
Abstract
Purpose of Review
Disaccharidase deficiency in adults causes carbohydrate malabsorption, resulting in symptoms which significantly overlap with irritable bowel syndrome (IBS). This article discusses the diagnosis and treatment of disaccharidase deficiency within the context of recent literature.
Recent Findings
Disaccharidase deficiency in adults is more common than previously thought, which includes lactase, sucrase, maltase and isomaltase enzymes. Deficiency in disaccharidases, which are produced by the intestinal brush border, will interfere with the breakdown and absorption of carbohydrates and may result in abdominal pain, gas, bloating and diarrhea. Patients deficient in all 4 disaccharidases are known as having “pan-disaccharidase” deficiency, which has a distinct phenotype with more reported weight loss than patients deficient in one enzyme. IBS patients who do not respond to low FODMAP dietary restriction may have undiagnosed disaccharidase deficiency and may benefit from testing. Diagnostic testing methods are limited to duodenal biopsies, which is the gold standard, and breath testing. Dietary restriction and enzyme replacement therapy have been shown to be effective treatments in these patients.
Summary
Disaccharidase deficiency is an underdiagnosed condition in adults with chronic GI symptoms. Patients who do not respond to traditional treatment strategies for DBGI may benefit from testing for disaccharidase deficiency. Further studies delineating the distinctions between disaccharidase deficient patients and those with other motility disorders are needed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The implications of disaccharidase deficiency as it pertains to neurogastroenterology and motility disorders has been a mystery for the past several decades. While congenital disaccharidase deficiency has been established as a cause of chronic abdominal pain in young children, it is more common in adults than previously thought [1, 2]. This review serves to summarize what is known thus far about disaccharidase deficiency in adults, how to evaluate adult patients for such deficiencies and treatment strategies.
Carbohydrate Digestion and DGBI
As carbohydrates comprise a large proportion of daily caloric consumption, the problem of carbohydrate intolerance is all the more significant due to the rising consumption of sugars by Americans [1, 3,4,−5]. The average American has a daily carbohydrate intake of 300 g and sugar intake of 140 g [6]. Lactose and sucrose are the most common disaccharides found in the diet [7]. Lactose is the main sugar in milk, and lactase non-persistence is the common phenotype in healthy humans, which accounts for the downregulation of lactose after infancy [7]. Diarrhea usually results if the load of lactose exceeds the colonic capacity for resorption but this is also dependent upon the intestinal microbiome, microbial function and small bowel bacterial overgrowth [8]. Lactase deficient patients with IBS also appear to express more severe symptoms associated with lactose intake, so visceral hypersensitivity also plays a role [8].
Sucrase is critical in metabolizing table sugar to glucose. The Western diet is high in sucrose, which is present in fruits such as peaches and cherries, desserts and sugary drinks [9]. Sucrase-Isomaltase (SI) gene product is located along the mucosal brush border which cleaves isomaltase and sucrose into sugar monomers. These cleaved monosaccharides are then transported across the epithelial brush border for absorption and metabolism [10]. Certain variants of congenital SI deficiency (CSID) result in sucrose and starch intolerance. CSID is an autosomal recessive disorder of carbohydrate metabolism involving the Sucrose-Isomaltase (SI) gene. CSID mutations are more commonly found in IBS patients vs. healthy controls, and symptom expression is heterogenous depending on the biochemical phenotype of the mutation [11, 12].
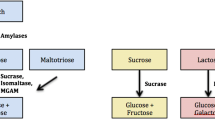
Carbohydrates are broken down in large part by disaccharidases found along the brush border of the small intestine into monosaccharides such as glucose, galactose and fructose which are transported across the epithelial brush border where they are absorbed and metabolized. The disaccharidases lactase, sucrase, maltase and isomaltase (also referred to as palatinase or trehalase) serve to break down sugars into monosaccharides, allowing for quick absorption [13]. Disaccharidase activity varies depending on location and is highest in the mid jejunum and lower in the proximal duodenum and terminal ileum [14]. A deficiency in any one disaccharidase can result in increased osmotic load from malabsorbed sugars in the small intestine (Fig. 1). This can result in abdominal pain, gas, bloating and diarrhea which are symptoms commonly reported by patients with Disorders of Gut-Brain Interaction (DGBI) [15, 16]. Diets low in fermentable substrates are associated with improved symptoms in Irritable Bowel Syndrome (IBS) patients [17]. A diagnosis of IBS may miss potential disaccharidase deficiencies, so earlier diagnosis of disaccharidase deficiencies may lead to more successful treatment of DGBI.

Effect of fermented starches in disaccharidase deficient patients compared to normal, healthy patients
Disaccharidase Deficiency in Adults
The incidence of sucrase-isomaltase deficiency has been estimated to be between 0.2% in North America and 10.0% in Greenland Eskimos [18]. Disaccharidase deficiencies have been well documented in children to explain abdominal pain. Prevalence of disaccharidase deficiencies within the pediatric population has been reported to be 37% lactase, 25% maltase, 21% sucrase and 8% palatinase [1]. Cases of children with all 4 disaccharidase deficiencies have not been reported in the literature. Viswanathan et al.. reported the first published prevalence data of disaccharidase deficiency in 120 adult patients with unexplained gastrointestinal symptoms. They found that 9.2% of patients had all 4 disaccharidase enzyme (pan-disaccharidase) deficiencies, 35.8% were lactase deficient, 0.8% were maltase deficient and 0.8% had combined sucrase, maltase and isomaltase deficiency [2] (Fig. 2). Figure 2b shows the proportional distribution of disaccharidase deficiencies in adults. Patients with single disaccharidase deficiency reported abdominal pain, bloating, fullness, gas, indigestion, cramping and nausea and reported less vomiting and weight loss [2]. The pan-disaccharidase cohort reported bloating and constipation, but reported relatively less cramping, pain, fullness, nausea, belching, diarrhea and gas, though these data were not significant [2]. Interestingly, they also reported more weight loss as compared to the single disaccharidase deficiency group [2]. This suggests that disaccharidase deficiencies occur often in combination, pan-disaccharidase deficiency is more common in adults than previously thought and this cohort of patients has a distinct clinical phenotype. Further characterization of the pan-disaccharidase group is needed, including possible etiology and its relationship with dysbiosis.

IBS affects 10% of the Western population, and its associated symptoms can mimic the post-prandial symptoms related to carbohydrate intake reported in disaccharidase deficient patients [18]. Thirty-one patients with presumed IBS-D/M based on symptoms of pain, bloating and diarrhea underwent disaccharidase assays [15]. Sucrase-isomaltase deficiency (SID) was present in 35% of patients, and these patients were less likely to report abdominal pain [15]. No difference in bloating or diarrhea was found between SID and IBS-D/M patients [15]. SIBO was not excluded in these 31 patients, which may have overestimated the prevalence of IBS. In another study of 82 patients with a mix of functional diarrhea and constipation, a majority of patients expressed disaccharidase deficiency: 86.5% were lactase deficient, 48.7% were maltase deficient, 50% were sucrase deficient and 84.1% were gluco-amylase deficient [19]. Interestingly, 31.7% of these patients were deficient in all enzymes [20].
Testing
Testing options for the diagnosis of disaccharidase deficiency include disaccharidase assay, genetic testing, breath testing and disaccharide challenge (Fig. 3). The diagnosis should be considered in patients complaining of lifelong postprandial abdominal pain, gas, bloating and/or diarrhea. Secondary causes of disaccharidase deficiency, such as Celiac or Crohn’s disease, chemoradiation therapy, small intestinal bacterial overgrowth (SIBO), acute gastroenteritis or any other condition which may damage the brush border, should be excluded.

The gold standard for testing disaccharidase deficiency is endoscopically obtaining two biopsies from the third portion of the duodenum [2, 15]. One biopsy is used to study the architecture of the mucosa and the second is used for analysis of sucrase, maltase, lactase and isomaltase levels per the Dahlqvist method. This method gives precise levels of disaccharidase levels; however, it can be limited by sample error, as two biopsies may not give a complete picture of brush border enzyme activity as enzyme distribution can be patchy [12]. Biopsies from distal duodenal provide higher yield than those from the proximal duodenum [14]. Figure 4 shows normative data of disaccharidase levels from duodenal biopsies in adults per the Dalqvist method. Current reports of disaccharidase assays from commercial labs also include alpha-glucosidase and alpha-amylase levels.

Reference levels of normal disaccharidase levels from duodenal biopsies in adults
Sucrose hydrogen breath testing is readily available both as in clinic or at home options and is a non-invasive surrogate method for testing for disaccharidase deficiency. This test can be performed either by ingesting a standard amount of 13 C-sucrose or sucrose (known as a direct sucrose challenge) and measuring the amount of expired 13 C-methane or hydrogen, respectively [21]. 13 C-methane breath testing measures sucrase activity without administering a sucrose load to the patient while the direct challenge sucrose test is cheaper but will provoke symptoms. Breath test results correlate well with intestinal sucrase activity [12]. However, it is important to note that in practice, SIBO should be ruled out as the presence of bacterial overgrowth can confound sucrose breath test results [22]. False positive results can also be a result of villous injury, such as Celiac disease and dumping syndrome [23]. A study of 258 adults with chronic GI symptoms who underwent both hydrogen-methane and 13 C sucrase breath testing reported positive yield for 13 C sucrase breath test of 26% [24]. The hydrogen-methane group had false positives due to underlying SIBO [24]. Patients with sucrose deficiency identified on 13 C sucrose breath testing reports functional diarrhea, flatulence, bloating and IBS-M and 60% of those patients reports improvement with dietary adjustment and/or enzyme replacement therapy [24]. Symptom severity is dependent upon several factors, to include sucrase-isomaltase activity, amount of sugar and starch intake and gastric and small bowel transit [23]. While all CSID patients have sucrase deficiency, they vary in degree of isomaltase activity [25]. Therefore, patients with disaccharidase deficiency can present with a varied constellation of symptoms so it is important to keep this diagnosis on your differential in the face of chronic, nonspecific GI symptoms.
Treatment
Patients with IBS-D with underlying SI deficiency did not experience any benefit from a low FODMAP (fermentable oligosaccharide, disaccharide, monosaccharide and polyols) diet when compared with non-SI carriers [26]. This may be because, in patients with SI, sucrose is metabolized as a FODMAP and fermented in the colon. Dietary exclusion of sucrose and/or starches can be helpful, though difficult to continue longterm. Given the difficulty of adherence to this diet, patients may benefit from formal consultation with a GI dietician, if available.
Enzyme supplementation, either with lactase or sacrosidase is another treatment option. It is unknown whether treatment of lactase deficiency will improve symptoms of other disaccharidase deficiencies. Sacrosidase, sucrose enzyme derived from Saccharomyces cervisiae, is the only FDA approved treatment for CSID/SI patients though there are other over the counter options as well (Table 1). Sacrosidase treatment in pediatric patients improved symptoms of diarrhea, gas, cramping and bloating at a diluted dose and completely resolved symptoms at the full dose [23]. Additionally, sacrosidase oral replacement normalized 13C-sucrose breath test results in deficient patients [10, 12]. Similar results of improved symptoms of abdominal pain, bloating, flatulence, borborygmi and diarrhea and lower hydrogen breath test scores were reported by lactase deficient patients receiving lactase supplementation [27].
Several other enzyme supplements are available over the counter, including vegan enzyme options, but these have not been evaluated in clinical trials. A recent product, FODZYME (inulinase enzyme powder), is now available over the counter, and is believed to break down fructans and other disaccharides. But its usefulness merits a randomized controlled trial.
Conclusions
Disaccharidase deficiency is an important and underappreciated clinical problem in the adult population although more readily diagnosed in pediatric patients. This is a disease entity which hides in plain sight but may often be overlooked in patients labeled with IBS. Awareness and education about the importance of carbohydrate malabsorption will benefit both the clinician and the patient, especially in the context of unexplained, chronic, post-prandial symptoms of abdominal pain, gas, bloating and diarrhea. It should be considered in patients who have longstanding symptoms which are unresponsive to a low FODMAP diet or to other common treatments for IBS. The gold standard assay for testing disaccharidase function involves an esophagogastroduodenoscopy with 2 biopsies from the third portion of the duodenum but can be limited by sample error. Less invasive surrogate tests include the direct sucrose challenge and the 13C-sucrose breath test. SIBO should be exclude first, when employing breath testing to avoid a false positive result. Once the diagnosis is confirmed, treatment options include dietary restrictions vs. enzyme replacement therapy.
While we now know more about this disease entity, there is much more to be discovered. Larger population studies are needed, as this epigenetic phenomenon is expressed differently, depending on the patient. Clinical stratification to predict who may develop disaccharidase deficiency and who may benefit from dietary restriction or enzyme replacement therapy would also be helpful. IBS patients who do not improve on a low FODMAP diet may benefit from enzyme replacement therapy, but there are no known distinguishing characteristics between IBS and disaccharidase deficiency. Increased screening and testing along with detailed characterization of the phenotypes will likely help us to learn more about this unique and often misunderstood condition and provide relief for many patients.
References
El-Chammas K, Williams SE, Miranda A. Disaccharidase deficiencies in children with chronic abdominal pain. JPEN J Parenter Enteral Nutr. 2017;41:463–9.
**Viswanathan L, Rao SSC, Kennedy K, Sharma A, Yan Y, Jimenez E. Prevalence of Disaccharidase Deficiency in adults with unexplained gastrointestinal symptoms. J Neurogastroenterol Motil. 2020;26(3):384–90. First reported disaccharidase deficiency prevalence data in adults.
Russo P, Ruchelle ED, Piccoli DA. Pathophysiology of pediatric gastrointestinal and liver disease. 2nd ed. New York, NY: Springer; 2014. pp. 19–54.
US Department of Agriculture and US Department of Health and Human Services. Dietary guidelines for Americans. 7th ed. Washington, DC:US Government Printing Office 20011.
Choi YK, Kraft N, Zimmerman B, Jackson M, Rao SS. Fructose intolerance in IBS and utility of fructose-restricted diet.J Clin Gastroenterol20008;42:233–238.
U.S. Department of Agriculture, Agricultural Research Service. 2014. Nutrient Intakes from Food and Beverages: Mean Amounts Consumed per Individual, by Gender and Age, What We Eat in America, NHANES 2011–2012.
Misselwitz B, Butter M, Verbeke K, Fox MR. Update on lactose malabsorption and intolerance: pathogenesis, diagnosis and clinical management. Gut. 2019;68:2080–91.
Ventura P, Gede N, et al. Sugar content of popular sweetened beverages based on objective laboratory analysis: focus on fructose content. Obesity. 2011;19:868–74.
Marcadier JL, Boland M, Scott CR, et al. Congenital sucrase-isomaltase deficiency: identification of a common inuit founder mutation. CMAJ. 2015;187:102–7.
Cohen SA. The clinical consequences of sucrase-isomaltase deficiency. Mol Cell Pediatr. 2016;3:5.
Husein DM, Rizh S, Naim HY. Differential effects of sucrase-isomaltase mutants on its trafficking and function of irritable bowel syndrome: similarities to congenital sucrase-isomaltase deficiency. Nutrients. 2021;13(1):9.
Opekun A, Chumpitazi BP, Abdulsada MM, Nichols BL. Routine disaccharidase testing: are we there yet? Curr Opin Gastroenterol. 2020;36(2):101–9.
Hauser H, Semenza G. Sucrase-isomaltase: a stalked intrinsic protein of the brush border membrane. CRC Crit Rev Biochem. 1983;14:319–45.
Chey WD, Cash B, Lembo A, Patel DB, Scarlata K. Congenital sucrase-isomaltase Deficiency: what, when, and how? Gastroenterol Hepatol. 2020;16(10):5.
Kim SB, Calmet FH, Garrido J, Garcia-Buitrago MT, Moshiree B. Sucrase-Isomaltase Deficiency as a potential masquerader in irritable bowel syndrome. Dig disease Sci. 2020;65(2):534–40.
Nichols BL, Adams B, Roach CM, Ma CX, Baker SS. Frequency of sucrase deficiency in mucosal biopsies. J Pediatr Gastroenterol Nutr. 2002;35:551–6.
Halmos EP, Power VA, Shepherd SJ, Gibson PR, Muir JG. A diet low in FODMAPs reduces symptoms of irritable bowel syndrome. Gastroenterology. 2014;146:67–75e5.
Chey WD, Kurlander J, Eswaran S. Irritable bowel syndrome: a clinical review. JAMA. 2015;313:949–58.
Henstrom M, et al. Functional variants in the sucrase-isomaltase gene associate with increased risk of irritable bowel syndrome. Gut. 2018;67:263–70.
Dbar S, Akhmadullina O, et al. Patients with functional bowel disorder have disaccharidase deficiency: a single-center study from Russia. World J Clin Cases. 2021;9(17):4178–87.
Robayo-Torres CC, Opekun AR, Quezada-Calvillo R, Villa X, Smith EO, Navarrete M, Baker SS, Nichols BL. 13 C-breath tests for sucrose digestion in congenital sucrase isomaltase-deficient and sacrosidase-supplemented patients. J Pediatr Gastroenterol Nutr. 2009;48:412–8.
Rezaie A, et al. Hydrogen and methane-based Breath Testing in Gastrointestinal Disorders: the North American Consensus. Am J Gastroenterol. 2017;112:775–84.
Treem WR. Clinical aspects and treatment of congenital sucrase-isomaltase Deficiency. J Pediatr Gastroenterol Nutrition: November. 2012;55:7–13.
Frissora CL, Rao SSC. Sucrose intolerance in adults with common functional gastrointestinal symptoms. Baylor University Medical Center Proceedings 2022;0(0):1–4.
Alfalah M, Keiser M, Leeb T, Zimmer KP, Naim HY. Compound heterozygous mutations affect protein folding and function in patients with congenital sucrase-isomaltase deficiency. Gastroenterology. 2009;136:883–92.
Zheng T, Eswaran S et al. Reduced efficacy of low FODMAPs diet in patients with IBS-D carrying sucrase-isomaltase (SI) hypomorphic variants. Gut 2019;pii:gutjnl-2018-3180036.
Baijal R, Tandon RK. Effect of lactase on symptoms and hydrogen breath levels in lactose intolerance: a crossover placebo-controlled study. J Gastroenterol Hepatol Foundation. 2020;5:143–8.
Puntis JW, Zamvar V. Congenital sucrase-isomaltase deficiency: diagnostic challenges and response to enzyme replacement therapy. Arch Dis Child. 2015;100:869–71.
Funding
Open access funding provided by SCELC, Statewide California Electronic Library Consortium
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Disclosures: Her views are her own and do not reflect those of the U.S. Department of the Air Force nor the U.S. Department of Defense.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Viswanathan, L., Rao, S. Intestinal Disaccharidase Deficiency in Adults: Evaluation and Treatment. Curr Gastroenterol Rep 25, 134–139 (2023). https://doi.org/10.1007/s11894-023-00870-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11894-023-00870-z