK1/2 via the V1aR-dependent effect. It was also shown that V1aR-deficient mice did not respond with cardiac hypertrophy to pressure overload [278•]. The role of AVP in the regulation of the cardiac functions partly depends on its interaction with NO. Vasopressin increases inducible nitric oxide synthase (iNOS) expression and NO production, and activates NF-κB. Moreover, blockade of NO synthase partly eliminated the effects of i.v. infusion of AVP in adult pigs [279•]. The profibrotic effect of AVP in the heart of the adult pig can be inhibited by stimulation of Mas receptors by Ang-(1-7). In AVP-stimulated cardiac fibroblasts, Ang-(1-7) was found to reduce activity of calcineurin and to inhibit nuclear translocation of NF-κB and its effect on transcriptional activity [280•, 281•].
Experimental and clinical studies have provided evidence that blood AVP concentration is significantly elevated in hypertension and in patients with post-infarct cardiac failure [282••, 283•]. Moreover, experimental studies revealed that, in post-myocardial infarction, cardiac dysfunction is associated with increased cardiac and plasma AVP and Ang II expressions, and that AVP expression is positively correlated with left ventricular end diastolic diameter (LVEDD) [284]. The same authors have shown that, under in vitro conditions, cardiac microvascular endothelial cells respond with increased expression of AVP mRNA and protein to the application of Ang II. It has also been shown that AVP promotes proliferation of those cells. Prolonged exposure of neonatal rat cardiac fibroblasts to elevated AVP levels promoted their proliferation and induced increased expression of matrix metalloproteinases (MMP2 and MMP9). These effects were mediated by the stimulation of V1aR and activation of G protein-coupled receptor kinase 2 (GRK2) that is followed by phosphorylation of ERK1/2 [285].
Effects of prolonged exposure of the heart to stimulation of V1aR were explored in the study of Li X et al. [286••], who found that mice with cardiac-targeted overexpression of V1aR develop cardiac hypertrophy associated with reduced cardiac contractility and dysfunction of isolated cardiomyocytes. The latter effect was manifested by reduced amplitude and elongated duration of action potentials of the myocytes, and this was associated with a reduction in [Ca2+]i. Insulin-mediated ERK1/2 phosphorylation was enhanced whereas Akt-induced and JNK-induced phosphorylations were not altered. The cardiac effects of overexpression of V1aR were reduced by inhibition of the Gαq/11 transduction pathway [286••].
Direct Renal Effects of Vasopressin
Stimulation of V1aR by AVP is involved in the regulation of glomerular filtration and renal circulation. Acting on V1aR, AVP enhances the calcium influx into the smooth muscle cells of the preglomerular arterioles, constricts the afferent and efferent arterioles, and contracts the mesangial cells [287•, 288•, 289•]. In the glomerular mesangial cells of the cultured rat, AVP stimulated mitosis and growth of the cells and increased the intracellular calcium content. The mitotic response to AVP was stimulated by insulin. Acidification followed by alkalinization was shown after exposure of the mesangial cells either to AVP or to Ang II and the alkalinization depended on the activation of the Na+,H+ exchange [290•, 291•]. It has been proven that the inner medullary region of the kidney circulation is more sensitive to V1aR stimulation than the cortical vessels and that AVP can regulate the renal papillary blood flow in physiological concentrations [292•, 293•, 294•].
Central Cardiovascular Effects of Vasopressin
Centrally projecting vasopressinergic neurons innervate several groups of neurons engaged in cardiovascular control such as the NTS, RVLM, DVMNc, PBN, amygdala, BNST, periaqueductal gray matter, striatum, and the prefrontal cortex [212•, 218•, 219•, 295•]. There is significant evidence that centrally acting AVP may evoke both pressor and hypotensive responses [137•, 145•, 296•, 297•] and that the final effect depends on the stimulation of specific groups of vasopressinergic fibers innervating special sets of cardiovascular neurons and activated by specific stimuli.
Vasopressin in the Regulation of the Autonomic Nervous System and Cardiovascular Reflexes
Similarly as the RAS, the vasopressinergic system closely cooperates with the autonomic nervous system by means of multifarious interactions. In the brain, the presympathetic neurons of PVN and RVLM are innervated by vasopressinergic fibers and are furnished with V1a receptors. Moreover, the vasopressinergic fibers project to the spinal cord [212•, 218•, 219•, 243•, 245•]. AVP-expressing neurons furnished with synaptic-like contacts were also identified in the dorsal vagal complex, specifically in the DVMNc, and in the NTS subnuclei. In the A2/C2 and A2 catecholaminergic neurons and in the nucleus ambiguus, AVP positive fibers make contacts with tyrosine hydroxylase (TH) positive neurons [295•]. Vasopressinergic neurons and V1R are present in the RVLM and the rostral ventral respiratory column (RVLC) and expression of V1aR in this region increases during hypoxia whereas blockade of V1aR there reduces pressor responses to hypoxia [298••].
Studies using viral transneuronal labeling and immunohistochemistry revealed that the PVN is the source of vasopressinergic and angiotensinergic neurons projecting to the stellate ganglion, which is the main source of sympathetic innervation of the heart [299••]. Furthermore, experiments on sympathoadrenal preganglionic neurons have provided evidence that some of the PVN neurons have terminal varicosities closely associated with SPNs and that vasopressin fibers are likely to form synaptic contacts with SPN [268•]. Vasopressin-like peptide and V1R were identified in the sympathetic ganglia, and application of AVP has been shown to stimulate inositol lipid breakdown in the ganglia [300••, 301••]. The functional significance of the presence of AVP in the peripheral autonomic nervous system is not yet fully clear. Iontophoresis of AVP on antidromically identified sympathetic preganglionic neurons in the second thoracic (T) segment of the spinal cord results in the inhibition of the majority of neurons, however, some of the neurons are excited [302••]. ICV administration of AVP causes c-fos induction in adrenergic and noradrenergic cells of the adrenal medulla, which may argue that centrally released vasopressin has an impact on the activity of the preganglionic sympathetic neurons innervating the catecholaminergic neurons of the adrenal glands [303••]. Peripheral administration of AVP significantly potentiated the vasoconstrictory effect of lumbar sympathetic nerve stimulation and of norepinephrine administered into the isolated perfused hind limb of the rabbit through an effect mediated by V1R [304••].
Several studies have provided evidence that AVP plays an essential role in the potentiation of the baroreflex, although there are interspecies differences in magnitude of this effect [266•, 305••, 306••, 307••]. Most of the studies have shown that AVP facilitates the baroreflex by means of V1aR located in the area postrema and the NTS [308••, 309••, 310••].
Vasopressin and the Water-Electrolyte Balance
Vasopressin participates in the regulation of the water-electrolyte balance through the regulation of water and electrolyte transport in the renal tubules and through the control of sodium and water intake. These effects are exerted either by means of its independent action or through cross-talks with Ang II and aldosterone.
Reabsorption of water in the collecting ducts requires activation of V2R and is the most prominent effect of AVP in the kidney. The effectiveness of this process is determined by the availability of AVP and active V2R; however, it also depends on the effectiveness of the transport of electrolytes and the regulation of renal blood flow. In the collecting ducts, AVP binds to V2R on the basolateral membrane of the principal cells and this results in the immediate activation of adenylyl cyclase and formation of cAMP. Subsequent stimulation of PKA results in the phosphorylation of AVP-sensitive water channels (AQP2) and their translocation to the apical tubular membrane. Apical insertion of AQP2 is coupled to calcium mobilization from ryanodine-gated intracellular calcium stores and to extracellular Ca2+ import via the capacitative calcium influx [311••]. Prolonged AVP-induced intracellular cAMP elevation also increases AQP2 gene transcription and AQP2 abundance [312••].
In the principal cells of the collecting ducts (mpkCCD, c14 cell model), AVP stimulates transepithelial Na+ transport and increases sodium absorption by means of active NKA and passive Na+,K+,2Cl− (NKCC2) transports. Activity of NKA is constitutively regulated by the ERK1/2 pathway (p42 and p44 mitogen-activated protein kinase pathways). It is suggested that the basal activity of the ERK1/2 pathway plays a permissive role in the activation of Na,K-ATPase. Vasopressin, like Ang II and aldosterone, induces the translocation of NKA from the cytoplasm to the plasma membrane and facilitates its activation [313••, 314••]. In the thick ascending limb of the loop of Henle, AVP enhances passive sodium transport by means of NKCC2 cotransport [315••]. In the collecting ducts, it facilitates permeability to urea and plays an essential role in the regulation of the epithelial sodium transport mediated by ENaC. The latter effect involves interaction of AVP with aldosterone [316•, 317•].
Interaction of AVP with Aldosterone
Vasopressin stimulates the release of aldosterone from the dispersed adrenal gland cells through an effect mediated by V1aR [318•]. The presence of AVP is also necessary for the effective action of aldosterone in the renal tubules. Acting on V2R in the collecting ducts, AVP increases the catalytic activity of 11β-HSD2, which causes cortisol to be converted into inactive cortisone. As a result, MR receptors can be stimulated more selectively by aldosterone [319•]. AVP is also necessary for full activation of the ENaC channels by aldosterone in the distal part of the nephron. The excessive activation of these channels during prolonged stimulation of MR results in sodium retention and the elevation of blood pressure while the inhibition of action of aldosterone causes a decrease in ENaC activity and sodium loss [320•]. There is evidence that, in some instances, AVP can partly compensate the inefficient action of aldosterone in the collecting ducts. This is particularly essential in adrenocortical insufficiency, during which the nonosmotic stimulation of AVP release is elevated because of body fluid depletion and the lack of inhibition of AVP release by glucocorticoids [273•, 321•]. Experiments on adrenalectomized mice, with an aldosterone concentration below the lower limit of quantification, revealed that those mice manifest a thriving expression of the ENaC channels, and that those channels can be stimulated by AVP acting on V2 receptors [322•, 323•]. Experiments on cells of the cortical collecting duct (CCD) have shown that AVP and aldosterone act synergistically to promote sodium reabsorption acting both at the apical and the basolateral membranes. In the epithelial membrane, AVP is necessary for full sensitivity of ENaC to aldosterone while, in the basolateral membrane, aldosterone and AVP cooperatively recruit NKA from the intracellular stores and promote their translocation to the membrane. It has been shown that aldosterone produces delayed increases of mRNAs encoding alpha subunits of the ENaC (sixfold), and of alpha1 subunits of NKA, whereas AVP induces rapid translocation of NKA into the basolateral membrane [324•]. It has been shown that vasopressin, similar to aldosterone, promotes the synthesis of α- and γ-ENaC subunits [325•].
Experiments using molecular engineering indicate that V1aRs are engaged in the regulation of the acid-base balance via aldosterone. Thus, mice deprived of V1aR developed type 4 renal tubular acidosis and manifested a reduced expression of MR and 11β-HSD2 in the kidney medulla. In addition, transgenic rats expressing MR but deprived of V2R did not respond with an increased expression of H+,K+-ATPase, H+-ATPase, and C glycoprotein of the Rh family to the administration of aldosterone after an additional knockdown of the V1aR gene. Moreover, it was shown that, in V1aR(+/+) mice, AVP is able to increase the expressions of protein kinase C alpha (PKCα) and protein kinase C beta1 (PKCβ1), whereas aldosterone elevates the expressions of protein kinase C delta (PKCδ) and protein kinase C zeta (PKC-ζ). These effects are abolished by the knockdown of V1aR. These studies indicate that, in the renal tubules, V1aR cooperates with aldosterone in a complex manner, which has a significant impact on sodium reabsorption and may include regulation of MR translocation from the cytoplasm to the nucleus [326•, 327•].
Significance of Central and Peripheral Interactions of the RAS and the VPS
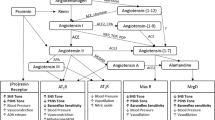
The above survey indicates that, in many instances, there are multiple interactions of angiotensin II and vasopressin and that the close cooperation of these two peptides is a condition of the fully efficient regulation of blood pressure and body fluid volume. As shown in Fig. 1, Ang II and AVP interact reciprocally at several levels through (1) mutual regulation of their own release, (2) targeting the same groups of cells, and (3) engaging the same intracellular pathways.

Interactions of the renin-angiotensin system (RAS) with the vasopressinergic system (VPS) in the regulation of blood pressure and body fluid volume. RAS and VPS closely cooperate in adjusting blood pressure to cardiovascular challenges. The cooperation takes place in the cardiovascular regions of the brain, in the cardiovascular and the sympathoadrenal systems, and in the kidney. Multiple synergistic and/or antagonistic actions of angiotensin peptides and vasopressin, as well as positive and negative feedbacks between RAS and VPS are involved in the regulation of cardiovascular functions. The figure shows that dysregulated interaction of RAS and VPS in the brain and in the peripheral tissues results in excessive stimulation of angiotensin AT1 receptors (AT1R), and vasopressin V1a (V1aR) and V2 (V2R) receptors, and in the development of hypertension and/or body fluid retention. AT2R, angiotensin AT2 receptors; AVP, arginine vasopressin; DVMNc/Nc Amb, complex of the dorsoventromedial nucleus of the vagus and the nucleus ambiguous; MasR, Mas receptor of angiotensin- (1-7); Post Pit, the posterior pituitary; PVN, the paraventricular nucleus of the hypothalamus; RVLM, the rostral ventrolateral medulla of the brain; UNaV, sodium excretion
Interactive Regulation of the RAS and the VPS
Angiotensin II is a potent stimulator of AVP release. The mechanism underlying this effect has been partly clarified as it has been shown that, in the PVN, Ang II interacts with nonselective cation channel TRPV4. Application of Ang II to the immortalized neuroendocrine rat hypothalamic 4B cell line (the line expressing transcripts of AVP, AT1R, AT1bR, and TRPV4) significantly elevates the expression of TRPV4 and this effect can be abolished by the blockade of AT1R with losartan. Acting on AT1R, Ang II also potentiated the calcium influx evoked by a selective TRPV4 agonist (GSK 1016790A) [328•]. Thus, it is likely that the release of AVP by Ang II in the PVN neurons involves the activation of TRPV4. In this line, it is interesting to note that rats with liver cirrhosis, manifesting symptoms of the syndrome of inappropriate antidiuretic hormone secretion (SIADH), show overexpression of the TRPV4 channels in the hypothalamic SON neurons immunopositive for AVP [329•].
There is also evidence for the regulation of the RAS by vasopressin. Acting directly on the macula densa cells, AVP stimulates the release of renin and can interact thereby with the RAS in a positive feedback loop. However, this action has a limit, because in high concentrations both Ang II and AVP elevate blood volume and blood pressure, which may in turn result in the stimulation of cardiopulmonary receptors and baroreceptors and in the inhibition of the release of renin and AVP (Fig. 1). The consequences of chronic deficiency of V1aR receptors for RAS were assessed in the study by Aoyagi et al. [330•], which was performed on a model of V1aR−/− mice. The mice manifested reduced concentrations of renin and Ang II in plasma, a decreased expression of renin in the renal granule cells, and lowered expressions of nNOS and COX-2 in the macula densa cells [330•]. The study has provided evidence that the stimulation of V1aR by AVP is necessary to maintain the appropriate release of renin from the granule cells of the kidney and that this action may be associated with altered activities of nNOS and COX-2.
Cooperative Regulation of the Cardiovascular System by Angiotensins and Vasopressin
Several studies have provided evidence that the joint action of Ang II and AVP is necessary for the appropriate regulation of the vascular tone, cardiac contractility, and the release of other cardiovascular factors (see above and Fig. 1). The cooperative action of Ang II and AVP in the regulation of blood pressure is particularly well observed at the level of the CNS and plays a role both in the maintenance of resting blood pressure and in adjustments to posthemorrhagic hypovolemia, hypoxia, cardiorespiratory disorders, and stress. Several studies have provided evidence that the central pressor action of Ang II can be abolished or significantly reduced by blockade of the brain V1aR and that the combined administration of Ang II and AVP exerts greater changes of blood pressure than those changes observed after the separate application of these peptides. Importantly, the interaction of Ang II and AVP is significantly enhanced in hypertension, post-infarct cardiac failure, and during chronic stress [137•, 145•, 331•, 332•, 333•, 334•]. The elevated release of AVP plays a major role in the development of hypertension in mice with brain-specific hyperactivity of the renin and angiotensinogen genes as it has been shown that the baseline blood pressure can be normalized in this strain by the administration of the nonselective AVP receptor antagonist conivaptan or the V2-selective antagonist tolvaptan [335•]. Electrophysiological studies have shown that AVP and Ang II increase the activity of the same rostrodiencephalic neurons and coactivate neurons in the SFO and organum vasculosum of the lamina terminalis (OVLT) [336•, 337•]. The biochemical background of this interaction is partly clarified by studies showing that Ang II and AVP may jointly regulate calcium transits. Studies on a culture of the area postrema/NTS cells have shown that Ang II and AVP evoke a transient increase in [Ca2+] and that this effect can be abolished by pretreatment with specific antagonists of AT1R or V1R [338•, 339•]. In in vivo experiments, blockade of central V1aR effectively abolished the central pressor action of Ang II [137•, 145•].
Although, under most circumstances, Ang II and AVP cooperate synergistically with the sympathetic nervous system, in some instances, they can act antagonistically. The most evident example of such counteraction is the inhibition of the baroreceptor reflex by Ang II and potentiation of this reflex by vasopressin [171•, 174•, 340•]. There is also evidence that Ang II and AVP play different roles in the regulation of respiration during acute hypercapnia. Namely, it has been shown that Ang II acting on AT1R stimulates ventilation and increases the metabolic rate during hypercapnia and that this effect is reduced by the simultaneous stimulation of V1aR by vasopressin [341•].
Cooperative Regulation of the Water and Electrolyte Balance by Angiotensin and Vasopressin
Deletion of V1aR results in a decrease of renin expression in the JGA cells and in a reduced release of renin and Ang II into the systemic circulation [330•]. There are also interactions between renin, AVP, and Ang II in the renal tubules. Renin is synthesized in the principal cells of the collecting duct (CD) and vasopressin stimulates this process by means of V2R and cAMP/PKA/CREB (cAMP response element-binding protein) pathway [342•]. Furthermore, exposure of the inner medulla collecting duct cells to Ang II increases V2R mRNA in these cells in a dose-dependent manner. This effect is stimulated by PKA and suppressed by PKC [343•]. Blockade of AT1R with losartan was shown to reduce the effect of AVP in the renal tubules [344•]. Specifically, a decrease of AVP-mediated cAMP accumulation in the thick ascending limb of the loop of Henle and restoration of a normal NKCC2 expression was found in rats with myocardial infarction [344•].
Some evidence indicates that chronic stimulation of ATR in the kidney may alter the responsiveness of the renal tubules to vasopressin and that stimulation of AT1R potentiates the effect of dDAVP on AQP2 plasma membrane targeting [345•, 346•]. This effect may play a significant role under some pathological circumstances. For example, rats exposed to severe sodium restriction show elevated concentration of Ang II and symptoms of acute renal failure associated with AVP-resistant urinary concentrating defects. These defects are associated with decreased expressions of AQP1, AQP2, AQP3, Na+/H+ exchanger, Na+,Cl− cotransporter, and Na+,K+-ATPase. The above abnormalities can be corrected by the administration of a V2 agonist (dDAVP). The authors showed that the beneficial effect of dDAVP crucially depends on cooperation of AT1R with V2R as blockade of AT1R with candesartan completely abolished the dDAVP urine concentrating effect. In a less severe NaCl-restriction rat model, blockade of AT1R increased the fractional excretion of sodium and decreased urine osmolality. It also reduced expression of AQP2 and AQP3 and downregulated phosphorylated AQP2 (p-AQP2) in the inner renal medulla. In vitro experiments performed in the same study revealed that both Ang II and dDAVP increase AQP2 and cAMP expressions [347•].
Conclusions and Perspectives
Altogether, precise cooperation of Ang II and AVP is necessary for the efficient regulation of blood pressure and body fluid volume. In many instances, the effects of both hormones are significantly modulated by the simultaneous action of Ang-(1-7), mineralocorticosteroids, and other cardiovascular factors. Close cooperation of the RAS and VPS with aldosterone inclines to regard the renin-angiotensin-aldosterone system and the vasopressinergic system as a functional unity. Under physiological conditions, cooperation of Ang II and AVP plays a significant role in the maintenance of cardiovascular homeostasis. Excessive stimulation of the RAS and the VPS may have prolonged negative consequences for the cardiovascular system, resulting from the formation of multiple positive feedbacks between renin, angiotensin II, vasopressin, and aldosterone.
Dysregulation of the interactions between the RAS and vasopressin under pathological conditions may significantly contribute to the generation of disturbances in blood pressure regulation and the development of cardiovascular diseases. A rationalized combined treatment with inhibitors of the RAS system and vasopressin antagonists appears to be a promising tool in the therapy of various forms of hypertension and cardiac failure, especially in patients manifesting evident overactivity of the sympathetic component of the autonomic nervous system or excessive stimulation of mineralocorticoid receptors.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Sequeira Lopez ML, Pentz ES, Nomasa T, Smithies O, Gomez RA. Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Dev Cell. 2004;6(5):719–28.
Hobart PM, Fogliano M, O'Connor BA, Schaefer IM, Chirgwin JM. Human renin gene: structure and sequence analysis. Proc Natl Acad Sci U S A. 1984;81(16):5026–30.
Morris BJ. Renin, genes, microRNAs, and renal mechanisms involved in hypertension. Hypertension. 2015;65(5):956–62.
Kuoppala A, Lindstedt KA, Saarinen J, Kovanen PT, Kokkonen JO. Inactivation of bradykinin by angiotensin-converting enzyme and by carboxypeptidase N in human plasma. Am J Physiol Heart Circ Physiol. 2000;278(4):H1069–74.
Soubrier F, Wei L, Hubert C, Clauser E, Alhenc-Gelas F, Corvol P. Molecular biology of the angiotensin I converting enzyme: II. Structure-function. Gene polymorphism and clinical implications. J Hypertens. 1993;11(6):599–604.
Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87(5):E1–9.
Gomez RA, Lynch KR, Chevalier RL, Wilfong N, Everett A, Carey RM, et al. Renin and angiotensinogen gene expression in maturing rat kidney. Am J Physiol Renal Physiol. 1988;254(4):F582–7.
Ingelfinger JR, Zuo WM, Fon EA, Ellison KE, Dzau VJ. In situ hybridization evidence for angiotensinogen messenger RNA in the rat proximal tubule. An hypothesis for the intrarenal renin angiotensin system. J Clin Invest. 1990;85(2):417.
Leyssac PP. Changes in single nephron renin release are mediated by tubular fluid flow rate. Kidney Int. 1986;30(3):332–9.
Prieto-Carrasquero MC, Botros FT, Kobori H, Navar LG. Collecting duct renin: a major player in angiotensin II–dependent hypertension. J Am Soc Hypertens. 2009;3(2):96–104.
Friis UG, Jensen BL, Sethi S, Andreasen D, Hansen PB, Skøtt O. Control of renin secretion from rat juxtaglomerular cells by cAMP-specific phosphodiesterases. Circ Res. 2002;90(9):996–1003.
Beierwaltes WH. The role of calcium in the regulation of renin secretion. Am J Physiol Renal Physiol. 2010;298(1):F1–11.
Klar J, Sigl M, Obermayer B, Schweda F, Krämer BK, Kurtz A. Calcium inhibits renin gene expression by transcriptional and posttranscriptional mechanisms. Hypertension. 2005;46(6):1340–6.
Demerath T, Staffel J, Schreiber A, Valletta D, Schweda F. Natriuretic peptides buffer renin-dependent hypertension. Am J Physiol Renal Physiol. 2014;306(12):F1489–98. https://doi.org/10.1152/ajprenal.00668.2013.
Schricker K, Kurtz A. Liberators of NO exert a dual effect on renin secretion from isolated mouse renal juxtaglomerular cells. Am J Physiol Renal Physiol. 1993;265(2):F180–6.
Isaksson G, Stubbe J, Lyngs Hansen P, Jensen B, Bie P. Salt sensitivity of renin secretion, glomerular filtration rate and blood pressure in conscious Sprague-Dawley rats. Acta Physiol. 2014;210(2):446–54.
Schweda F, Friis U, Wagner C, Skott O, Kurtz A. Renin release. Physiology (Bethesda). 2007;22:310–9. https://doi.org/10.1152/physiol.00024.2007.
• Quadri SS, Culver SA, Li C, Siragy HM. Interaction of the renin angiotensin and cox systems in the kidney. Front Biosci (Schol Ed). 2016;8:215.
Aoyagi T, Izumi Y, Hiroyama M, Matsuzaki T, Yasuoka Y, Sanbe A, et al. Vasopressin regulates the renin-angiotensin-aldosterone system via V1a receptors in macula densa cells. Am J Physiol Renal Physiol. 2008;295(1):F100–7. https://doi.org/10.1152/ajprenal.00088.2008.
• Fujita T. Mechanism of salt-sensitive hypertension: focus on adrenal and sympathetic nervous systems. J Am Soc Nephrol. 2014;25(6):1148–55.
Dostal DE, Baker KM. The cardiac renin-angiotensin system. Conceptual, or a regulator of cardiac function? Circ Res. 1999;85(7):643–50. https://doi.org/10.1161/01.res.85.7.643.
Lazartigues E, Feng Y, Lavoie JL. The two fACEs of the tissue renin-angiotensin systems: implication in cardiovascular diseases. Curr Pharm Des. 2007;13(12):1231–45.
Sawa H, Tokuchi F, Mochizuki N, Endo Y, Furuta Y, Shinohara T, et al. Expression of the angiotensinogen gene and localization of its protein in the human heart. Circulation. 1992;86(1):138–46.
Dell'Italia LJ, Meng QC, Balcells E, Wei CC, Palmer R, Hageman GR, et al. Compartmentalization of angiotensin II generation in the dog heart. Evidence for independent mechanisms in intravascular and interstitial spaces. J Clin Invest. 1997;100(2):253–8.
Sugiyama T, Yoshimoto T, Tsuchiya K, Gochou N, Hirono Y, Tateno T, et al. Aldosterone induces angiotensin converting enzyme gene expression via a JAK2-dependent pathway in rat endothelial cells. Endocrinology. 2005;146(9):3900–6.
Sadoshima J-I, Xu Y, Slayter HS, Izumo S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75(5):977–84.
Bader M, Ganten D. Editorial: it’s renin in the brain. Circ Res. 2002;90:8–10.
Bader M, Ganten D. Update on tissue renin–angiotensin systems. J Mol Med. 2008;86(6):615.
Tan J, Wang H, Leenen FH. Increases in brain and cardiac AT 1 receptor and ACE densities after myocardial infarct in rats. Am J Physiol Heart Circ Physiol. 2004;286(5):H1665–71.
Szczepanska-Sadowska E, Cudnoch-Jedrzejewska A, Ufnal M, Zera T. Brain and cardiovascular diseases: common neurogenic background of cardiovascular, metabolic and inflammatory diseases. J Physiol Pharmacol. 2010;61(5):509–21.
Cousin C, Bracquart D, Contrepas A, Nguyen G. Potential role of the (pro) renin receptor in cardiovascular and kidney diseases. J Nephrol. 2010;23(5):508.
Kubo T, Ikezawa A, Kambe T, Hagiwara Y, Fukumori R. Renin antisense injected intraventricularly decreases blood pressure in spontaneously hypertensive rats. Brain Res Bull. 2001;56(1):23–8.
Qadri F, Edling O, Wolf A, Gohlke P, Culman J, Unger T. Release of angiostensin in the paraventricular nucleus in response to hyperosmotic stimulation in conscious rats: a microdialysis study. Brain Res. 1994;637(1):45–9.
Wang JM, Slembrouck D, Tan J, Arckens L, Leenen FH, Courtoy PJ, et al. Presence of cellular renin-angiotensin system in chromaffin cells of bovine adrenal medulla. Am J Physiol Heart Circ Physiol. 2002;283(5):H1811–8.
Mulrow PJ, Franco-Saenz R. The adrenal renin-angiotensin system: a local hormonal regulator of aldosterone production. J Hypertens. 1996;14(2):173–6.
Doi Y, Atarashi K, Franco-Saenz R, Mulrow PJ. Effect of changes in sodium or potassium balance, and nephrectomy, on adrenal renin and aldosterone concentrations. Hypertension. 1984;6(2 Pt 2):I124–9.
Gupta P, Franco-Saenz R, Mulrow PJ. Locally generated angiotensin II in the adrenal gland regulates basal, corticotropin-, and potassium-stimulated aldosterone secretion. Hypertension. 1995;25(3):443–8.
Griendling KK, Murphy T, Alexander RW. Molecular biology of the renin-angiotensin system. Circulation. 1993;87(6):1816–28.
Ishizaka N, Alexander RW, Laursen JB, Kai H, Fukui T, Oppermann M, et al. G protein-coupled receptor kinase 5 in cultured vascular smooth muscle cells and rat aorta regulation by angiotensin II and hypertension. J Biol Chem. 1997;272(51):32482–8.
Shenoy SK, Lefkowitz RJ. Angiotensin II-stimulated signaling through G proteins and β-arrestin. Sci Signal. 2005;2005(311):cm14-cm.
Griendling K, Delafontaine P, Rittenhouse S, Gimbrone M, Alexander R. Correlation of receptor sequestration with sustained diacylglycerol accumulation in angiotensin II-stimulated cultured vascular smooth muscle cells. J Biol Chem. 1987;262(30):14555–62.
Bottari SP, de Gasparo M, Steckelings UM, Levens NR. Angiotensin II receptor subtypes: characterization, signalling mechanisms, and possible physiological implications. Front Neuroendocrinol. 1993;14(2):123–71. https://doi.org/10.1006/frne.1993.1005.
Karamyan VT, Speth RC. Distribution of the non-AT1, non-AT2 angiotensin-binding site in the rat brain: preliminary characterization. Neuroendocrinology. 2008;88(4):256–65.
Timmermans PB, Wong PC, Chiu AT, Herblin WF, Benfield P, Carini D, et al. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol Rev. 1993;45(2):205–51.
Shin H-M, Je H-D, Gallant C, Tao TC, Hartshorne DJ, Ito M, et al. Differential association and localization of myosin phosphatase subunits during agonist-induced signal transduction in smooth muscle. Circ Res. 2002;90(5):546–53.
• Touyz R, Schiffrin E. Reactive oxygen species in vascular biology: implications in hypertension. Histochem Cell Biol. 2004;122(4):339–52.
Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52(4):639–72.
Brand S, Amann K, Mandel P, Zimnol A, Schupp N. Oxidative DNA damage in kidneys and heart of hypertensive mice is prevented by blocking angiotensin II and aldosterone receptors. PLoS One. 2014;9(12):e115715. https://doi.org/10.1371/journal.pone.0115715.
Schiffrin EL, Touyz RM. From bedside to bench to bedside: role of renin-angiotensin-aldosterone system in remodeling of resistance arteries in hypertension. Am J Physiol Heart Circ Physiol. 2004;287(2):H435–46.
Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402(6764):884–8. https://doi.org/10.1038/47260.
Kim J, Ahn S, Rajagopal K, Lefkowitz RJ. Independent beta-arrestin2 and Gq/protein kinase Czeta pathways for ERK stimulated by angiotensin type 1A receptors in vascular smooth muscle cells converge on transactivation of the epidermal growth factor receptor. J Biol Chem. 2009;284(18):11953–62. https://doi.org/10.1074/jbc.M808176200.
Mederos y Schnitzler M, Storch U, Meibers S, Nurwakagari P, Breit A, Essin K, et al. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 2008;27(23):3092–103. https://doi.org/10.1038/emboj.2008.233.
• Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, et al. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004;6(6):499.
Lassegue B, Alexander RW, Nickenig G, Clark M, Murphy T, Griendling KK. Angiotensin II down-regulates the vascular smooth muscle AT1 receptor by transcriptional and post-transcriptional mechanisms: evidence for homologous and heterologous regulation. Mol Pharmacol. 1995;48(4):601–9.
•• Mitra AK, Gao L, Zucker IH. Angiotensin II-induced upregulation of AT 1 receptor expression: sequential activation of NF-κB and Elk-1 in neurons. Am J Physiol Cell Physiol. 2010;299(3):C561–9.
Huwiler A, van Rossum G, Wartmann M, Pfeilschifter J. Angiotensin II stimulation of the stress-activated protein kinases in renal mesangial cells is mediated by the angiotensin AT 1 receptor subtype. Eur J Pharmacol. 1998;343(2):297–302.
Sanvitto GL, Johren O, Hauser W, Saavedra JM. Water deprivation upregulates ANG II AT1 binding and mRNA in rat subfornical organ and anterior pituitary. Am J Physiol Endocrinol Metab. 1997;273(1):E156–63.
Burnier M, Hagman M, Nussberger J, Biollaz J, Armagnac C, Brouard R, et al. Short-term and sustained renal effects of angiotensin II receptor blockade in healthy subjects. Hypertension. 1995;25(4 Pt 1):602–9.
Aguilera G, Kiss A, Luo X. Increased expression of type 1 angiotensin II receptors in the hypothalamic paraventricular nucleus following stress and glucocorticoid administration. J Neuroendocrinol. 1995;7(10):775–83.
Bhatt SR, Lokhandwala MF, Banday AA. Vascular oxidative stress upregulates angiotensin II type I receptors via mechanisms involving nuclear factor kappa B. Clin Exp Hypertens. 2014;36(6):367–73.
Kang Y-M, Wang Y, Yang L-M, Elks C, Cardinale J, Yu X-J, et al. TNF-α in hypothalamic paraventricular nucleus contributes to sympathoexcitation in heart failure by modulating AT1 receptor and neurotransmitters. Tohoku J Exp Med. 2010;222(4):251–63.
Milik E, Szczepanska-Sadowska E, Cudnoch-Jedrzejewska A. Upregulation of angiotensin AT1a receptors mRNA in the heart and renal medulla after myocardial infarction in rats. J Physiol Pharmacol. 2006;57(3):375–88.
Milik E, Cudnoch-Jedrzejewska A, Szczepanska-Sadowska E. Effect of chronic mild stress on AT1 receptor messenger RNA expression in the brain and kidney of rats. Psychosom Med. 2016;78(2):208–20.
Wei S-G, Yu Y, Zhang Z-H, Felder RB. Angiotensin II upregulates hypothalamic AT 1 receptor expression in rats via the mitogen-activated protein kinase pathway. Am J Physiol Heart Circ Physiol. 2009;296(5):H1425–33.
Zucker IH, Schultz HD, Patel KP, Wang W, Gao L. Regulation of central angiotensin type 1 receptors and sympathetic outflow in heart failure. Am J Physiol Heart Circ Physiol. 2009;297(5):H1557–66.
Jesmin S, Sakuma I, Togashi H, Yoshioka M, Hattori Y, Kitabatake A, et al. Effects of endothelin receptor antagonist on expression of AT1 and AT2 receptors in the heart of SHR-SP. J Cardiovasc Pharmacol. 2004;44:S59–63.
Clark MA, Diz DI, Tallant EA. Angiotensin-(1-7) downregulates the angiotensin II type 1 receptor in vascular smooth muscle cells. Hypertension. 2001;37(4):1141–6.
Macova M, Pavel J, Saavedra JM. A peripherally administered, centrally acting angiotensin II AT 2 antagonist selectively increases brain AT 1 receptors and decreases brain tyrosine hydroxylase transcription, pituitary vasopressin and ACTH. Brain Res Bull. 2009;1250:130–40.
Van Linthout S, Spillmann F, Lorenz M, Meloni M, Jacobs F, Egorova M, et al. Vascular-protective effects of high-density lipoprotein include the downregulation of the angiotensin II type 1 receptor. Hypertension. 2009;53(4):682–7.
Feldstein JB, Sumners C, Raizada MK. Sodium increases angiotensin II receptors in neuronal cultures from brains of normotensive and hypertensive rats. Brain Res. 1986;370(2):265–72.
• Kishi T, Hirooka Y, Konno S, Ogawa K, Sunagawa K. Angiotensin II type 1 receptor-activated caspase-3 through ras/mitogen-activated protein kinase/extracellular signal-regulated kinase in the rostral ventrolateral medulla is involved in sympathoexcitation in stroke-prone spontaneously hypertensive rats. Hypertension. 2010;55(2):291–7.
Kagiyama S, Varela A, Phillips MI, Galli SM. Antisense inhibition of brain renin-angiotensin system decreased blood pressure in chronic 2-kidney, 1 clip hypertensive rats. Hypertension. 2001;37(2 Pt 2):371–5.
Huang BS, Ganten D, Leenen FH. Responses to central Na+ and ouabain are attenuated in transgenic rats deficient in brain angiotensinogen. Hypertension. 2001;37(2):683–6.
Pachori AS, Wang H, Gelband CH, Ferrario CM, Katovich MJ, Raizada MK. Inability to induce hypertension in normotensive rat expressing AT1 receptor antisense. Circ Res. 2000;86(11):1167–72.
• Cudnoch-Jedrzejewska A, Czarzasta K, Puchalska L, Dobruch J, Borowik O, Pachucki J, et al. Angiotensin converting enzyme inhibition reduces cardiovascular responses to acute stress in myocardially infarcted and chronically stressed rats. Biomed Res Int. 2014;2014 https://doi.org/10.1155/2014/385082.
Hamaguchi R, Takemori K, Inoue T, Masuno K, Ito H. Short-term treatment of stroke-prone spontaneously hypertensive rats with an at1 receptor blocker protects against hypertensive end-organ damage by prolonged inhibition of the renin–angiotensin system. Clin Exp Pharmacol Physiol. 2008;35(10):1151–5.
Seltzer A, Bregonzio C, Armando I, Baiardi G, Saavedra JM. Oral administration of an AT 1 receptor antagonist prevents the central effects of angiotensin II in spontaneously hypertensive rats. Brain Res. 2004;1028(1):9–18.
Strawn WB, Chappell MC, Dean RH, Kivlighn S, Ferrario CM. Inhibition of early atherogenesis by losartan in monkeys with diet-induced hypercholesterolemia. Circulation. 2000;101(13):1586–93.
Takaya T, Kawashima S, Shinohara M, Yamashita T, Toh R, Sasaki N, et al. Angiotensin II type 1 receptor blocker telmisartan suppresses superoxide production and reduces atherosclerotic lesion formation in apolipoprotein E-deficient mice. Atherosclerosis. 2006;186(2):402–10.
Lazard D, Briend-Sutren M, Villageois P, Mattei M, Strosberg A, Nahmias C. Molecular characterization and chromosome localization of a human angiotensin II AT2 receptor gene highly expressed in fetal tissues. Receptors Channels. 1994;2(4):271–80.
Mukoyama M, Nakajima M, Horiuchi M, Sasamura H, Pratt RE, Dzau VJ. Expression cloning of type 2 angiotensin II receptor reveals a unique class of seven-transmembrane receptors. J Biol Chem. 1993;268(33):24539–42.
Horiuchi M, Akishita M, Dzau VJ. Recent progress in angiotensin II type 2 receptor research in the cardiovascular system. Hypertension. 1999;33(2):613–21.
• Coleman CG, Anrather J, Iadecola C, Pickel VM. Angiotensin II type 2 receptors have a major somatodendritic distribution in vasopressin-containing neurons in the mouse hypothalamic paraventricular nucleus. Neuroscience. 2009;163(1):129–42.
Bregonzio C, Seltzer A, Armando I, Pavel J, Saavedra JM. Angiotensin II AT(1) receptor blockade selectively enhances brain AT(2) receptor expression, and abolishes the cold-restraint stress-induced increase in tyrosine hydroxylase mRNA in the locus coeruleus of spontaneously hypertensive rats. Stress. 2008;11(6):457–66. https://doi.org/10.1080/10253890801892040.
Ozono R, Wang Z-Q, Moore AF, Inagami T, Siragy HM, Carey RM. Expression of the subtype 2 angiotensin (AT2) receptor protein in rat kidney. Hypertension. 1997;30(5):1238–46.
Wang Z-Q, Moore AF, Ozono R, Siragy HM, Carey RM. Immunolocalization of subtype 2 angiotensin II (AT2) receptor protein in rat heart. Hypertension. 1998;32(1):78–83.
Ichiki T, Kambayashi Y, Inagami T. Multiple growth factors modulate mRNA expression of angiotensin II type-2 receptor in R3T3 cells. Circ Res. 1995;77(6):1070–6.
Kambayashi Y, Nagata K, Ichiki T, Inagami T. Insulin and insulin-like growth factors induce expression of angiotensin type-2 receptor in vascular-smooth-muscle cells. FEBS J. 1996;239(3):558–65.
Siragy HM, Inagami T, Carey RM. NO and cGMP mediate angiotensin AT 2 receptor-induced renal renin inhibition in young rats. Am J Physiol Regul Integr Comp Physiol. 2007;293(4):R1461–7.
Su J-Z, Fukuda N, Jin X-Q, Lai Y-M, Suzuki R, Tahira Y, et al. Effect of AT2 receptor on expression of AT1 and TGF-β receptors in VSMCs from SHR. Hypertension. 2002;40(6):853–8.
Ohkubo N, Matsubara H, Nozawa Y, Mori Y, Murasawa S, Kijima K, et al. Angiotensin type 2 receptors are reexpressed by cardiac fibroblasts from failing myopathic hamster hearts and inhibit cell growth and fibrillar collagen metabolism. Circulation. 1997;96(11):3954–62.
Kurisu S, Ozono R, Oshima T, Kambe M, Ishida T, Sugino H, et al. Cardiac angiotensin II type 2 receptor activates the kinin/NO system and inhibits fibrosis. Hypertension. 2003;41(1):99–107.
Santos RA, Ferreira AJ, Simões e Silva AC. Recent advances in the angiotensin-converting enzyme 2-angiotensin (1-7)-Mas axis. Exp Physiol. 2008;93(5):519–27.
Chappell MC. Emerging evidence for a functional angiotensin-converting enzyme 2-angiotensin-(1-7)-Mas receptor axis. Hypertension. 2007;50(4):596–9.
Santos RA, Ferreira AJ, Verano-Braga T, Bader M. Angiotensin-converting enzyme 2, angiotensin-(1–7) and Mas: new players of the renin–angiotensin system. J Endocrinol. 2013;216(2):R1–R17.
Ferreira AJ, Jacoby BA, Araújo CA, Macedo FA, Silva GA, Almeida AP, et al. The nonpeptide angiotensin-(1–7) receptor Mas agonist AVE-0991 attenuates heart failure induced by myocardial infarction. Am J Physiol Heart Circ Physiol. 2007;292(2):H1113–9.
Giani JF, Gironacci MM, Muñoz MC, Turyn D, Dominici FP. Angiotensin-(1–7) has a dual role on growth-promoting signalling pathways in rat heart in vivo by stimulating STAT3 and STAT5a/b phosphorylation and inhibiting angiotensin II-stimulated ERK1/2 and Rho kinase activity. Exp Physiol. 2008;93(5):570–8.
Tallant EA, Ferrario CM, Gallagher PE. Angiotensin-(1–7) inhibits growth of cardiac myocytes through activation of the Mas receptor. Am J Physiol Heart Circ Physiol. 2005;289(4):H1560–6.
Lara LS, Vives D, Correa JS, Cardozo FP, Marques-Fernades MF, Lopes AG, et al. PKA-mediated effect of MAS receptor in counteracting angiotensin II-stimulated renal Na+-ATPase. Arch Biochem Biophys. 2010;496(2):117–22.
Magaldi AJ, Cesar KR, de Araújo M, e Silva ACS, Santos RA. Angiotensin-(1–7) stimulates water transport in rat inner medullary collecting duct: evidence for involvement of vasopressin V2 receptors. Pflugers Arch. 2003;447(2):223–30.
Zhang J, Noble NA, Border WA, Huang Y. Infusion of angiotensin-(1–7) reduces glomerulosclerosis through counteracting angiotensin II in experimental glomerulonephritis. Am J Physiol Renal Physiol. 2010;298(3):F579–88.
Dobruch J, Paczwa P, Lon S, Khosla MC, Szczepanska-Sadowska E. Hypotensive function of the brain angiotensin-(1-7) in Sprague Dawley and renin transgenic rats. J Physiol Pharmacol. 2003;54(3):371–81.
Potts P, Horiuchi J, Coleman M, Dampney R. The cardiovascular effects of angiotensin-(1–7) in the rostral and caudal ventrolateral medulla of the rabbit. Brain Res. 2000;877(1):58–64.
Schiavone MT, Santos R, Brosnihan KB, Khosla MC, Ferrario CM. Release of vasopressin from the rat hypothalamo-neurohypophysial system by angiotensin-(1-7) heptapeptide. Proc Natl Acad Sci. 1988;85(11):4095–8.
Moriguchi A, Ferrario CM, Brosnihan KB, Ganten D, Morris M. Differential regulation of central vasopressin in transgenic rats harboring the mouse Ren-2 gene. Am J Physiol Regul Integr Comp. 1994;267(3):R786–91.
• Zhou L-M, Shi Z, Gao J, Han Y, Yuan N, Gao X-Y, et al. Angiotensin-(1–7) and angiotensin II in the rostral ventrolateral medulla modulate the cardiac sympathetic afferent reflex and sympathetic activity in rats. Pflugers Arch. 2010;459(5):681–8.
• Feng Y, Xia H, Cai Y, Halabi CM, Becker LK, Santos RA, et al. Brain-selective overexpression of human angiotensin-converting enzyme type 2 attenuates neurogenic hypertension. Circ Res. 2010;106(2):373–82.
da Silva AQG, dos Santos RAS, Fontes MAP. Blockade of endogenous angiotensin-(1–7) in the hypothalamic paraventricular nucleus reduces renal sympathetic tone. Hypertension. 2005;46(2):341–8.
• Burgelova M, Vanourkova Z, Thumova M, Dvorak P, Opocensky M, Kramer HJ, et al. Impairment of the angiotensin-converting enzyme 2-angiotensin-(1-7)-Mas axis contributes to the acceleration of two-kidney, one-clip Goldblatt hypertension. J Hypertens. 2009;27(10):1988–2000. https://doi.org/10.1097/HJH.0b013e32832f0d06.
Pinheiro SV, Ferreira AJ, Kitten GT, Da Silveira KD, Da Silva DA, Santos SH, et al. Genetic deletion of the angiotensin-(1–7) receptor Mas leads to glomerular hyperfiltration and microalbuminuria. Kidney Int. 2009;75(11):1184–93.
Yugandhar VG, Clark MA. Angiotensin III: a physiological relevant peptide of the renin angiotensin system. Peptides. 2013;46:26–32.
Yang R, Walther T, Gembardt F, Smolders I, Vanderheyden P, Albiston AL, et al. Renal vasoconstrictor and pressor responses to angiotensin IV in mice are AT1a-receptor mediated. J Hypertens. 2010;28(3):487–94.
Fung M-L, Lam S-Y, Wong T-P, Tjong Y-W, Leung P-S. Carotid body AT4 receptor expression and its upregulation in chronic hypoxia. Open Cardiovasc Med J. 2007;1:1.
Touyz RM. The role of angiotensin II in regulating vascular structural and functional changes in hypertension. Curr Hypertens Rep. 2003;5(2):155–64.
Gohla A, Schultz G, Offermanns S. Role for G12/G13 in agonist-induced vascular smooth muscle cell contraction. Circ Res. 2000;87(3):221–7.
Inagami T. Molecular biology and signaling of angiotensin receptors: an overview. J Am Soc Nephrol. 1999;10(Suppl 11):S2–7.
De Mello WC. Intracellular angiotensin II increases the total potassium current and the resting potential of arterial myocytes from vascular resistance vessels of the rat. Physiological and pathological implications. J Am Soc Hypertens. 2013;7(3):192–7.
Alvarez A, Cerda-Nicolas M, Naim Abu Nabah Y, Mata M, Issekutz AC, Panes J, et al. Direct evidence of leukocyte adhesion in arterioles by angiotensin II. Blood. 2004;104(2):402–8. https://doi.org/10.1182/blood-2003-08-2974.
Tummala PE, Chen X-L, Sundell CL, Laursen JB, Hammes CP, Alexander RW, et al. Angiotensin II induces vascular cell adhesion molecule-1 expression in rat vasculature. Circulation. 1999;100(11):1223–9.
Gosgnach W, Challah M, Coulet F, Michel J-B, Battle T. Shear stress induces angiotensin converting enzyme expression in cultured smooth muscle cells: possible involvement of bFGF. Cardiovasc Res. 2000;45(2):486–92.
Diez-Freire C, Vazquez J, Correa de Adjounian MF, Ferrari MF, Yuan L, Silver X, et al. ACE2 gene transfer attenuates hypertension-linked pathophysiological changes in the SHR. Physiol Genomics. 2006;27(1):12–9. https://doi.org/10.1152/physiolgenomics.00312.2005.
Peters J, Farrenkopf R, Clausmeyer S, Zimmer J, Kantachuvesiri S, Sharp MG, et al. Functional significance of prorenin internalization in the rat heart. Circ Res. 2002;90(10):1135–41.
Dugourd C, Gervais M, Corvol P, Monnot C. Akt is a major downstream target of PI3-kinase involved in angiotensin II-induced proliferation. Hypertension. 2003;41(4):882–90. https://doi.org/10.1161/01.hyp.0000060821.62417.35.
Rogers TB, Lokuta AJ. Angiotensin II signal transduction pathways in the cardiovascular system. Trends Cardiovasc Med. 1994;4(3):110–6.
• García-Hoz C, Sánchez-Fernández G, García-Escudero R, Fernández-Velasco M, Palacios-García J, Ruiz-Meana M, et al. Protein kinase C (PKC) ζ-mediated Gαq stimulation of ERK5 protein pathway in cardiomyocytes and cardiac fibroblasts. J Biol Chem. 2012;287(10):7792–802.
Thibault G, Lacombe M-J, Schnapp LM, Lacasse A, Bouzeghrane F, Lapalme G. Upregulation of α 8 β 1-integrin in cardiac fibroblast by angiotensin II and transforming growth factor-β1. Am J Physiol Cell Physiol. 2001;281(5):C1457–67.
Thomas WG, Brandenburger Y, Autelitano DJ, Pham T, Qian H, Hannan RD. Adenoviral-directed expression of the type 1A angiotensin receptor promotes cardiomyocyte hypertrophy via transactivation of the epidermal growth factor receptor. Circ Res. 2002;90(2):135–42.
• De Mello WC. Chemical communication between heart cells is disrupted by intracellular renin and angiotensin II: implications for heart development and disease. Front Endocrinol (Lausanne). 2015;6:72. https://doi.org/10.3389/fendo.2015.00072.
Banerjee I, Yekkala K, Borg TK, Baudino TA. Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Ann N Y Acad Sci. 2006;1080(1):76–84.
• Kakishita M, Nakamura K, Asanuma M, Morita H, Saito H, Kusano K, et al. Direct evidence for increased hydroxyl radicals in angiotensin II-induced cardiac hypertrophy through angiotensin II type 1a receptor. J Cardiovasc Pharmacol. 2003;42:S67–70.
Everett AD, Tufro-McReddie A, Fisher A, Gomez RA. Angiotensin receptor regulates cardiac hypertrophy and transforming growth factor-beta 1 expression. Hypertension. 1994;23(5):587–92.
Lal A, Veinot JP, Ganten D, Leenen FH. Prevention of cardiac remodeling after myocardial infarction in transgenic rats deficient in brain angiotensinogen. J Mol Cell Cardiol. 2005;39(3):521–9. https://doi.org/10.1016/j.yjmcc.2005.05.002.
Miguel-Carrasco JL, Monserrat MT, Mate A, Vázquez CM. Comparative effects of captopril and l-carnitine on blood pressure and antioxidant enzyme gene expression in the heart of spontaneously hypertensive rats. Eur J Pharmacol. 2010;632(1):65–72.
Tsutsumi Y, Matsubara H, Ohkubo N, Mori Y, Nozawa Y, Murasawa S, et al. Angiotensin II type 2 receptor is upregulated in human heart with interstitial fibrosis, and cardiac fibroblasts are the major cell type for its expression. Circ Res. 1998;83(10):1035–46.
Smookler HH, Severs WB, Kinnard WJ, Buckley JP. Centrally mediated cardiovascular effects of angiotensin II. J Pharmacol Exp Ther. 1966;153(3):485–94.
Buckley JP. Actions of angiotensin on the central nervous system. Fed Proc. 1972;31(4):1332–7.
• Lon S, Szczepanska-Sadowska E, Szczypaczewska M. Evidence that centrally released arginine vasopressin is involved in central pressor action of angiotensin II. Am J Physiol Heart Circ Physiol. 1996;270(1):H167–73.
Schölkens B, Jung W, Rascher W, Schömig A, Ganten D. Brain angiotensin II stimulates release of pituitary hormones, plasma catecholamines and increases blood pressure in dogs. Clin Sci (Lond). 1980;59:53s–6s.
• Allen AM, O’Callaghan E, Chen D, Bassi J. Central neural regulation of cardiovascular function by angiotensin: a focus on the rostral ventrolateral medulla. Neuroendocrinology. 2009;89(4):361–9.
Chen C-Y, Huang W-C. Pressor and renal effects of intracerebroventricularly administered angiotensins II and III in rats. Kidney Blood Press Res. 2000;23(2):95–105.
Ferguson AV, Washburn DL. Angiotensin II: a peptidergic neurotransmitter in central autonomic pathways. Prog Neurobiol. 1998;54(2):169–92.
•• Nasimi A, Kafami M. Vasopressin and sympathetic system mediate the cardiovascular effects of the angiotensin II in the bed nucleus of the stria terminalis in rat. Neurosci Res. 2016;108:34–9.
Dampney RA, Tan PS, Sheriff MJ, Fontes MA, Horiuchi J. Cardiovascular effects of angiotensin II in the rostral ventrolateral medulla: the push-pull hypothesis. Curr Hypertens Rep. 2007;9(3):222–7.
Saad WA, de Arruda Camargo LA, Guarda IF, dos Santos TA, Guarda RS, Saad WA, et al. Interaction between supraoptic nucleus and septal area in the control of water, sodium intake and arterial blood pressure induced by injection of angiotensin II. Pharmacol Biochem Behav. 2004;77(4):667–74. https://doi.org/10.1016/j.pbb.2004.01.013.
• Szczepanska-Sadowska E, Paczwa P, Lon S, Ganten D. Increased pressor function of central vasopressinergic system in hypertensive renin transgenic rats. J Hypertens. 1998;16(10):1505–14.
Chan SH, Hsu KS, Huang CC, Wang LL, Ou CC, Chan JY. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced pressor effect via activation of p38 mitogen-activated protein kinase in the rostral ventrolateral medulla. Circ Res. 2005;97(8):772–80. https://doi.org/10.1161/01.res.0000185804.79157.c0.
DiBona GF. Central sympathoexcitatory actions of angiotensin II: role of type 1 angiotensin II receptors. J Am Soc Nephrol. 1999;10(Suppl 11):S90–4.
Sagara Y, Hirooka Y, Nozoe M, Ito K, Kimura Y, Sunagawa K. Pressor response induced by central angiotensin II is mediated by activation of Rho/Rho-kinase pathway via AT1 receptors. J Hypertens. 2007;25(2):399–406.
Chen Q, Pan H-L. Signaling mechanisms of angiotensin II-induced attenuation of GABAergic input to hypothalamic presympathetic neurons. J Neurophysiol. 2007;97(5):3279–87.
Zhu G-Q, Patel KP, Zucker IH, Wang W. Microinjection of ANG II into paraventricular nucleus enhances cardiac sympathetic afferent reflex in rats. Am J Physiol Heart Circ Physiol. 2002;282(6):H2039–45.
Gao J, Zhang H, Le KD, Chao J, Gao L. Activation of central angiotensin type 2 receptors suppresses norepinephrine excretion and blood pressure in conscious rats. Am J Hypertens. 2011;24(6):724–30.
Li Z, Iwai M, Wu L, Shiuchi T, Jinno T, Cui TX, et al. Role of AT2 receptor in the brain in regulation of blood pressure and water intake. Am J Physiol Heart Circ Physiol. 2003;284(1):H116–21. https://doi.org/10.1152/ajpheart.00515.2002.
Gao L, Wang W, Wang W, Li H, Sumners C, Zucker IH. Effects of angiotensin type 2 receptor overexpression in the rostral ventrolateral medulla on blood pressure and urine excretion in normal rats. Hypertension. 2008;51(2):521–7.
He X, Zhao M, Bi X, Sun L, Yu X, Zhao M, et al. Novel strategies and underlying protective mechanisms of modulation of vagal activity in cardiovascular diseases. Br J Pharmacol. 2015;172(23):5489–500.
Grassi G, Ram VS. Evidence for a critical role of the sympathetic nervous system in hypertension. J Am Soc Hypertens. 2016;10(5):457–66. https://doi.org/10.1016/j.jash.2016.02.015.
Oliveira-Sales EB, Colombari DS, Davisson RL, Kasparov S, Hirata AE, Campos RR, et al. Kidney-induced hypertension depends on superoxide signaling in the rostral ventrolateral medulla. Hypertension. 2010;56(2):290–6.
Bunag RD, Page IH, McCubbin JW. Neural stimulation of release of renin. Circ Res. 1966;19(4):851–8.
Liu J-L, Murakami H, Sanderford M, Bishop VS, Zucker IH. ANG II and baroreflex function in rabbits with CHF and lesions of the area postrema. Am J Physiol Heart Circ Physiol. 1999;277(1):H342–50.
Llewellyn TL, Sharma NM, Zheng H, Patel KP. Effects of exercise training on SFO-mediated sympathoexcitation during chronic heart failure. Am J Physiol Heart Circ Physiol. 2014;306(1):H121–31.
Strömberg C, Tsutsumi K, Viswanathan M, Saavedra JM. Angiotensin II AT1 receptors in rat superior cervical ganglia: characterization and stimulation of phosphoinositide hydrolysis. Eur J Pharmacol. 1991;208(4):331–6.
Dendorfer A, Thornagel A, Raasch W, Grisk O, Tempel K, Dominiak P. Angiotensin II induces catecholamine release by direct ganglionic excitation. Hypertension. 2002;40(3):348–54.
FARR WC, GRUPP G. Ganglionic stimulation: mechanism of the positive inotropic and chronotropic effects of angiotensin. J Pharmacol Exp Ther. 1971;177(1):48–55.
Castren E, Kurihara M, Gutkind JS, Saavedra JM. Specific angiotensin II binding sites in the rat stellate and superior cervical ganglia. Brain Res. 1987;422(2):347–51.
Ma X, Chapleau MW, Whiteis CA, Abboud FM, Bielefeldt K. Angiotensin selectively activates a subpopulation of postganglionic sympathetic neurons in mice. Cir Res. 2001;88(8):787–93.
Shapiro MS, Wollmuth LP, Hille B. Angiotensin II inhibits calcium and M current channels in rat sympathetic neurons via G proteins. Neuron. 1994;12(6):1319–29.
Acosta E, Mendoza V, Castro E, Cruzblanca H. Modulation of a delayed-rectifier K+ current by angiotensin II in rat sympathetic neurons. J Neurophysiol. 2007;98(1):79–85.
Allen AM, Lewis SJ, Verberne AJ, Mendelsohn FA. Angiotensin receptors and the vagal system. Clin Exp Hypertens A. 1988;10(6):1239–49.
Du XJ, Cox HS, Dart AM, Esler MD. Depression of efferent parasympathetic control of heart rate in rats with myocardial infarction: effect of losartan. J Cardiovasc Pharmacol. 1998;31(6):937–44.
Flapan AD, Nolan J, Neilson JM, Ewing DJ. Effect of captopril on cardiac parasympathetic activity in chronic cardiac failure secondary to coronary artery disease. Am J Cardiol. 1992;69(5):532–5.
Osterziel KJ, Dietz R. Improvement of vagal tone by ACE inhibition: a mechanism of cardioprotection in patients with mild-to-moderate heart failure. J Cardiovasc Pharmacol. 1996;27:25–30.
• Allen AM, Moeller I, Jenkins TA, Zhuo J, Aldred GP, Chai SY, et al. Angiotensin receptors in the nervous system. Brain Res Bull. 1998;47(1):17–28.
Barron KW, Trapani AJ, Gordon FJ, Brody MJ. Baroreceptor denervation profoundly enhances cardiovascular responses to central angiotensin II. Am J Physiol Heart Circ Physiol. 1989;257(1):H314–23.
Brooks VL, Ell KR, Wright RM. Pressure-independent baroreflex resetting produced by chronic infusion of angiotensin II in rabbits. Am J Physiol Heart Circ Physiol. 1993;265(4):H1275–82.
• Head G, Saigusa T, Mayorov D. Angiotensin and baroreflex control of the circulation. Braz J Med Biol Res. 2002;35(9):1047–59.
Saigusa T, Iriki M, Arita J. Brain angiotensin II tonically modulates sympathetic baroreflex in rabbit ventrolateral medulla. Am J Physiol Heart Circ Physiol. 1996;271(3):H1015–21.
Polson JW, Dampney RA, Boscan P, Pickering AE, Paton JF. Differential baroreflex control of sympathetic drive by angiotensin II in the nucleus tractus solitarii. Am J Physiol Regul Integr Comp Physiol. 2007;293(5):R1954–60.
Tan PS, Killinger S, Horiuchi J, Dampney RA. Baroreceptor reflex modulation by circulating angiotensin II is mediated by AT 1 receptors in the nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol. 2007;293(6):R2267–78.
Palma-Rigo K, Bassi JK, Nguyen-Huu T-P, Jackson KL, Davern PJ, Chen D, et al. Angiotensin 1A receptors transfected into caudal ventrolateral medulla inhibit baroreflex gain and stress responses. Cardiovasc Res. 2012;96(2):330–9.
Wang W-Z, Gao L, Wang H-J, Zucker IH, Wang W. Interaction between cardiac sympathetic afferent reflex and chemoreflex is mediated by the NTS AT 1 receptors in heart failure. Am J Physiol Heart Circ Physiol. 2008;295(3):H1216–26.
Murakami H, Liu J-L, Zucker IH. Angiotensin II enhances baroreflex control of sympathetic outflow in heart failure. Hypertension. 1997;29(2):564–9.
Zhang W, Huang BS, Leenen FH. Brain renin-angiotensin system and sympathetic hyperactivity in rats after myocardial infarction. Am J Physiol Heart Circ Physiol. 1999;276(5):H1608–15.
Abdulla MH, Johns EJ. Nitric oxide impacts on angiotensin AT2 receptor modulation of high-pressure baroreflex control of renal sympathetic nerve activity in anaesthetized rats. Acta Physiol (Oxf). 2014;210(4):832–44. https://doi.org/10.1111/apha.12207.
Diz DI, Garcia-Espinosa MA, Gegick S, Tommasi EN, Ferrario CM, Ann Tallant E, et al. Injections of angiotensin-converting enzyme 2 inhibitor MLN4760 into nucleus tractus solitarii reduce baroreceptor reflex sensitivity for heart rate control in rats. Exp Physiol. 2008;93(5):694–700. https://doi.org/10.1113/expphysiol.2007.040261.
Cangussu LM, de Castro UG, do Pilar Machado R, Silva ME, Ferreira PM, dos Santos RA, et al. Angiotensin-(1-7) antagonist, A-779, microinjection into the caudal ventrolateral medulla of renovascular hypertensive rats restores baroreflex bradycardia. Peptides. 2009;30(10):1921–7. https://doi.org/10.1016/j.peptides.2009.06.028.
Yamazato M, Ferreira AJ, Yamazato Y, Diez-Freire C, Yuan L, Gillies R, et al. Gene transfer of angiotensin-converting enzyme 2 in the nucleus tractus solitarius improves baroreceptor heart rate reflex in spontaneously hypertensive rats. J Renin-Angiotensin-Aldosterone Syst. 2011;12(4):456–61.
Li P, Sun H-J, Cui B-P, Zhou Y-B, Han Y. Angiotensin-(1–7) in the rostral ventrolateral medulla modulates enhanced cardiac sympathetic afferent reflex and sympathetic activation in renovascular hypertensive rats. Hypertension 2013;62(3):e11. https://doi.org/10.1161/HYP.0b013e3182a65fc8.
Bacal K, Kunze D. Dual effects of angiotensin II on calcium currents in neonatal rat nodose neurons. J Neurosci. 1994;14(11):7159–67.
Takahashi K, Hiraishi K, Hirose T, Kato I, Yamamoto H, Shoji I, et al. Expression of (pro) renin receptor in the human brain and pituitary, and co-localisation with arginine vasopressin and oxytocin in the hypothalamus. J Neuroendocrinol. 2010;22(5):453–9.
Fyhrquist F, Eriksson L, Wallenius M. Plasma vasopressin in conscious goats after cerebroventricular infusions of angiotensins, sodium chloride, and fructose. Endocrinology. 1979;104(4):1091–5.
Zini S, Fournie-Zaluski M-C, Chauvel E, Roques BP, Corvol P, Llorens-Cortes C. Identification of metabolic pathways of brain angiotensin II and III using specific aminopeptidase inhibitors: predominant role of angiotensin III in the control of vasopressin release. Proc Natl Acad Sci. 1996;93(21):11968–73.
Foster RH, MacFarlane CH, Bustamante MO. Recent progress in understanding aldosterone secretion. Gen Pharmacol. 1997;28(5):647–51.
Rautureau Y, Paradis P, Schiffrin EL. Cross-talk between aldosterone and angiotensin signaling in vascular smooth muscle cells. Steroids. 2011;76(9):834–9.
Schiffrin EL, Gutkowska J, Genest J. Effect of angiotensin II and deoxycorticosterone infusion on vascular angiotensin II receptors in rats. Am J Physiol Heart Circ Physiol. 1984;246(4):H608–14.
Virdis A, Neves MF, Amiri F, Viel E, Touyz RM, Schiffrin EL. Spironolactone improves angiotensin-induced vascular changes and oxidative stress. Hypertension. 2002;40(4):504–10.
Suzuki J, Iwai M, Mogi M, Oshita A, Yoshii T, Higaki J, et al. Eplerenone with valsartan effectively reduces atherosclerotic lesion by attenuation of oxidative stress and inflammation. Arterioscler Thromb Vasc Biol. 2006;26(4):917–21.
Coghlan J, Considine P, Denton D, Fei D, Leksell L, McKinley M, et al. Sodium appetite in sheep induced by cerebral ventricular infusion of angiotensin: comparison with sodium deficiency. Science. 1981;214(4517):195–7.
Fitzsimons J. Angiotensin, thirst, and sodium appetite. Physiol Res. 1998;78(3):583–686.
Morris MJ, Wilson WL, Starbuck EM, Fitts DA. Forebrain circumventricular organs mediate salt appetite induced by intravenous angiotensin II in rats. Brain Res. 2002;949(1):42–50.
Shade RE, Blair-West JR, Carey KD, Madden LJ, Weisinger RS, Denton DA. Synergy between angiotensin and aldosterone in evoking sodium appetite in baboons. Am J Physiol Regul Integr Comp Physiol. 2002;283(5):R1070–8.
Abrao Saad W, Antonio De Arruda Camargo L, Sergio Cerri P, Simoes S, Abrao Saad W, Garcia G, et al. Influence of arginine vasopressin receptors and angiotensin receptor subtypes on the water intake and arterial blood pressure induced by vasopressin injected into the lateral septal area of the rat. Auton Neurosci. 2004;111(1):66–70. https://doi.org/10.1016/j.autneu.2003.08.013.
de Arruda Camargo LA, Saad WA, Cerri PS. Effects of V1 and angiotensin receptor subtypes of the paraventricular nucleus on the water intake induced by vasopressin injected into the lateral septal area. Brain Res Bull. 2003;61(5):481–7.
McKinley M, Cairns M, Denton D, Egan G, Mathai M, Uschakov A, et al. Physiological and pathophysiological influences on thirst. Physiol Behav. 2004;81(5):795–803.
Szczepanska-Sadowska E. Hormonal inputs to thirst. In: Ramsay DJ, Booth D, editors. Thirst: physiological and psychological aspects. London: Springer London; 1991. p. 110–30.
Wright JW, Sullivan MJ, Quirk WS, Batt CM, Harding JW. Heightened blood pressure and drinking responsiveness to intracerebroventricularly applied angiotensins in the spontaneously hypertensive rat. Brain Res. 1987;420(2):289–94.
Miyata N, Park F, Li XF, Cowley AW. Distribution of angiotensin AT 1 and AT 2 receptor subtypes in the rat kidney. Am J Physiol Renal Physiol. 1999;277(3):F437–46.
Navar LG, Kobori H, Prieto-Carrasquero M. Intrarenal angiotensin II and hypertension. Curr Hypertens Rep. 2003;5(2):135–43.
Oliverio MI, Best CF, Smithies O, Coffman TM. Regulation of sodium balance and blood pressure by the AT1A receptor for angiotensin II. Hypertension. 2000;35(2):550–4.
Oliverio MI, Delnomdedieu M, Best CF, Li P, Morris M, Callahan MF, et al. Abnormal water metabolism in mice lacking the type 1A receptor for ANG II. Am J Physiol Renal Physiol. 2000;278(1):F75–82.
Casare FAM, Thieme K, Costa-Pessoa JM, Rossoni LV, Couto GK, Fernandes FB, et al. Renovascular remodeling and renal injury after extended angiotensin II infusion. Am J Physiol Regul Integr Comp Physiol. 2016;310(11):F1295–307.
Kihara M, Umemura S, Sumida Y, Yokoyama N, Yabana M, Nyui N, et al. Genetic deficiency of angiotensinogen produces an impaired urine concentrating ability in mice. Kidney Int. 1998;53(3):548–55.
Li XC, Shao Y, Zhuo JL. AT1a receptor knockout in mice impairs urine concentration by reducing basal vasopressin levels and its receptor signaling proteins in the inner medulla. Kidney Int. 2009;76(2):169–77. https://doi.org/10.1038/ki.2009.134.
• Szczepanska-Sadowska E, Zera T, Sosnowski P, Cudnoch-Jedrzejewska A, Puszko A, Misicka A. Vasopressin and related peptides; potential value in diagnosis, prognosis and treatment of clinical disorders. Curr Drug Metab. 2017;18(4):306–45.
Buijs R, Swaab D. Immuno-electron microscopical demonstration of vasopressin and oxytocin synapses in the limbic system of the rat. Cell Tissue Res. 1979;204(3):355–65.
van Leeuwen F, Caffé R. Vasopressin-immunoreactive cell bodies in the bed nucleus of the stria terminalis of the rat. Cell Tissue Res. 1983;228(3):525–34.
Wacker DW, Tobin VA, Noack J, Bishop VR, Duszkiewicz AJ, Engelmann M et al. inventors; Expression of early growth response protein 1 in vasopressin neurones of the rat anterior olfactory nucleus following social odour exposure patent 1469–7793. 2010.
Ginsberg SD, Hof PR, Young WG, Morrison JH. Noradrenergic innervation of vasopressin-and oxytocin-containing neurons in the hypothalamic paraventricular nucleus of the macaque monkey: quantitative analysis using double-label immunohistochemistry and confocal laser microscopy. J Comp Neurol. 1994;341(4):476–91.
Sawchenko PE, Swanson LW. Immunohistochemical identification of neurons in the paraventricular nucleus of the hypothalamus that project to the medulla or to the spinal cord in the rat. J Comp Neurol. 1982;205(3):260–72.
• Hallbeck M, Hermanson O, Blomqvist A. Distribution of preprovasopressin mRNA in the rat central nervous system. J Comp Neurol. 1999;411(2):181–200.
• Hallbeck M, Blomqvist A. Spinal cord-projecting vasopressinergic neurons in the rat paraventricular hypothalamus. J Comp Neurol. 1999;411(2):201–11.
Ludwig M, Stern J. Multiple signalling modalities mediated by dendritic exocytosis of oxytocin and vasopressin. Phil Trans R Soc B. 2015;370(1672):20140182.
Hupf H, Grimm D, Riegger GA, Schunkert H. Evidence for a vasopressin system in the rat heart. Circ Res. 1999;84(3):365–70.
Ravid R, Oosterbaan H, Hansen B, Swaab D. Localisation of oxytocin, vasopressin and parts of precursors in the human neonatal adrenal. Histochem Cell Biol. 1986;84(4):401–7.
Yibchok-anun S, Abu-Basha EA, Yao C-Y, Panichkriangkrai W, Hsu WH. The role of arginine vasopressin in diabetes-associated increase in glucagon secretion. Regul Pept. 2004;122(3):157–62.
Bichet DG. Vasopressin at central levels and consequences of dehydration. Ann Nutr Metab. 2016;68(Suppl 2):19–23. https://doi.org/10.1159/000446200.
Janas S, Seghers F, Schakman O, Alsady M, Deen P, Vriens J, et al. TRPV4 is associated with central rather than nephrogenic osmoregulation. Pflugers Arch. 2016;468(9):1595–607.
Dawson CA, Jhamandas JH, Krukoff TL. Activation by systemic angiotensin II of neurochemically identified neurons in rat hypothalamic paraventricular nucleus. J Neuroendocrinol. 1998;10(6):453–9.
Kageyama K, Kumata Y, Akimoto K, Takayasu S, Tamasawa N, Suda T. Ghrelin stimulates corticotropin-releasing factor and vasopressin gene expression in rat hypothalamic 4B cells. Stress. 2011;14(5):520–9.
Sladek CD, Kapoor JR. Neurotransmitter/neuropeptide interactions in the regulation of neurohypophyseal hormone release. Exp Neurol. 2001;171(2):200–9.
Ganella DE, Ma S, Gundlach AL. Relaxin-3/RXFP3 signaling and neuroendocrine function—a perspective on extrinsic hypothalamic control. Front Endocrinol. 2013;4:128. https://doi.org/10.3389/fendo.2013.00128
Sladek CD, Armstrong WE. Gamma-aminobutyric acid antagonists stimulate vasopressin release from organ-cultured hypothalamo-neurohypophyseal explants. Endocrinology. 1987;120(4):1576–80. https://doi.org/10.1210/endo-120-4-1576.
Tobin VA, Bull PM, Arunachalam S, O'Carroll A-M, Ueta Y, Ludwig M. The effects of apelin on the electrical activity of hypothalamic magnocellular vasopressin and oxytocin neurons and somatodendritic peptide release. Endocrinology. 2008;149(12):6136–45.
Zhang L, Tong M, Xiao M, Li L, Ding J. Nitric oxide mediates feedback inhibition in angiotensin II-induced upregulation of vasopressin mRNA. Peptides. 2009;30(5):913–7.
Decavel C, Van den Pol AN. GABA: a dominant neurotransmitter in the hypothalamus. J Comp Neurol. 1990;302(4):1019–37. https://doi.org/10.1002/cne.903020423.
Haam J, Popescu IR, Morton LA, Halmos KC, Teruyama R, Ueta Y, et al. GABA is excitatory in adult vasopressinergic neuroendocrine cells. J Neurosci. 2012;32(2):572–82.
Kim Y-B, Kim YS, Kim WB, Shen F-Y, Lee SW, Chung HJ, et al. GABAergic excitation of vasopressin neurons novelty and significance. Circ Res. 2013;113(12):1296–307.
Jorgensen H, Knigge U, Kjaer A, Warberg J. Serotonergic involvement in stress-induced vasopressin and oxytocin secretion. Eur J Endocrinol. 2002;147(6):815–24.
Haque M, Wilson R, Sharma K, Mills N, Teruyama R. Localisation of 11β-hydroxysteroid dehydrogenase type 2 in mineralocorticoid receptor expressing magnocellular neurosecretory neurones of the rat supraoptic and paraventricular nuclei. J Neuroendocrinol. 2015;27(11):835–49.
• Pietranera L, Saravia F, Roig P, Lima A, De Nicola AF. Mineralocorticoid treatment upregulates the hypothalamic vasopressinergic system of spontaneously hypertensive rats. Neuroendocrinology. 2004;80(2):100–10.
Amin MS, Wang H-W, Reza E, Whitman SC, Tuana BS, Leenen FH. Distribution of epithelial sodium channels and mineralocorticoid receptors in cardiovascular regulatory centers in rat brain. Am J Physiol Regul Integr Comp Physiol. 2005;289(6):R1787–97.
Teruyama R, Sakuraba M, Wilson LL, Wandrey NE, Armstrong WE. Epithelial Na+ sodium channels in magnocellular cells of the rat supraoptic and paraventricular nuclei. Am J Physiol Endocrinol Metab. 2012;302(3):E273–85.
Abel A, Wittau N, Wieland T, Schultz G, Kalkbrenner F. Cell cycle-dependent coupling of the vasopressin V1a receptor to different G proteins. J Biol Chem. 2000;275(42):32543–51.
Schoneberg T, Kostenis E, Liu J, Gudermann T, Wess J. Molecular aspects of vasopressin receptor function. Adv Exp Med Biol. 1998;449:347–58.
• Phillips PA, Kelly JM, Abrahams JM, Grzonka Z, Paxinos G, Mendelsohn FA, et al. Vasopressin receptors in rat brain and kidney: studies using a radio-iodinated V1 receptor antagonist. J Hypertens. 1988;6(4):S550–3.
• Góźdź A, Szczepańska-Sadowska E, Maśliński W, Kumosa M, Szczepańska K, Dobruch J. Differential expression of vasopressin V1a and V1b receptors mRNA in the brain of renin transgenic TGR (mRen2) 27 and Sprague–Dawley rats. Brain Res Bull. 2003;59(5):399–403.
• Young L, Toloczko D, Insel R. Localization of vasopressin (V~ 1~ a) receptor binding and mRNA in the rhesus monkey brain. J Neuroendocrinol. 1999;11:291–8.
Moriya T, Kayano T, Kitamura N, Hosaka YZ, Asano A, Forostyak O, et al. Vasopressin-induced intracellular Ca 2+ concentration responses in non-neuronal cells of the rat dorsal root ganglion. Brain Res. 2012;1483:1–12.
Arpin-Bott MP, Waltisperger E, Freund-Mercier MJ, Stoeckel ME. Historadioautographic localization, pharmacology and ontogeny of V(1a) vasopressin binding sites in the rat kidney. Nephron. 1999;83(1):74–84.
Koshimizu T-A, Nakamura K, Egashira N, Hiroyama M, Nonoguchi H, Tanoue A. Vasopressin V1a and V1b receptors: from molecules to physiological systems. Physiol Rev. 2012;92(4):1813–64.
• Park F, Mattson DL, Skelton MM, Cowley AW. Localization of the vasopressin V1a and V2 receptors within the renal cortical and medullary circulation. Am J Physiol Regul Integr Comp Physiol. 1997;273(1):R243–51.
• Gozdz A, Szczepanska-Sadowska E, Szczepanska K, Maslinski W, Luszczyk B. Vasopressin V1a, V1b and V2 receptors mRNA in the kidney and heart of the renin transgenic TGR(mRen2)27 and Sprague Dawley rats. J Physiol Pharmacol. 2002;53(3):349–57.
Hernando F, Schoots O, Lolait SJ, Burbach JPH. Immunohistochemical localization of the vasopressin V1b receptor in the rat brain and pituitary gland: anatomical support for its involvement in the central effects of vasopressin. Endocrinology. 2001;142(4):1659–68.
Roper JA, O'Carroll A, Young W III, Lolait S. The vasopressin Avpr1b receptor: molecular and pharmacological studies. Stress. 2011;14(1):98–115.
• Zhang J, Sato M, Duzic E, Kubalak SW, Lanier SM, Webb JG. Adenylyl cyclase isoforms and vasopressin enhancement of agonist-stimulated cAMP in vascular smooth muscle cells. Am J Physiol-Heart Circ Physiol. 1997;273(2):H971–80.
• Henderson KK, Byron KL. Vasopressin-induced vasoconstriction: two concentration-dependent signaling pathways. J Appl Physiol. 2007;102(4):1402–9.
• Brueggemann LI, Moran CJ, Barakat JA, Yeh JZ, Cribbs LL, Byron KL. Vasopressin stimulates action potential firing by protein kinase C-dependent inhibition of KCNQ5 in A7r5 rat aortic smooth muscle cells. Am J Physiol Heart Circ Physiol. 2007;292(3):H1352–H63.
• Byron KL, Lucchesi PA. Signal transduction of physiological concentrations of vasopressin in A7r5 vascular smooth muscle cells. A role for PYK2 and tyrosine phosphorylation of K+ channels in the stimulation of Ca2+ spiking. J Biol Chem. 2002;277(9):7298–307. https://doi.org/10.1074/jbc.M104726200.
• Nishiwaki-Yasuda K, Suzuki A, Kakita A, Sekiguchi S, Asano S, Nishii K, et al. Vasopressin stimulates Na-dependent phosphate transport and calcification in rat aortic smooth muscle cells. Endocr J. 2007;54(1):103–12.
• Jackiewicz E, Szczepanska-Sadowska E, Dobruch J. Altered expression of angiotensin AT1a and vasopressin V1a receptors and nitric oxide synthase mRNA in the brain of rats with renovascular hypertension. J Physiol Pharmacol. 2004;55(4):725–37.
•• Milik E, Szczepanska-Sadowska E, Dobruch J, Cudnoch-Jedrzejewska A, Maslinski W. Altered expression of V1a receptors mRNA in the brain and kidney after myocardial infarction and chronic stress. Neuropeptides. 2014;48(5):257–66.
• Mutig K, Paliege A, Kahl T, Jöns T, Müller-Esterl W, Bachmann S. Vasopressin V 2 receptor expression along rat, mouse, and human renal epithelia with focus on TAL. Am J Physiol Renal Physiol. 2007;293(4):F1166–77.
• Hayashi M, Sasaki S, Tsuganezawa H, Monkawa T, Kitajima W, Konishi K, et al. Role of vasopressin V2 receptor in acute regulation of aquaporin-2. Kidney Blood Press Res. 1996;19(1):32–7.
• Schrier RW. Vasopressin and aquaporin 2 in clinical disorders of water homeostasis. Semin Nephrol. 2008;28(3):289–96. https://doi.org/10.1016/j.semnephrol.2008.03.009.
• Schrier RW. Interactions between angiotensin II and arginine vasopressin in water homeostasis. Kidney Int. 2009;76(2):137–9.
Szalai B, Hoffmann P, Prokop S, Erdélyi L, Várnai P, Hunyady L. Improved methodical approach for quantitative BRET analysis of G protein coupled receptor dimerization. PLoS One. 2014;9(10):e109503.
• Gonzalez CB, Herrera VL, Ruiz-Opazo N. Renal immunocytochemical distribution and pharmacological properties of the dual angiotensin II/AVP receptor. Hypertension. 1997;29(4):957–61.
• Cowley AW Jr, Monos E, Guyton AC. Interaction of vasopressin and the baroreceptor reflex system in the regulation of arterial blood pressure in the dog. Circ Res. 1974;34(4):505–14.
• Szczepańska-Sadowska E. Hemodynamic effects of moderate increase of the plasma vasopressin level in conscious dogs. Pflugers Arch. 1973;338(4):313–22.
• Motawei K, Pyner S, Ranson RN, Kamel M, Coote JH. Terminals of paraventricular spinal neurones are closely associated with adrenal medullary sympathetic preganglionic neurones: immunocytochemical evidence for vasopressin as a possible neurotransmitter in this pathway. Exp Brain Res. 1999;126(1):68–76.
• Altura BM, Altura BT. Actions of vasopressin, oxytocin, and synthetic analogs on vascular smooth muscle. Fed Proc. 1984;43(1):80–6.
• Hidaka T, Tsuneyoshi I, Boyle WA 3rd, Onomoto M, Yonetani S, Hamasaki J, et al. Marked synergism between vasopressin and angiotensin II in a human isolated artery. Crit Care Med. 2005;33(11):2613–20.
• Kaplan-Albuquerque N, Garat C, Van Putten V, Nemenoff RA. Regulation of SM22α expression by arginine vasopressin and PDGF-BB in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2003;285(4):H1444–52.
• Nishihashi T, Trandafir CC, Wang A, Ji X, Kurahashi K. Enhanced reactivity to vasopressin in rat basilar arteries during vasospasm after subarachnoid hemorrhage. Eur J Pharmacol. 2005;513(1):93–100.
• Friedman SM. Additive effects of aldosterone with vasopressin or angiotensin. Hypertension. 1984;6(2 Pt 1):242–8.
• Leung E, Johnston CI, Woodcock EA. Stimulation of phosphatidylinositol metabolism in the heart. Clin Exp Pharmacol Physiol. 1986;13(4):359–63.
• Gutkowska J, Miszkurka M, Danalache B, Gassanov N, Wang D, Jankowski M. Functional arginine vasopressin system in early heart maturation. Am J Physiol Heart Circ Physiol. 2007;293(4):H2262–70.
• Weig H-J, Laugwitz K-L, Moretti A, Kronsbein K, Städele C, Brüning S, et al. Enhanced cardiac contractility after gene transfer of V2 vasopressin receptors in vivo by ultrasound-guided injection or transcoronary delivery. Circulation. 2000;101(13):1578–85.
• Pelletier J-S, LaBossiere J, Dicken B, Gill RS, Sergi C, Tahbaz N, et al. Low-dose vasopressin improves cardiac function in newborn piglets with acute hypoxia-reoxygenation. Shock. 2013;40(4):320–6.
• Hiroyama M, Wang S, Aoyagi T, Oikawa R, Sanbe A, Takeo S, et al. Vasopressin promotes cardiomyocyte hypertrophy via the vasopressin V 1A receptor in neonatal mice. Eur J Pharmacol. 2007;559(2):89–97.
• Mayr VD, Wenzel V, Wagner-Berger HG, Stadlbauer KH, Cavus E, Raab H, et al. Arginine vasopressin during sinus rhythm: effects on haemodynamic variables, left anterior descending coronary artery cross sectional area and cardiac index, before and after inhibition of NO-synthase, in pigs. Resuscitation. 2007;74(2):366–71.
• Fan Y-H, Zhao L-Y, Zheng Q-S, Dong H, Wang H-C. Yang X-d. Arginine vasopressin increases iNOS–NO system activity in cardiac fibroblasts through NF-κB activation and its relation with myocardial fibrosis. Life Sci. 2007;81(4):327–35.
• Niu X, Xue Y, Li X, He Y, Zhao X, Xu M, et al. Effects of angiotensin-(1-7) on the proliferation and collagen synthesis of arginine vasopressin–stimulated rat cardiac fibroblasts: role of mas receptor-calcineurin-NF-κB signaling pathway. J Cardiovasc Pharmacol. 2014;64(6):536–42.
•• Ebert TJ, Cowley AW, Skelton M. Vasopressin reduces cardiac function and augments cardiopulmonary baroreflex resistance increases in man. J Clin Invest. 1986;77(4):1136–42.
• Goldsmith SR. The role of vasopressin in congestive heart failure. Cleve Clin J Med. 2006;73(Suppl 3):S19–23.
• Chen X, Lu G, Tang K, Li Q, Gao X. The secretion patterns and roles of cardiac and circulating arginine vasopressin during the development of heart failure. Neuropeptides. 2015;51:63–73.
• Chen Y, Xu F, Zhang L, Wang X, Wang Y, Woo AY, et al. GRK2/beta-arrestin mediates arginine vasopressin-induced cardiac fibroblast proliferation. Clin Exp Pharmacol Physiol. 2017;44(2):285–93. https://doi.org/10.1111/1440-1681.12696.
•• Li X, Chan TO, Myers V, Chowdhury I, Zhang XQ, Song J, et al. Controlled and cardiac-restricted overexpression of the arginine vasopressin V1A receptor causes reversible left ventricular dysfunction through Galphaq-mediated cell signaling. Circulation. 2011;124(5):572–81. https://doi.org/10.1161/circulationaha.111.021352.
• Iversen BM, Arendshorst WJ. ANG II and vasopressin stimulate calcium entry in dispersed smooth muscle cells of preglomerular arterioles. Am J Physiol Renal Physiol. 1998;274(3):F498–508.
• Okuda T, Yamashita N, Kurokawa K. Angiotensin II and vasopressin stimulate calcium-activated chloride conductance in rat mesangial cells. J Clin Invest. 1986;78(6):1443.
• Weihprecht H, Lorenz JN, Briggs JP, Schnermann J. Vasoconstrictor effect of angiotensin and vasopressin in isolated rabbit afferent arterioles. Am J Physiol Renal Physiol. 1991;261(2):F273–82.
• Ganz M, Boyarsky G, Boron W, Sterzel R. Effects of angiotensin II and vasopressin on intracellular pH of glomerular mesangial cells. Am J Physiol Renal Physiol. 1988;254(6):F787–94.
• Ganz M, Pekar S, Perfetto M, Sterzel R. Arginine vasopressin promotes growth of rat glomerular mesangial cells in culture. Am J Physiol Renal Physiol. 1988;255(5):F898–906.
• Evans RG, Bergstrom G, Lawrence AJ. Effects of the vasopressin V1 agonist [Phe2,Ile3,Orn8] vasopressin on regional kidney perfusion and renal excretory function in anesthetized rabbits. J Cardiovasc Pharmacol. 1998;32(4):571–81.
• Franchini KG, Cowley A. Renal cortical and medullary blood flow responses during water restriction: role of vasopressin. Am J Physiol Regul Integr Comp Physiol. 1996;270(6):R1257–64.
• Szczepanska-Sadowska E, Stepniakowski K, Skelton M, Cowley A. Prolonged stimulation of intrarenal V1 vasopressin receptors results in sustained hypertension. Am J Physiol Regul Integr Comp Physiol. 1994;267(5):R1217–25.
• Goncharuk VD, Buijs RM, Jhamandas JH, Swaab DF. Vasopressin (VP) and neuropeptide FF (NPFF) systems in the normal and hypertensive human brainstem. J Comp Neurol. 2011;519(1):93–124.
• Martin S, Malkinson T, Veale W, Pittman Q. The action of centrally administered arginine vasopressin on blood pressure in the conscious rabbit. Brain Res. 1985;348(1):137–45.
• Noszczyk B, Łon S, Szczepańska-Sadowska E. Central cardiovascular effects of AVP and AVP analogs with V 1, V 2 and ‘V 3’agonistic or anatagonistic properties in conscious dog. Brain Res. 1993;610(1):115–26.
•• Kc P, Balan KV, Tjoe SS, Martin RJ, LaManna JC, Haxhiu MA, et al. Increased vasopressin transmission from the paraventricular nucleus to the rostral medulla augments cardiorespiratory outflow in chronic intermittent hypoxia-conditioned rats. J Physiol. 2010;588(4):725–40.
• Jansen A, Wessendorf M, Loewy A. Transneuronal labeling of CNS neuropeptide and monoamine neurons after pseudorabies virus injections into the stellate ganglion. Brain Res. 1995;683(1):1–24.
• Hanley MR, Benton HP, Lightman SL, Todd K, Bone EA, Fretten P, et al. A vasopressin-like peptide in the mammalian sympathetic nervous system. Nature. 1984;309(5965):258–61.
• Horn AM, Lightman SL. Vasopressin-stimulated turnover of phosphatidylinositol in the decentralised superior cervical ganglion of the rat. Brain Res. 1988;455(1):18–23.
• Gilbey MP, Coote JH, Fleetwood-Walker S, Peterson DF. The influence of the paraventriculo-spinal pathway, and oxytocin and vasopressin on sympathetic preganglionic neurones. Brain Res. 1982;251(2):283–90.
• Yamaguchi-Shima N, Okada S, Shimizu T, Usui D, Nakamura K, Lu L, et al. Adrenal adrenaline-and noradrenaline-containing cells and celiac sympathetic ganglia are differentially controlled by centrally administered corticotropin-releasing factor and arginine–vasopressin in rats. Eur J Pharmacol. 2007;564(1):94–102.
• Patel K, Schmid P. Vasopressin inhibits sympathetic ganglionic transmission but potentiates sympathetic neuroeffector responses in hindlimb vasculature of rabbits. J Pharmacol Exp Ther. 1988;245(3):779–85.
• Brizzee BL, Walker BR. Vasopressinergic augmentation of cardiac baroreceptor reflex in conscious rats. Am J Phys. 1990;258(4 Pt 2):R860–8.
• Cowley AW Jr, Liard JF. Vasopressin and arterial pressure regulation. Special lecture. Hypertension. 1988;11(2 Pt 2):I25–32.
• Guo G, Schmid PG, Abboud FM. Sites at which vasopressin facilitates baroreflex inhibition of lumbar sympathetic nerve activity. Am J Physiol Heart Circ Physiol. 1986;251(3):H644–55.
• Cox BF, Hay M, Bishop VS. Neurons in area postrema mediate vasopressin-induced enhancement of the baroreflex. Am J Phys. 1990;258(6 Pt 2):H1943–6.
• Hegarty AA, Felder RB. Vasopressin and V1-receptor antagonists modulate the activity of NTS neurons receiving baroreceptor input. Am J Physiol Regul Integr Comp Physiol. 1997;273(1):R143–52.
• Oikawa R, Nasa Y, Ishii R, Kuwaki T, Tanoue A, Tsujimoto G, et al. Vasopressin V1A receptor enhances baroreflex via the central component of the reflex arc. Eur J Pharmacol. 2007;558(1):144–50.
• Balasubramanian L, Sham JS, Yip K-P. Calcium signaling in vasopressin-induced aquaporin-2 trafficking. Pflugers Arch. 2008;456(4):747–54.
• Nielsen S, Frøkiær J, Marples D, Kwon T-H, Agre P, Knepper MA. Aquaporins in the kidney: from molecules to medicine. Physiol Rev. 2002;82(1):205–44.
• Michlig S, Mercier A, Doucet A, Schild L, Horisberger J-D, Rossier BC, et al. ERK1/2 controls Na, K-ATPase activity and transepithelial sodium transport in the principal cell of the cortical collecting duct of the mouse kidney. J Biol Chem. 2004;279(49):51002–12.
• Mordasini D, Bustamante M, Rousselot M, Martin P-Y, Hasler U, Féraille E. Stimulation of Na+ transport by AVP is independent of PKA phosphorylation of the Na-K-ATPase in collecting duct principal cells. Am J Physiol Renal Physiol. 2005;289(5):F1031–9.
•• Ares GR, Caceres PS, Ortiz PA. Molecular regulation of NKCC2 in the thick ascending limb. Am J Physiol Renal Physiol. 2011;301(6):F1143–59. https://doi.org/10.1152/ajprenal.00396.2011.
• Kato A, Klein JD, Zhang C, Sands JM. Angiotensin II increases vasopressin-stimulated facilitated urea permeability in rat terminal IMCDs. Am J Physiol Renal Physiol. 2000;279(5):F835–40.
• Nicco C, Wittner M, DiStefano A, Jounier S, Bankir L, Bouby N. Chronic exposure to vasopressin upregulates ENaC and sodium transport in the rat renal collecting duct and lung. Hypertension. 2001;38(5):1143–9.
• Birumachi J, Hiroyama M, Fujiwara Y, Aoyagi T, Sanbe A, Tanoue A. Impaired arginine-vasopressin-induced aldosterone release from adrenal gland cells in mice lacking the vasopressin V1A receptor. Eur J Pharmacol. 2007;566(1–3):226–30. https://doi.org/10.1016/j.ejphar.2007.03.022.
• Alfaidy N, Blot-Chabaud M, Bonvalet JP, Farman N. Vasopressin potentiates mineralocorticoid selectivity by stimulating 11 beta hydroxysteroid deshydrogenase in rat collecting duct. J Clin Invest. 1997;100(10):2437–42. https://doi.org/10.1172/jci119785.
• White PC. Disorders of aldosterone biosynthesis and action. N Engl J Med. 1994;331(4):250–8.
• Ahmed AB, George BC, Gonzalez-Auvert C, Dingman JF. Increased plasma arginine vasopressin in clinical adrenocortical insufficiency and its inhibition by glucosteroids. J Clin Invest. 1967;46(1):111.
• Bugaj V, Pochynyuk O, Stockand JD. Activation of the epithelial Na+ channel in the collecting duct by vasopressin contributes to water reabsorption. Am J Physiol Renal Physiol. 2009;297(5):F1411–8.
• Mironova E, Bugaj V, Roos KP, Kohan DE, Stockand JD. Aldosterone-independent regulation of the epithelial Na+ channel (ENaC) by vasopressin in adrenalectomized mice. Proc Natl Acad Sci U S A. 2012;109(25):10095–100. https://doi.org/10.1073/pnas.1201978109.
• Djelidi S, Beggah A, Courtois-Coutry N, Fay M, Cluzeaud F, Viengchareun S, et al. Basolateral translocation by vasopressin of the aldosterone-induced pool of latent Na-K-ATPases is accompanied by alpha1 subunit dephosphorylation: study in a new aldosterone-sensitive rat cortical collecting duct cell line. J Am Soc Nephrol. 2001;12(9):1805–18.
• Perlewitz A, Nafz B, Skalweit A, Fähling M, Persson PB, Thiele B-J. Aldosterone and vasopressin affect α-and γ-ENaC mRNA translation. Nucleic Acids Res. 2010;38(17):5746–60.
• Hori K, Nagai T, Izumi Y, Kimura M, Hasuike Y, Nakayama Y, et al. Vasopressin V1a receptor is required for nucleocytoplasmic transport of mineralocorticoid receptor. Am J Physiol Renal Physiol. 2012;303(7):F1080–8. https://doi.org/10.1152/ajprenal.00052.2012.
• Izumi Y, Hori K, Nakayama Y, Kimura M, Hasuike Y, Nanami M, et al. Aldosterone requires vasopressin V1a receptors on intercalated cells to mediate acid-base homeostasis. J Am Soc Nephrol. 2011;22(4):673–80.
• Saxena A, Bachelor M, Park YH, Carreno FR, Nedungadi TP, Cunningham JT. Angiotensin II induces membrane trafficking of natively expressed transient receptor potential vanilloid type 4 channels in hypothalamic 4B cells. Am J Physiol Regul Integr Comp Physiol. 2014;307(8):R945–55.
• Carreno FR, Ji LL, Cunningham JT. Altered central TRPV4 expression and lipid raft association related to inappropriate vasopressin secretion in cirrhotic rats. Am J Physiol Regul Integr Comp Physiol. 2009;296(2):R454–66. https://doi.org/10.1152/ajpregu.90460.2008.
• Aoyagi T, Koshimizu T-A, Tanoue A. Vasopressin regulation of blood pressure and volume: findings from V1a receptor-deficient mice. Kidney Int. 2009;76(10):1035–9.
• Cudnoch-Jedrzejewska A, Dobruch J, Puchalska L, Szczepanska-Sadowska E. Interaction of AT1 receptors and V1a receptors-mediated effects in the central cardiovascular control during the post-infarct state. Regul Pept. 2007;142(3):86–94. https://doi.org/10.1016/j.regpep.2007.01.010.
• Cudnoch-Jedrzejewska A, Szczepanska-Sadowska E, Dobruch J, Puchalska L, Ufnal M, Kowalewski S, et al. Differential sensitisation to central cardiovascular effects of angiotensin II in rats with a myocardial infarct: relevance to stress and interaction with vasopressin. Stress. 2008;11(4):290–301. https://doi.org/10.1080/10253890701794445.
• Ito S, Komatsu K, Tsukamoto K, Kanmatsuse K, Sved AF. Ventrolateral medulla AT1 receptors support blood pressure in hypertensive rats. Hypertension. 2002;40(4):552–9.
• Szczepanska-Sadowska E. Role of neuropeptides in central control of cardiovascular responses to stress. J Physiol Pharmacol. 2008;59(Suppl 8):61–89.
• Littlejohn NK, Siel RB Jr, Ketsawatsomkron P, Pelham CJ, Pearson NA, Hilzendeger AM, et al. Hypertension in mice with transgenic activation of the brain renin-angiotensin system is vasopressin dependent. Am J Physiol Regul Integr Comp Physiol. 2013;304(10):R818–28. https://doi.org/10.1152/ajpregu.00082.2013.
• Gebke E, Müller A, Jurzak M, Gerstberger R. Angiotensin II-induced calcium signalling in neurons and astrocytes of rat circumventricular organs. Neuroscience. 1998;85(2):509–20.
• Jeulin A, Nicolaidis S. Evidence for vasopressin V1 receptors of rostrodiencephalic neurons: iontophoretic studies in the in vivo rat. Responses to oxytocin and to angiotensin. Brain Res Bull. 1988;20(6):817–23.
• Consolim-Colombo FM, Hay M, Smith TC, Elizondo-Fournier M, Bishop VS. Subcellular mechanisms of angiotensin II and arginine vasopressin activation of area postrema neurons. Am J Physiol Regul Integr Comp Physiol. 1996;271(1):R34–41.
• Hay M, Edwards G, Lindsley K, Murphy S, Sharma R, Bhalla R, et al. Increases in cytosolic Ca 2+ in rat area postrema/mNTS neurons produced by angiotensin II and arginine-vasopressin. Neurosci Lett. 1993;151(2):121–5.
• Hasser EM, Bishop VS, Hay M. Interactions between vasopressin and baroreflex control of the sympathetic nervous system. Clin Exp Pharmacol Physiol. 1997;24(1):102–8.
• Walker J, Jennings DB. During acute hypercapnia vasopressin inhibits an angiotensin drive to ventilation in conscious dogs. J Appl Physiol. 1995;79(3):786–94.
• Gonzalez AA, Cifuentes-Araneda F, Ibaceta-Gonzalez C, Gonzalez-Vergara A, Zamora L, Henriquez R, et al. Vasopressin/V2 receptor stimulates renin synthesis in the collecting duct. Am J Physiol Renal Physiol. 2016;310(4):F284–93.
• Wong NL, Tsui JK. Angiotensin II upregulates the expression of vasopressin V2 mRNA in the inner medullary collecting duct of the rat. Metabolism. 2003;52(3):290–5.
• Torp M, Brønd L, Hadrup N, Nielsen J, Prætorius J, Nielsen S, et al. Losartan decreases vasopressin-mediated cAMP accumulation in the thick ascending limb of the loop of Henle in rats with congestive heart failure. Acta Physiol. 2007;190(4):339–50.
• Lee Y-J, Song I-K, Jang K-J, Nielsen J, Frøkiær J, Nielsen S, et al. Increased AQP2 targeting in primary cultured IMCD cells in response to angiotensin II through AT 1 receptor. Am J Physiol Renal Physiol. 2007;292(1):F340–50.
• Wang W, Li C, Summer S, Falk S, Schrier RW. Interaction between vasopressin and angiotensin II in vivo and in vitro: effect on aquaporins and urine concentration. Am J Physiol Renal Physiol. 2010;299(3):F577–84.
• Kwon T-H, Nielsen J, Knepper MA, Frøkiær J, Nielsen S. Angiotensin II AT 1 receptor blockade decreases vasopressin-induced water reabsorption and AQP2 levels in NaCl-restricted rats. Am J Physiol Renal Physiol. 2005;288(4):F673–84.
Acknowledgements
The study was supported by the Medical University of Warsaw (Project 1MA/2017). The authors wish to express their gratitude to Mrs. Edita Lalik and Mr. Marcin Kumosa for the secretarial and technical cooperation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest relevant to this manuscript
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Mechanisms of Hypertension
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Szczepanska-Sadowska, E., Czarzasta, K. & Cudnoch-Jedrzejewska, A. Dysregulation of the Renin-Angiotensin System and the Vasopressinergic System Interactions in Cardiovascular Disorders. Curr Hypertens Rep 20, 19 (2018). https://doi.org/10.1007/s11906-018-0823-9
Published:
DOI: https://doi.org/10.1007/s11906-018-0823-9