
Abstract
Purpose of Review
To revise experimental and clinical data supporting a less traditional role of anticoagulation for treating portal hypertension in patients with cirrhosis.
Recent Findings
Portal hypertension is the main driver of complications such as ascites, variceal hemorrhage, and hepatic encephalopathy, with inflammation as a key component. The traditional view of cirrhosis as a pro-hemorrhagic condition has recently changed, prothrombotic complications being recognized as frequently as the hemorrhagic ones. Several data indicate a close relationship between inflammation, prothrombotic status, worsening of hepatic fibrosis, and portal hypertension both in animal models and in patients with chronic liver disease. These findings indicate that anticoagulation may represent a potent tool to act on fibrogenesis and therefore consequently to treat portal hypertension. All anticoagulants have good to optimal safety profiles and can be used in patients with advanced chronic liver disease with confidence.
Summary
Anticoagulation has a role as a pleiotropic treatment of portal hypertension in cirrhosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
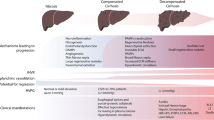
Portal hypertension (PH) is the most common complication of cirrhosis and the main determinant of clinical liver decompensation, including ascites, variceal bleeding, and hepatic encephalopathy. PH initially results from increased intrahepatic resistance to blood flow. Later, it is worsened by splanchnic vasodilation (and increased blood flow to the portal vein) [1, 2••], with systemic inflammation acting as a trigger for the shift from compensated to decompensated status [2••]. Increased resistances to portal blood flow derive from both architectural alterations of the liver parenchyma and dynamic increase in the hepatic vascular tone [3, 4]. Architectural distortion of the cirrhotic liver is partly due to excessive synthesis of extracellular matrix components due to dysregulated activation of fibrogenic cells such as hepatic stellate cells (HSC) and portal myofibroblasts [5]. Several data suggest that liver fibrogenesis is greatly influenced by lobular inflammation [6] and thrombosis [7].
Coagulative Balance in Patients with Liver Cirrhosis and Its Role in Fibrogenesis
Patients with liver cirrhosis were traditionally considered at high risk of bleeding episodes for the concurrent presence of thrombocytopenia, prolongation of prothrombin time (PT), and of activated partial thromboplastin time. Accordingly, guidelines recommended in patients undergoing invasive procedures the use of replacement therapy with platelets (PLT) and/or transfusion of fresh frozen plasma (FFP) [8, 9]. More recently, it has become quite clear that in patients with uncomplicated liver cirrhosis, bleeding episodes are the direct consequence of PH and not of a coagulative imbalance [9]. Indeed, they are mostly represented by hemorrhage due to ruptured varices or bleeding from portal hypertensive gastropathy/enteropathy while spontaneous bleeding from other sources is rare [10•]. This revised view of coagulation in patients with compensated cirrhosis shows that they are in a situation of “balanced hemostasis” as both procoagulant and anticoagulant factors are proportionately decreased [11]. When the coagulative balance is disrupted, e.g., by infection and acute kidney failure, it can hang towards either bleeding or thrombosis. One important implication of this view is that cirrhotic patients undergoing invasive procedures should receive prophylactic treatment with PLT or FFP not according to INR or platelet levels but on the basis of more comprehensive tests [12, 13, 14•]. Thromboelastography (TEG) or thromboelastometry (ROTEM), which assess the different phases of the coagulation cascade, are indeed much more informative by simultaneously assessing anticoagulant and procoagulant factors [13, 15].
Apart from bleeding risk assessment, this modified view of coagulation in cirrhosis has other implications. Several data have shown a close relationship between activation of coagulation and a more rapid progression of liver fibrosis [16,17,18]. Although non-novel, most appealing and informative regarding the relationship of procoagulative status and liver damage progression are the studies by Wanless et al. [7, 19], who showed frequent thrombotic occlusion of small intrahepatic veins and sinusoids in cirrhosis. This leads to intimal fibrosis and vein obstruction, with ensuing “parenchymal extinction” defined as the irreversible loss of contiguous hepatocytes and their replacement by fibrous tissue. Another hypothesis that involves thrombin generation considers functional activation of hepatic stellate cells by protease-activated receptors as the underlying mechanism leading to extracellular matrix deposition [20]. All these findings suggest that anticoagulation may interfere with liver fibrogenesis and consequently may be a potent tool for treating portal hypertension. In the following paragraphs, we will review experimental and clinical data supporting this less traditional role of anticoagulation.
Experimental Studies
A relationship between coagulation and establishment and/or progression of fibrosis has been suggested since long [21, 22]. In a murine model of acute hepatitis virus infection, the authors were able to demonstrate the presence of microthrombi in the areas of tissue necrosis, with ensuing ischemic damage, injury by inflammatory mediators, and repair by scarring [23]. A further support to this hypothesis was given by Neubauer et al. [24] who suggested that during acute liver damage, a “clotting-like process” involving fibrinogen/fibrin deposition would create a “provisional matrix” attracting inflammatory and matrix-producing cells, then favoring proliferation of fibroblasts and collagen deposition. The evidence that a procoagulant state strongly favors progression of hepatic fibrosis also came from the studies by Anstee et al. [25] in the prothrombotic factor V Leiden mutant mice. These mice developed significantly higher hepatic fibrosis than control mice after chronic carbon tetrachloride (CCl4) exposure.
More recent studies focused on the role of coagulative abnormalities in experimental models of non-alcoholic steatohepatitis (NASH), the etiology, which is going to become the predominant cause of chronic liver disease (CLD) in the future years. Geys et al. [26] showed that the Adamts13 deficiency in a murine model of diet-induced liver steatosis is associated with development of hepatic microthrombosis in obese mice. Nault et al. [27] in a murine model exposed to the environmental contaminant 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), which triggers the progression of non-alcoholic fatty liver disease (NAFLD) to NASH, showed that activation of the coagulation cascade can drive hepatic inflammation and fibrosis independently of hepatic lipid accumulation.
As the evidence of the link between procoagulant status and development of fibrosis was strong, the evaluation of anticoagulation as a preventive or therapeutic measure was an obvious consequence (Table 1). Early in vitro experimental data had shown that heparin is able to inhibit both gene and protein expressions of type I procollagen and fibronectin [28]. In vivo long-term administration of low anticoagulant activity heparins (LAAH) was able to suppress the hepatic fibrosis induced by CCl4 or injection of porcine serum [29]. Several other studies, in CCl4 or thioacetamide murine models, using different drugs (dalteparin, enoxaparin, aspirin) confirmed these early results [25, 28,29,30,31,32]. Interestingly, the study from Cerini et al. [32] showed that anticoagulation was effective when given both prophylactically or therapeutically. The effect of enoxaparin was multifaceted. A significant improvement in HSC phenotype, as shown by decreased alpha smooth muscle actin (α-SMA) expression and liver desmin content, associated with a notable reduction in liver fibrosis was observed. Furthermore, enoxaparin-treated rats showed a marked and significant decrease in fibrin deposition in the liver in comparison with vehicle-treated rats, suggesting a reduction of thrombotic events within the liver. These findings were associated with a significant increase in portal blood flow reflecting a marked and significant reduction in hepatic vascular resistance.
More recently, rivaroxaban, a direct factor Xa inhibitor, was also shown to markedly reduce fibrin deposition and intrahepatic microthrombosis in CCl4- and thioacetamide-treated rats. Rivaroxaban also significantly decreased portal pressure in both models of cirrhosis without changes in portal blood flow, suggesting a reduction in intrahepatic vascular resistance [33••]. Another interesting study employing one of the new direct-acting oral anticoagulants (DOACs), dabigatran was performed in C57BL/6 J mice fed a high-fat diet [34]. Dabigatran significantly reduced hepatic fibrin deposition, hepatic inflammation, hepatocellular injury, and steatosis. Interestingly, weight gain in dabigatran-treated mice on high-fat diet was significantly lower than in control animals. The authors, on the basis of transcriptomic analysis that showed that thrombin inhibition with dabigatran significantly suppressed genes associated with lipid metabolism, indicated a primary role for thrombin as driver of NAFLD pathogenesis.
The only contrasting study on the role of anticoagulation for preventing fibrosis comes from Fortea et al. [35]. This study evaluated several different doses of enoxaparin as well as diverse protocols of administration in three different models of advanced cirrhosis in rats. The study was totally negative. Enoxaparin did not improve liver fibrosis, portal hypertension, or endothelial dysfunction. The authors hypothesized that these negative results were possibly due to the use of experimental models in a late stage of advanced cirrhosis, and therefore too advanced to be significantly influenced by therapeutic interventions. It should be underlined, however, that the experimental models used were different from those of the other studies: in this study, CCl4 was administered by inhalation or gavage instead than intra-peritoneally. Last but not least, recent data from Yan et al. [36•] indicate that different molecules of low molecular weight heparin (LMWH) might have different effects. These authors created a depolymerized LMWH library, using a specific or a combination of them (Hep I, II, or III). Only LMWH depolymerized by heparinase I (Hep I-11) was able to significantly decrease liver inflammation by suppressing TNF-α and IL-1β secretion, and minimizing hepatic fibrogenesis. Other LMWHs, with lower molecular weights than that obtained by heparinase I (Hep I-11) had scarce or absent antifibrogenic activity. This suggests that only drugs with larger molecular weight have relevant antifibrogenic activity. This could help interpreting the data obtained by Abdel-Salam et al. [37] in a bile duct-ligated rat model with three different LMWHs. While nadroparin and enoxaparin showed a beneficial effect, obtaining a significant decrease of fibrosis in comparison with controls, tinzaparin, which has a lower molecular weight than the other two, had, as a paradoxical effect, an increase in fibrosis. Overall, these data underline the need for further investigation of the relationship between LMWH structure and their activity as modulators of liver fibrosis [36•].
Clinical Studies
Only few studies in humans have focused on the relationship between activation of coagulation, hepatic fibrosis, and subsequent development of portal hypertension (Table 1). Marra et al. [38] showed that thrombin could stimulate proliferation of cultured HSC and secretion of monocyte chemo-attractant protein-1 (MCP-1) through proteolytic activation of its receptor [38]. Upregulation of thrombin receptors was shown to occur in hepatitis C virus (HCV) and hepatitis B virus (HBV)—related acute or chronic liver disease—and at the same time, decreased plasma levels of antithrombin were shown to be correlated with more severe fibrosis in HCV infection [39]. As already shown in the experimental model by Anstee et al. [25], patients with HCV infection, when carrier of factor V Leiden mutation, have a fourfold risk of rapidly progressive fibrosis [40]. In human, it has been shown that patients with HCV carrying the prothrombin G20210A mutation (a pro-thrombotic condition) have a fivefold risk of liver fibrosis progression [41]. Factor V Leiden or prothrombin G20210A mutations were associated with an increased risk of clinically relevant liver fibrosis (defined as a liver stiffness > 8 kPa estimated by transient elastography) in the general population [42], corroborating the important role played by pro-thrombotic condition on liver fibrosis progression. On the opposite, patients with hemophilia and hepatitis C have a less severe hepatic disease than HCV-infected patients without hemophilia: in the study by Assy et al., [43] none of the hemophiliacs had histological evidence of advanced disease (bridging fibrosis and/or cirrhosis) as compared to controls.
After the increasing recognition of NASH as an etiologic factor for CLD, several studies have focused on the coagulative features in this condition, demonstrating that indeed, NASH is a prothrombotic condition. Wanless et al. [44] were among the first to show that in the progression from NAFLD to NASH-cirrhosis, the necrotic damage induced by lipid toxicity, leading to a direct inflammatory injury to hepatic veins, is followed by venous obstruction with secondary collapse, fibrous septation, and cirrhosis. Assy et al. [45] showed that about half of patients with NASH have genetic or acquired prothrombotic risk factors and that this is related with the severity of liver fibrosis. Tripodi et al. [46] later confirmed that the procoagulant imbalance in NASH is characterized by increased factor VIII and decreased protein C and antithrombin levels.
As in the animal models, the evaluation of the role in humans of anticoagulation for the treatment of fibrosis and hence of portal hypertension has a sound rationale. A study with short-term treatment with heparin or LMWH was performed a few years ago in patients with chronic hepatitis B [47]. Both treatments improved serum levels of (ALT), bilirubin, and reduced hyaluronic acid and type IV collagen concentrations compared with controls [47]. After treatment, the deposition of collagen in liver tissue was significantly decreased. These results indicate that anticoagulation inhibits hepatic stellate cells and it can be considered an applicable option for patients with CLD. In a later prospective randomized controlled clinical study [48], 75 patients were randomly divided into three groups: conventional treatment (n = 15), conventional treatment plus imported LMWH (n = 30), and conventional treatment plus indigenous LMWH (n = 30). Despite the short course of treatment (3 weeks), both LMWH treatment groups showed a distinct improvement in liver function tests and a marked decrease of propeptide of procollagen type III (PIIIP) and type IV collagen levels in comparison with conventional treatment. Most importantly, portal vein blood flow velocity markedly improved.
An interesting phase II study undergoing in the UK is evaluating the efficacy of anticoagulation with warfarin for prevention of fibrosis in liver transplant (LT) patients with recurrent HCV-infection [49]. The interim analysis showed a significant reduction of fibrosis 1 year after LT in anticoagulated patients. Although the recent availability of extremely effective and potent direct-acting antivirals against HCV, which can obtain sustained viral response (SVR) rate close to 100% and extensive regression of fibrosis, will probably make this therapeutic option obsolete, still, these results are extremely relevant as a proof of concept of the role of anticoagulation as antifibrotic therapy.
An indication of the results attainable with anticoagulation in terms of prevention of complications associated with portal hypertension comes from the randomized control trial performed by our group [50] for the prevention of portal vein thrombosis with LMWH. In this trial, we randomized 70 patients with cirrhosis, moderate liver failure (Child B7-C10), and no portal vein thrombosis (PVT) to receive prophylactic doses of enoxaparin (treatment arm) or no treatment (control arm) for 48 weeks. Patients treated with enoxaparin had a significantly lower incidence of PVT and had a significantly lower incidence of liver decompensation (mainly ascites) and of mortality than controls. While the prevention of PVT (9% in enoxa-treated vs. 28% in controls at 2 years) could be expected, much less obvious was the prevention of the other complications, namely decompensation (12% in enoxa-treated vs. 59% at 1 year) which was associated with a significantly improved overall survival (60 vs. 40%). In the anticoagulated group, both the incidence of bacterial infection and the markers of bacterial translocation and of systemic inflammation were significantly lower than in the control group. We hypothesized that anticoagulation acted by improving intestinal microcirculation, diminishing bacterial translocation, and the consequent systemic inflammation, one of the main drivers of decompensation [1, 2••]. Most relevant was the fact that the positive effect associated with enoxaparin was maintained for a prolonged period of time after stopping treatment, which is consistent with a mechanism of slow return to the baseline conditions of altered intestinal microcirculation. These observations provide a rationale to suggest that anticoagulation in patients with cirrhosis may have beneficial effects on liver function and portal hemodynamics beyond prevention of portal vein thrombosis, by counteracting the mechanisms of PH worsening, which develop outside the liver when splanchnic vasodilation takes place. The lack so far of other effective antifibrotic treatment indicates in anticoagulation a possibility ready at hand, although prospective randomized confirmatory studies would be necessary. Ongoing registered trials exploring the effect of anticoagulation on hard clinical outcomes in cirrhotic patients are reported in Table 2. Among these studies, a multicenter trial proposed in France with LMWH (Childbenox) was prematurely discontinued because of low rate of enrollment. The others are still recruiting, but no preliminary data are available.
Safety
Safety of anticoagulation in patients with cirrhosis has always been a matter of debate. However, the change of view, which has occurred in the last years, regarding hemorrhagic risk due to altered coagulative balance of patients with cirrhosis, has also greatly modified the attitude toward anticoagulation. Among the various categories of anticoagulants, only vitamin K antagonists (VKA such as warfarin and acenocoumarol) are still considered to have a narrower therapeutic window and to carry higher risk of bleeding complications in patients with liver disease. Indeed, a retrospective analysis of 63 patients with cirrhosis and PVT treated with VKA showed an increased risk of minor bleeding episodes [51]. On the whole, however, this did not negatively influence survival, and portal hypertension, rather than anticoagulants, was found as the major driver of major bleedings in these patients [51].
For unfractioned heparin (UH) and LMWHs, there is ample evidence of their safety, either when used for treating PVT [52] or for preventing it [50]. A recent meta-analysis on the effects of anticoagulants in patients with cirrhosis and PVT showed that anticoagulated patients did not have an excess of major and minor bleedings and, most importantly, had less incidence of variceal bleeding [52]. When used prophylactically [50], enoxaparin, 4000 IU daily, was well tolerated and showed a very good safety profile. No significant difference during active period between treatment group and controls was found in terms of bleeding events (only 3 out of 70 patients experienced bleeding events from esophageal varices, 2 in the treatment arm and 1 in control arm without significant difference), or in terms of hemoglobin levels. Safety of anticoagulation has been demonstrated also in patients with cirrhosis undergoing invasive procedures [12, 53, 54•]. Anticoagulation did not impact outcome in patients with active upper gastrointestinal bleeding [53] or in those undergoing elective variceal band ligation procedures for primary or secondary prophylaxis [54•]. The bleeding risk of these patients, rather than to anticoagulation, is related to the severity of portal hypertension (larger size of esophageal varices, higher number of bands used for variceal band ligation) or to the concomitant presence of multiorgan dysfunction expressed by SOFA score and of comorbidities [10, 53, 54•].
Less information on safety in patients with cirrhosis is available on direct oral anti coagulants (DOACs) as registration studies excluded patients with liver disease, for concern about increased risk of bleeding in the absence of reversal agents. De Gottardi et al. [55] retrospectively evaluated antithrombotic treatment with DOACs in patients with splanchnic vein thrombosis and cirrhosis and concluded for a substantial effectiveness and safety of these drugs. Furthermore, for all DOACs, reversal agents are now available.
Conclusions
The available data, both experimental and clinical, strongly support the link between altered coagulation, occurrence of microvascular thrombosis (“parenchymal extinction”) [7, 19], inflammation, and fibrotic evolution of the liver parenchyma. In patients with chronic liver disease, alterations of coagulation are usually not early detectable due to their occurrence after the establishment of cirrhosis and severe portal hypertension. Therefore, for earlier stages, etiology-targeted therapies, like direct-acting antiviral for HCV or nucleoside analogues for HBV, are the treatment of choice to prevent occurrence and progression of fibrosis and consequent onset of portal hypertension. However, for patients with more advanced disease or those with NASH, effective antifibrotic therapies are still lacking and therefore therapeutic anticoagulation could be a relevant option to be validated. A drawback so far for an extensive application of anticoagulation as antifibrotic therapy and therefore as a treatment for portal hypertension was the fact that the most evaluated category of drugs and the one with an optimal safety profile (i.e., LMWHs) required parenteral administration, thus decreasing compliance in patients often already overloaded with medicines. The availability of the new DOACs will hopefully allow overcoming this difficulty. The placebo-controlled prospective trial with rivaroxaban expected to be completed in 2019 (Table 2) will constitute an extremely relevant opportunity to confirm the potential role of anticoagulation as a therapeutic modality to decrease progression of fibrosis and to treat established portal hypertension.
Change history
16 November 2018
The article The Role of Anticoagulation in Treating Portal Hypertension, written by Laura Turco, Filippo Schepis and Erica Villa, was originally published electronically on the publisher’s internet portal (currently SpringerLink) on 18 June 2018 without open access.
Abbreviations
- ALT:
-
Alanine aminotransferase
- CCl4 :
-
Carbon tetrachloride
- CLD:
-
Chronic liver disease
- DOACs:
-
Direct-acting oral anticoagulants
- FFP:
-
Fresh frozen plasma
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- Hep:
-
Heparinase
- HSC:
-
Hepatic stellate cells
- HVPG:
-
Hepatic venous pressure gradient
- IL-1β:
-
Interleukin-1 beta
- LAAH:
-
Low anticoagulant activity heparin
- LMWH:
-
Low molecular weight heparin
- LT:
-
Liver transplant
- MCP-1:
-
Monocyte chemo-attractant protein-1
- NAFLD:
-
Non-alcoholic fatty liver disease
- NASH:
-
Non-alcoholic steatohepatitis
- PAR-1:
-
Protease activated receptor-1
- PH:
-
Portal hypertension
- PLT:
-
Platelets
- PIIIP:
-
Propeptide of procollagen type III
- PT:
-
Prothrombin time
- PVT:
-
Portal vein thrombosis
- ROTEM:
-
Thromboelastometry
- α-SMA:
-
Alpha smooth muscle actin
- SVR:
-
Sustained viral response
- TEG:
-
Thromboelastography
- TCDD:
-
2,3,7,8-Tetrachlorodibenzo-p-dioxin
- TNF-α:
-
Tumor necrosis factor alpha
- VKA:
-
Vitamin K antagonists
- UH:
-
Unfractioned heparin
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Bernardi M, Moreau R, Angeli P, Schnabl B, Arroyo V. Mechanisms of decompensation and organ failure in cirrhosis: from peripheral arterial vasodilation to systemic inflammation hypothesis. J Hepatol. 2015;63:1272–84.
•• Turco L, Garcia-Tsao G, Magnani I, Bianchini M, Costetti M, Caporali C, et al. Cardiopulmonary hemodynamics and C-reactive protein as prognostic indicators in compensated and decompensated cirrhosis. J Hepatol. 2018;68:949–58.This study demonstrates the relevant role of inflammation as a key factor involved both in decompensation and death of cirrhotic patients.
Bosch J, Garcia-Pagan JC. Complications of cirrhosis I. Portal hypertension. J Hepatol. 2000;32:141–56.
Garcia-Pagan JC, Gracia-Sancho J, Bosch J. Functional aspects on the pathophysiology of portal hypertension in cirrhosis. J Hepatol. 2012;57:458–61.
Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol. 2013 Oct;3(4):1473–92.
Tacke F, Luedde T, Trautwein C. Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol. 2009;36:4–12.
Wanless IR, Wong F, Blendis LM, Greig P, Heathcote EJ, Levy G. Hepatic and portal vein thrombosis in cirrhosis: possible role in development of parenchymal extinction and portal hypertension. Hepatology. 1995;21:1238–47.
Liumbruno GM, Bennardello F, Lattanzio A, Piccoli P, Rossetti G. Italian Society of Transfusion Medicine and Immunohaematology (STMI) Working Party. Recommendations for the transfusion management of patients in the peri-operative period. III. The post-operative period. Blood Transfus. 2011;9:320–35.
Patel IJ, Davidson JC, Nikolic B, Salazar GM, Schwartzberg MS, Walker TG, et al. Consensus guidelines for periprocedural management of coagulation status and hemostasis risk in percutaneous image-guided interventions. J Vasc Interv Radiol. 2012;23:727–36.
• de Franchis R. Baveno VI Faculty. Expanding consensus in portal hypertension: report of the Baveno VI Consensus Workshop: stratifying risk and individualizing care for portal hypertension. J Hepatol. 2015;63:743–52. Reference text for all aspects of pathogenesis, treatment, and complications of portal hypertension.
Weeder PD, Porte RJ, Lisman T. Hemostasis in liver disease: implications of new concepts for perioperative management. Transfus Med Rev. 2014;28:107–13.
Schepis F, Turco L, Bianchini M, Villa E. Prevention and management of bleeding risk related to invasive procedures in cirrhosis. Semin Liver Dis 2018.
De Pietri L, Bianchini M, Montalti R, De Maria N, Di Maira T, Begliomini B, et al. Thrombelastography-guided blood product use before invasive procedures in cirrhosis with severe coagulopathy: a randomized, controlled trial. Hepatology. 2016;63:566–73.
• Hung A, Garcia-Tsao G. Acute kidney injury, but not sepsis, is associated with higher procedure-related bleeding in patients with decompensated cirrhosis. Liver Int 2018;2. Ahead of Print. This study identifies a novel risk factor involved in derangement of coagulation in patients with liver cirrhosis.
Andriulli A, Tripodi A, Angeli P, Senzolo M, Primignani M, Giannini EG, et al. Hemostatic balance in patients with liver cirrhosis: report of a consensus conference. Dig Liver Dis. 2016;48:455–67.
Anstee QM. Coagulation status modulates murine hepatic fibrogenesis: implications for the development of novel therapies. J Thromb Haemost. 2008;6:1336–43.
Northup PG, Sundaram V, Fallon MB, Reddy KR, Balogun RA, Sanyal AJ, et al. Hypercoagulation and thrombophilia in liver disease. J Thromb Haemost. 2008;6:2–9.
Duplantier JG. A role for thrombin in liver fibrosis. Gut. 2004;53:1682–7.
Wanless IR. Micronodular transformation (nodular regenerative hyperplasia) of the liver: a report of 64 cases among 2,500 autopsies and a new classification of benign hepatocellular nodules. Hepatology. 1990;11:787–97.
Anstee QM, Dhar A, Thursz MR. The role of hypercoagulability in liver fibrogenesis. Clin Res Hepatol Gastroenterol. 2011;35:526–33.
MacPhee PJ, Dindzans VJ, Fung LS, Levy GA. Acute and chronic changes in the microcirculation of the liver in inbred strains of mice following infection with mouse hepatitis virus type 3. Hepatology. 1985;5:649–60.
MacPhee PJ, Schmidt EE, Keown PA, Groom AC. Microcirculatory changes in livers of mice infected with murine hepatitis virus: evidence from microcorrosion casts and measurements of red cell velocity. Microvasc Res. 1988;36:140–9.
Levy GA, MacPhee PJ, Fung LS, Fisher MM, Rappaport AM. The effect of mouse hepatitis virus infection on the microcirculation of the liver. Hepatology. 1983;3:964–73.
Neubauer K, Knittel T, Armbrust T, Ramadori G. Accumulation and cellular localization of fibrinogen/fibrin during short-term and long-term rat liver injury. Gastroenterology. 1995;108:1124–35.
Anstee QM, Wright M, Goldin R, Martinelli A, Cox R, Thursz MR. Coagulation status modulates hepatic fibrosis: implications for the development of novel therapies. J Thromb Haemost. 2008;6:1336–43.
Geys L, Bauters D, Roose E, Tersteeg C, Vanhoorelbeke K, Hoylaerts MF, et al. ADAMTS13 deficiency promotes microthrombosis in a murine model of diet-induced liver steatosis. Thromb Haemost. 2017;117:19–26.
Nault R, Fader KA, Kopec AK, Harkema JR, Zacharewski TR, Luyendyk JP, et al. From the cover: coagulation-driven hepatic fibrosis requires protease activated receptor-1 (PAR-1) in a mouse model of TCDD-elicited steatohepatitis. Toxicol Sci. 2016;154:381–91.
Li W, Zhang J, Huang G, Zhu H, Zhang X, Zhang Y. Effects of heparin on the growth, extracellular matrix and matrix metalloproteinase gene expression in rat hepatic stellate cells. Zhonghua Gan Zang Bing Za Zhi (Chin J Hepatol). 2000;8:200–2.
Li W, Zhang J, Huang Q, Zhu H, Zhang X. Long-term administering low anticoagulant activity heparin can lessen rat hepatic fibrosis induced by either CCl4 or porcine serum injection. Hepatol Res. 2006;36:115–23.
Abe W, Ikejima K, Lang T, Okumura K, Enomoto N, Kitamura T, et al. Low molecular weight heparin prevents hepatic fibrogenesis caused by carbon tetrachloride in the rat. J Hepatol. 2007;46:286–94.
Assy N, Hussein O, Khalil A, Luder A, Szvalb S, Paizi M, et al. The beneficial effect of aspirin and enoxaparin on fibrosis progression and regenerative activity in a rat model of cirrhosis. Dig Dis Sci. 2007;52:1187–93.
Cerini F, Vilaseca M, Lafoz EE, García-Irigoyen O, García-Calderó H, Tripathi DM, et al. Enoxaparin reduces hepatic vascular resistance and portal pressure in cirrhotic rats. J Hepatol. 2016 Apr;64(4):834–42.
•• Vilaseca M, García-Calderó H, Lafoz E, García-Irigoyen O, Avila MA, Reverter JC, et al. The anticoagulant rivaroxaban lowers portal hypertension in cirrhotic rats mainly by deactivating hepatic stellate cells. Hepatology. 2017;65:2031–44. First demonstration that also DOACs have not only anticoagulant activity but also antifibrotic properties.
Kopec AK, Joshi N, Towery KL, Kassel KM, Sullivan BP, Flick MJ, et al. Thrombin inhibition with dabigatran protects against high-fat diet-induced fatty liver disease in mice. J Pharmacol Exp Ther. 2014;351:288–97.
Fortea JI, Zipprich A, Fernandez-Mena C, Puerto M, Bosoi CR, Almagro J, et al. Enoxaparin does not ameliorate liver fibrosis or portal hypertension in rats with advanced cirrhosis. Liver Int. 2018;38:102–12.
• Yan Y, Guan C, Du S, Zhu W, Ji Y, Su N, et al. Effects of enzymatically depolymerized low molecular weight heparins on CCl4-induced liver fibrosis. Front Pharmacol. 2017;8:514. Relevant study that underlines the possible different role of LMWH with different molecular weight and composition.
Abdel-Salam OM, Baiuomy AR, Ameen A, Hassan NS. A study of unfractionated and low molecular weight heparins in a model of cholestatic liver injury in the rat. Pharmacol Res. 2005;51:59–67.
Marra F, Grandaliano G, Valente AJ, Abboud HE. Thrombin stimulates proliferation of liver fat-storing cells and expression of monocyte chemotactic protein-1: potential role in liver injury. Hepatology. 1995;22:780–7.
Poujol-Robert A, Rosmorduc O, Serfaty L, Coulet F, Poupon R, Robert A. Genetic and acquired thrombotic factors in chronic hepatitis C. Am J Gastroenterol. 2004;99:527–31.
Wright M, Goldin R, Hellier S, Knapp S, Frodsham A, Hennig B, et al. Factor V Leiden polymorphism and the rate of fibrosis development in chronic hepatitis C virus infection. Gut. 2003;52:1206–10.
Maharshak N, Halfon P, Deutsch V, Peretz H, Berliner S, Fishman S, et al. Increased fibrosis progression rates in hepatitis C patients carrying the prothrombin G20210A mutation. World J Gastroenterol. 2011 Dec 7;17(45):5007–13.
Plompen EP, Darwish Murad S, Hansen BE, Loth DW, Schouten JN, Taimr P, et al. Prothrombotic genetic risk factors are associated with an increased risk of liver fibrosis in the general population. Rotterdam Study J Hepatol. 2015;63:1459–65.
Assy N, Pettigrew N, Lee SS, Chaudhary RK, Johnston J, Minuk GY. Are chronic hepatitis C viral infections more benign in patients with hemophilia? Am J Gastroenterol. 2007;102:1672–6.
Wanless IR, Shiota K. The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases: a four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis. Semin Liver Dis. 2004;24:99–106.
Assy N, Bekirov I, Mejritsky Y, Solomon L, Szvalb S, Hussein O. Association between thrombotic risk factors and extent of fibrosis in patients with non-alcoholic fatty liver diseases. World J Gastroenterol. 2005;11:5834–9.
Tripodi A, Fracanzani AL, Primignani M, Chantarangkul V, Clerici M, Mannucci PM, et al. Procoagulant imbalance in patients with non-alcoholic fatty liver disease. J Hepatol. 2014;61:148–54.
Shi J, Hao JH, Ren WH. Effects of heparin on liver fibrosis in patients with chronic hepatitis B. World J Gastroenterol. 2003;9:1611–4.
Huang JS, Luo X, Yu JX, Liu W, Chen XW, Xie L, et al. Indigenous and imported low molecular weight heparin in the treatment of chronic hepatitis B and cirrhosis with hepatitis B virus: a prospective randomized controlled clinical study. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2007;19:408–11.
Dhar A, Tschotazis E, Brown R. Warfarin anticoagulation for liver fibrosis in patients transplanted for hepatitis C (WAFT-C): results at one year. J Hepatol. 2015;62:s268–9.
Villa E, Cammà C, Marietta M, Luongo M, Critelli R, Colopi S, et al. Enoxaparin prevents portal vein thrombosis and liver decompensation in patients with advanced cirrhosis. Gastroenterology. 2012;143:1253–60.
La Mura V, Braham S, Tosetti G, Branchi F, Bitto N, Moia M, et al. Harmful and beneficial effects of anticoagulants in patients with cirrhosis and portal vein thrombosis. Clin Gastroenterol Hepatol 2017.
Loffredo L, Pastori D, Farcomeni A, Violi F. Effects of anticoagulants in patients with cirrhosis and portal vein thrombosis: a systematic review and meta-analysis. Gastroenterology. 2017;153:480–7.
Cerini F, Gonzalez JM, Torres F, Puente Á, Casas M, Vinaixa C, et al. Impact of anticoagulation on upper-gastrointestinal bleeding in cirrhosis. A retrospective multicenter study. Hepatology. 2015;62:575–83.
• Bianchini M, Cavani G, Bonaccorso A, Turco L, Vizzutti F, Sartini A, et al. Low molecular weight heparin does not increase bleeding and mortality post endoscopic variceal band ligation in cirrhotic patients. Liver Int. 2018; https://doi.org/10.1111/liv.13728. Relevant study that underlines the safety of LMWH in cirrhotic patients undergoing variceal band ligation.
De Gottardi A, Trebicka J, Klinger C, Plessier A, Seijo S, Terziroli B, et al. Antithrombotic treatment with direct-acting oral anticoagulants in patients with splanchnic vein thrombosis and cirrhosis. Liver Int. 2017;37:694–9.
Funding
This paper was partially supported by unrestricted grants from COOK Medical and W.L. Gore.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Laura Turco, Filippo Schepis, and Erica Villa declare no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
The original version of this article was revised due to a retrospective Open Access order.
This article is part of the Topical Collection on Portal Hypertension
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
About this article

Cite this article
Turco, L., Schepis, F. & Villa, E. The Role of Anticoagulation in Treating Portal Hypertension. Curr Hepatology Rep 17, 200–208 (2018). https://doi.org/10.1007/s11901-018-0406-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11901-018-0406-x