
Abstract
Luminal amino acids and lack of luminal acidity as a result of acid neutralization by intragastric foodstuffs are powerful signals for acid secretion. Although the hormonal and neural pathways underlying this regulatory mechanism are well understood, the nature of the gastric luminal pH sensor has been enigmatic. In clinical studies, high pH, tryptic peptides, and luminal divalent metals (Ca2+ and Mg2+) increase gastrin release and acid production. The calcium-sensing receptor (CaSR), first described in the parathyroid gland but expressed on gastric G cells, is a logical candidate for the gastric acid sensor. Because CaSR ligands include amino acids and divalent metals, and because extracellular pH affects ligand binding in the pH range of the gastric content, its pH, metal, and nutrient-sensing functions are consistent with physiologic observations. The CaSR is thus an attractive candidate for the gastric luminal sensor that is part of the neuroendocrine negative regulatory loop for acid secretion.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastric acid secretion, like any other bodily function, is regulated according to need. Because acid secretion is needed most just prior to a meal, neural and other mechanisms triggered by smell and taste increase the rate of acid secretion (cephalic phase) followed by meal-stimulated secretion. Through acid neutralization, food in the stomach increases intragastric pH, which is sensed by antral sensors, releasing gastrin, which increases the rate of parietal cell acid secretion. This acid secretion helps initiate meal digestion, which then acidifies the gastric lumen after the food empties, lowering gastric pH and decreasing gastrin release, impairing acid secretion. Feedback regulation of gastric acid secretion was confirmed several decades ago, based on the work of Andersson and Olbe [1], and other researchers, who described that the rate of acid secretion was decreased when acid was introduced into an isolated antral pouch in dogs. Formal gastric analysis in humans also was consistent with antral acid negatively feeding back to the oxyntic mucosa. The mechanism of this regulation includes activation of afferent nerves by antral acid with consequent local somatostatin release, decreasing acid secretion. Although the neuronal circuitry and hormonal inputs to parietal cell secretion are relatively well understood, the nature of the antral acid sensor has been elusive. Although many molecular acid sensors are expressed in the gastrointestinal tract, such as the acid-sensing ion channels (ASICs) and transient receptor potential (TRP) channels, few data supported their antral localization and function as antral acid sensors.
One early observation that may provide a tantalizing hint to the nature of the antral sensor is the function of luminal calcium as a potent acid secretagogue. Before the advent of current histamine type-2 receptor antagonist (H2RA) and proton pump inhibitor (PPI)-based antisecretory therapies, the primary method of ulcer treatment was acid neutralization, which was achieved by the administration of dairy products or by the use of alkaline calcium salts. Research in the 1970s revealed that luminal calcium significantly increased gastrin release and thus could paradoxically increase acid secretion [2, 3]. Magnesium and aluminum-based antacids also increase serum gastrin, although to a lesser extent [4, 5]. Interestingly, the gastrin response was pH modulated, with increased secretion noted in an alkaline environment [4] and calcium effects abolished when administered in a pH 1 solution [3]. These data, however, supported the presence of a gastric mucosal calcium sensor, the molecular nature of which was identified only recently.
Gastric Anatomy
Before discussing the current understanding of intragastric pH sensing mechanisms, it is helpful to quickly review the basic principles of gastric anatomy and acid secretion. The stomach is composed of two major regions, the oxyntic and the antral mucosae. The oxyntic mucosa, which is the locus of the acid-secreting parietal cells and constitutes 80% of the acid-secreting gastric mucosa, is located proximally in the gastric body and fundus. The oxyntic mucosa also contains histamine-secreting enterochromaffin-like (ECL) cells and pepsinogen-secreting chief cells. The distal 20% of gastric mucosa contains antral pyloric glands with gastrin-secreting G cells. Both regions include surface mucous cells, mucous neck cells, enterochromaffin (EC) cells, and D cells, which secrete the important regulatory peptide somatostatin [6••, 7].
Regulation of Acid Secretion
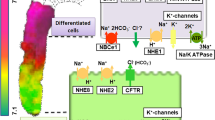
The control of gastric acid secretion from parietal cells is an orchestration of neuronal, hormonal, and paracrine signaling that regulates the secretion of hydrochloric acid through the H+/K+ ATPase pump. Three distinct phases of acid secretion exist. Basal acid secretion is under a relative tonic inhibition of somatostatin on parietal, EC, and G cells. During the cephalic phase, the preparation for eating—including thought and sensory cues from the sight, smell, and taste of food—stimulate the central nervous system. Activated vagal efferent preganglionic neurons synapse with postganglionic neurons of the enteric nervous system located in the stomach wall, leading to the innervation of target cells in gastric mucosa [6••, 7]. Cholinergic stimulation releases acetylcholine, which activates muscarinic M3 receptors on parietal cells. Furthermore, cholinergic stimulation of G cells releases gastrin, and activates gastric acid secretion directly and indirectly because gastrin also releases histamine, a direct parietal cell agonist, from ECL cells. In parallel, cholinergic stimulation inhibits somatostatin release. Relieved of its suppression, the parietal cell is enabled to increase acid production. Moreover, the enteric nervous system releases neurotransmitters, including gastrin-releasing peptide (GRP) [8] and pituitary adenylate cyclase-activating polypeptide (PACAP), further increasing local concentrations of gastrin and histamine, respectively (Fig. 1) [9, 10].

Hormonal and paracrine signaling regulating gastric acid with respective receptors. Antral gastric glands (A) contain G cells (G), mucous cells (M), and open type D cells (D). Fundic oxyntic glands (B) contain enterochromaffin-like cells (ECL), mucous cells (M), parietal cells (P), and closed-type D cells. The neuronal circuits from the enteric nervous system (ENS) with acetylcholine (Ach) release and selected neuropeptide output are also shown. Feedback control of acid secretion by extrinsic sensory afferent nerves (SA) of nodose and dorsal root ganglion origin includes calcitonin gene-related peptide (CGRP)–related somatostatin release, which indirectly decreases gastrin release (shown in A). The SA nerves monitor luminal acidity, decreasing acid output as pH decreases below 2.5 with reciprocal effects as pH increases. The calcium-sensing receptor (CaSR) depicted on the luminal surface of a G cell is in the family C of G-protein coupled 7 transmembrane receptors (A inset). Agonists include calcium and polyvalent cations such as gadolinium with activity modulated allosterically by L-amino acids and luminal pH. CCK2 cholecystokinin receptor 2; GRP gastrin-releasing peptide; GRPR GRP receptor; H 2 histamine receptor type 2; M3 muscarinic receptor type 3; PACAP pituitary adenylate cyclase-activating polypeptide; PAC1 PACAP receptor type 1; SST somatostatin; SST2 SST receptor type 2; (+) stimulating action; (−) inhibitory action. (Adapted from Wank [10].)
A rapid increase in gastric acid secretion occurs during the gastric phase as a result of the meal effects of gastric wall distension and luminal nutrient exposure. Sensory stretch receptors in the body and antrum activate vagal reflexes, which increase acid secretion as described above. Luminal exposure to amino acids, ethanol, calcium, and coffee contribute to further gastrin release [11]. During a meal, foodstuffs buffer the gastric content, raising luminal pH, and then enabling gastric acid secretion to continue. Finally, the intestinal phase occurs when stomach contents reach the duodenum. The general response of this phase is inhibition of gastric acid through the release of enterogastrones, such as gastric inhibitory peptide, cholecystokinin, secretin, and glucagon-like peptide-1. In response to intestinal luminal fat, these hormones activate the inhibitory somatostatin pathway.
Feedback Control
Antral control of acid secretion is under the hormonal direction of gastrin and negatively regulated by the paracrine effects of somatostatin. Feedback mechanisms adjust acid output by modulating somatostatin-related inhibition of gastrin release. One feedback pathway is the direct stimulation of D-cell somatostatin release by gastrin [12], which, in turn, indirectly inhibits further gastric release from G cells. Nevertheless, the main feedback control mechanism monitors luminal acidity and adjusts acid secretion and gastrin release accordingly, with the observed decrease in gastrin and acid secretion at low antral pH and increased acid secretion as intragastric pH rises and luminal acidity wanes.
The regulation of gastrin and gastric acid secretion by intra-luminal pH was noted more than 40 years ago. In canine and human subjects, the increase of gastrin and acid secretion was observed at an intragastric pH of 2.5 or higher [13] with abolishment of acid output at pH 1.0 [1]. One identified pathway by which this feedback control is carried out is via the release of neuropeptide calcitonin gene-related peptide (CGRP) by gastric sensory afferent nerves. CGRP increases somatostatin release, indirectly decreasing gastrin and acid secretion (Fig. 1A). The release of CGRP and inhibition of gastrin were reported to be pH dependent in rats [14]. Yet, the pathway by which acid secretion is enhanced in the setting of high intragastric pH is still incompletely understood [6••].
As discussed, CGRP contributes to the feedback control of acid secretion, although there undoubtedly are other pathways yet to be elucidated. D cells present in antral mucosa and in fundic gastric glands have distinct characteristics. Antral D cells have apical processes that contact the gastric lumen, enabling response to acidity, peptides, and neuronal mediators (open type) (Fig. 1A). Fundic D cells, however, lack these sampling probes and therefore the ability to sense luminal content (closed type) (Fig. 1B) [15]. In rat studies, antral D cells were identified as acid sensitive because of decreased somatostatin production measured after treatment with proton-pump inhibitors, whereas corpus D cells, likely comparable to D cells in the gastric glands of the fundus, are CGRP sensitive and therefore under the influence of the gastric sensory afferent nerves rather than direct luminal content [16]. Although these observations have not been consistent with other studies that reported a CGRP effect on antral D cells [14, 17], they provide insight into the existence of alternate mechanisms of pH sensing.
Sensory Afferent Nerves
The sensory afferent nerves of the stomach respond to changes in luminal pH and acid content. These chemosensitive neurons are present in the dorsal root ganglion of the spinal nerves and the nodose ganglion of the visceral sensory nerves [18]. Visceral afferent pathways in rat mucosa can be sensitized by acid and inflammation to trigger enhanced nociceptive visceromotor responses, which may contribute to symptoms of dyspepsia [19].
The exact molecular sensors encoding gastric acid chemonociception are not known although several candidates have been identified [20]. Capsaicin-sensitive TRP vanilloid subtype 1 (TRPV1) is also activated by protons and thermal stimuli, giving it the potential to transduce polymodal nociception [21]. In rat studies, TRPV1 activity was more prevalent in the visceral nodose ganglion than in the spinal afferents [20], and thus might act as the acid sensor for the visceral afferent nerves.
Several classes of ASICs have differential expression on somatic and visceral sensory neurons [20], although the contributions from the different channels to gastric mechano- and chemosensation are still being elucidated. In the visceral afferent neurons of the nucleus tractus solitarii in mice, ASIC2 attenuates afferent signaling after gastric acid insult. Similar methods have identified TRPV1 and ASIC3 as the predominant proton-sensitive ion channels in gastric sensory neurons [22••]. In particular, ASIC3 may play a key role in gastritis-evoked hyperresponsiveness to acid [23•].
The sensation of dyspepsia may correlate with activation of afferent sensory nerves by gastric acid that has diffused or is transported through the mucosal barrier. Once the afferent nerves sense an acid load, the conducted signals mainly function to increase mucosal protective factors via gastric vasodilation, hyperemia, and increased bicarbonate secretion and mucus production. Additionally, afferent sensory nerve activation contributes to feedback modulation of gastrin and somatostatin levels. As previously discussed, this process has been demonstrated by the action of CGRP, which is a marker for sensory afferent neurons. A recent study confirmed the linear relationship between increasing CGRP levels and decreasing gastric pH from 3.0 to 0.5 that was also dependent on the presence of extracellular calcium, but independently of TRPV1 or ASIC3 function. Although the respective channel antagonists had inhibitory effects on CGRP release, the level of inhibition was not different in the stomach of receptor knockout mice [24••]. These findings suggest the existence of a different calcium-dependent receptor for acid sensing, requiring further investigation.
Calcium-Sensing Receptor: Candidate for Novel Gastric Chemosensor
The calcium-sensing receptor (CaSR) is a member of the G-protein coupled receptor family C with widespread distribution, most notably in tissues of the parathyroid and kidney, where it helps maintain extracellular calcium homeostasis [25]. The gastric mucosal CaSR is localized to the basolateral membrane of parietal cells and mucous cells [26, 27], the apical and basolateral surface of antral G cells [28], and most recently its presence was detected on D cells [29••]. Although the function of CaSR in the gastric mucosa is a subject of ongoing research, the multiple substances that modulate its activity, including cations, amino acids, and pH, make it an intriguing candidate for gastric chemosensing [30••] (Fig. 1A, inset).
Polyvalent Agonists
CaSR present on parietal cells in intact rat gastric glands [26] and human gastric tissue [31] is activated with the divalent cations Ca2+ and Mg2+, which enhances histamine-induced, H+/K+ ATPase-mediated acid secretion. Using the potent stimulant Gd3+, CaSR activation can also occur without histamine [26]. In addition, CaSR is also present on antral G cells [28]. The stimulation of CaSR in human cultured antral G cells releases gastrin. The receptor relies on extracellular calcium binding and phospholipase C-mediated opening of nonspecific cation channel and elevating intracellular calcium [32]. Stimulation of CaSR in vivo with the allosteric agonist cinacalcet supports the above findings, because significant increases in serum gastrin levels and basal gastric acid secretion have been reported [33•]. Because CaSR has been identified on the apical surface of G cells by immunohistochemical staining [28], it is possible that luminal sensing of calcium by CaSR triggers the increase in gastrin and acid secretion noted after ingestion of calcium-containing antacids.
Amino Acid Agonists
Intravenously administered amino acids and high-protein diets stimulate gastric acid secretion [34] during the gastric phase of acid secretion because luminal food content leads to rising gastrin levels. The CaSR has the ability to detect nutrient amino acids, and thus may serve as the mediator of the gastric phase response. In rat ex vivo whole stomach and parietal cells studied in vitro, exposure to L-Phe and L-Trp in the presence of physiologic levels of extracellular calcium augments acid secretion in a pattern most consistent with allosteric activation [35], independent of secretagogue pathways such as gastrin or histamine.
Heteromeric L-type amino acid transporters are present on the basolateral surface of mice and rat parietal cells. These transporters consist of 4F2hc and LAT2 heavy chains, functioning as exchange transporters for neutral amino acids. Administration of glutamine and phenylalanine stimulates H+/K+ ATPase-mediated acid secretion, presumably by the intracellular exchange of amino acids and through a histamine-independent pathway [36]. Together, these two amino acid-responsive systems may account for the increase of acid secretion after high-protein diets and may contribution to gastroesophageal reflux disease symptoms.
Modulation by pH
The CaSR is activated by polyvalent cations and polycationic molecules as described above. One stipulation is that rather than using specific motifs, the ligand binding on CaSR may occur through electrostatic interactions that screen charged side chains of amino acids or cationic charges. CaSR activation may be modified by extracellular pH because the acid–base balance of ligands and receptor binding sites is pH-dependent [37]. Modulation of CaSR-mediated responses by luminal pH and the augmentation of acid secretion by CaSR ligands, combined with its strategic localization on the cells of the gastric secretory regulatory pathway, suggests that the CaSR may serve as a gastric luminal pH sensor.
In vitro studies on CaSR-transfected HEK 293 cell line by Quinn et al. [37] confirmed an extracellular pH-dependent activation of CaSR, as manifest by increased intracellular Ca2+ in the presence of extracellular Ca2+ and Mg2+. From pH 7.5 to 9.0, CaSR activation was enhanced, shifting the threshold concentration and effective concentration for 50% curve for Ca2+ to the left, whereas the opposite was observed with pH 5.5 to 7.5. Interestingly, activity of the CaSR was markedly increased below pH 5.5 independent of agonist stimulation. In accordance with these findings, when exposed to a constant extracellular calcium concentration, CaSR activation had a linear correlation with extracellular pH, demonstrating one manner in which it may serve as a pH sensor. Because the CaSR is present on the basolateral surface of G cells and parietal cells, one consideration is that exposure to the alkaline environments (eg, the alkaline tide after gastric secretion) would further enhance CaSR activation. Surprisingly, the opposite relationship of pH-dependent CaSR sensitization was reported when examining organic polycationic agonist molecules, such as spermidine, neomycin, or histatine-5, each containing multiple titratable amine groups [37]. For these ligands, acidic pH enhanced activation of CaSR. The varying response may result from key differences in protonation states, because the polyvalent molecules have dissociation constants within the pH range of the gastric lumen, whereas the inorganic cations Ca2+ and Mg2+ have a high negative logarithm of the acid ionization constant such that their net charge would not have been affected in this study.
Conclusions
Our evolving knowledge regarding the physiology of acid secretion and its regulation has produced progressively more effective treatments for peptic ulcer disease and acid reflux. Early therapeutic approaches, including surgical antrectomy, vagatomy, and simple antacids, have primarily been replaced by H2RAs (developed in 1972) and PPIs (introduced in 1989). Despite these highly effective treatments, many people still suffer persistent symptoms and new therapeutic targets are sought to control acid secretion.
Regulation of gastric acid secretion by luminal pH is a potentially important control mechanism. Convergent clinical, biochemical, molecular, and physiologic data support the CaSR as the putative gastric acid sensor in addition to a neural CGRP-mediated mechanism. Recently, the CaSR was identified on isolated rat gastric D cells, with CaSR ligands increasing somatostatin release [29••]. In light of prior known actions of CaSR activation increasing gastrin and acid secretion, the net effect of CaSR ligands may depend on its relative expression in each cell type, whether stimulatory G cells or inhibitory D cells, and whether the endocrine cells are of the closed or open type. CaSR knockout mice lack gastrin release in response to luminal Ca2+, peptone, or rise of pH [30••], supporting CaSR as a luminal acid sensor. With continued studies, we may soon better understand the nature of intragastric chemosensing and design therapies based on CaSR ligands.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Andersson S, Olbe L: Inhibition of gastric acid response to sham feeding in pavlov pouch dogs by acidification of antrum. Acta Physiol Scand 1964, 61:55–64.
Barreras RF: Calcium and gastric secretion. Gastroenterology 1973, 64:1168–1184.
Behar J, Hitchings M, Smyth RD: Calcium stimulation of gastrin and gastric acid secretion: effect of small doses of calcium carbonate. Gut 1977, 18:442–448.
Feurle GE: Effect of rising intragastric pH induced by several antacids on serum gastrin concentrations in duodenal ulcer patients and in a control group. Gastroenterology 1975, 68:1–7.
Mazzacca G, Cascione F, Budillon G, et al.: Parietal cell hyperplasia induced by long-term administration of antacids to rats. Gut 1978, 19:798–801.
•• Schubert ML, Peura DA: Control of gastric acid secretion in health and disease. Gastroenterology 2008, 134:1842–1860. This article provides a comprehensive review of cellular anatomy and physiologic control of gastric acid secretion in the normal state and describes the pathologic clinical presentations of abnormal acid states.
Shulkes A, Baldwin GS, Giraud AS: Regulation of gastric acid secretion. In Physiology of the Gastrointestinal Tract. Edited by Johnson LR. New York: Academic Press; 2006:1223–1258.
Campos RV, Buchan AM, Meloche RM, et al.: Gastrin secretion from human antral G cells in culture. Gastroenterology 1990, 99:36–44.
Zeng N, Athmann C, Kang T, et al.: PACAP type I receptor activation regulates ECL cells and gastric acid secretion. J Clin Invest 1999, 104:1383–1391.
Wank SA: PACAP upsets stomach theory. J Clin Invest 1999, 104:1341–1342.
Richardson CT, Walsh JH, Hicks MI, Fordtran JS: Studies on the mechanisms of food-stimulated gastric acid secretion in normal human subjects. J Clin Invest 1976, 58:623–631.
Schubert ML, Jong MJ, Makhlouf GM: Bombesin/GRP-stimulated somatostatin secretion is mediated by gastrin in the antrum and intrinsic neurons in the fundus. Am J Physiol 1991, 261:G885–G889.
Walsh JH, Richardson CT, Fordtran JS: pH dependence of acid secretion and gastrin release in normal and ulcer subjects. J Clin Invest 1975, 55:462–468.
Manela FD, Ren J, Gao J, et al.: Calcitonin gene-related peptide modulates acid-mediated regulation of somatostatin and gastrin release from rat antrum. Gastroenterology 1995, 109:701–706.
Holst JJ: Differences in the control of somatostatin release from antrum and fundus. In The Stomach as an Endocrine Organ. Edited by Hankanson R, Sundler F. Amsterdam: Elsevier Science Publishers; 1991:244–250.
Sandvik AK, Dimaline R, Forster ER, et al.: Differential control of somatostatin messenger RNA in rat gastric corpus and antrum. Role of acid, food, and capsaicin-sensitive afferent neurons. J Clin Invest 1993, 91:244–250.
Ren J, Dunn ST, Tang Y, et al.: Effects of calcitonin gene-related peptide on somatostatin and gastrin gene expression in rat antrum. Regul Pept 1998, 73:75–82.
Holzer P: Sensory neurone responses to mucosal noxae in the upper gut: relevance to mucosal integrity and gastrointestinal pain. Neurogastroenterol Motil 2002, 14:459–475.
Lamb K, Kang YM, Gebhart GF, Bielefeldt K: Gastric inflammation triggers hypersensitivity to acid in awake rats. Gastroenterology 2003, 125:1410–1418.
Sugiura T, Dang K, Lamb K, et al.: Acid-sensing properties in rat gastric sensory neurons from normal and ulcerated stomach. J Neurosci 2005, 25:2617–2627.
Caterina MJ, Rosen TA, Tominaga M, et al.: A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 1999, 398:436–441.
•• Bielefeldt K, Davis BM: Differential effects of ASIC3 and TRPV1 deletion on gastroesophageal sensation in mice. Am J Physiol Gastrointest Liver Physiol 2008, 294:G130–G138. This in vitro study of knock-out mice examined vagal afferent nerve activation and demonstrated both TRPV1 and ASIC3 play an important role in luminal chemosensing of gastric acidification. TRPV1 also is important in mechanosensation of luminal distension, whereas the ASIC3 contribution appears to be less significant.
• Wultsch T, Painsipp E, Shahbazian A, et al.: Deletion of the acid-sensing ion channel ASIC3 prevents gastritis-induced acid hyperresponsiveness of the stomach-brainstem axis. Pain 2008, 134:245–253. This study of ASIC2 and ASIC3 knock-out mice demonstrated the ASIC3 l to play an important role in gastric acid hyperresponsiveness after experimentally induced gastritis. The ASIC2 function appears to attenuate gastric acid signaling by sensory neurons to the NTS.
•• Auer J, Reeh PW, Fischer MJM: Acid-induced CGRP release from the stomach does not depend on TRPV1 or ASIC3. Neurogastroenterol Motil 2010, 22:680–687. This study investigated the sensory response to acid load in the isolated everted mouse stomach by measuring CGRP as a marker for neuronal activation in wild-type and TRPV1 or ASIC3 knock-outs. A direct linear relationship was found between rising CGRP levels and decreasing extracellular pH. However, this response was found to be independent of TRPV1 or ASIC3 channels, suggesting existence of another acid-sensing pathway yet to be determined.
Brown EM, Gamba G, Riccardi D, et al.: Cloning and characterization of an extracellular Ca 2+ -sensing receptor from bovine parathyroid. Nature 1993, 366:575–580.
Geibel JP, Wagner CA, Caroppo R, et al.: The stomach divalent ion-sensing receptor scar is a modulator of gastric acid secretion. J Biol Chem 2001, 276:39549–39552.
Rutten MJ, Bacon KD, Marlink KL, et al.: Identification of a functional Ca 2+ -sensing receptor in normal human gastric mucous epithelial cells. Am J Physiol 1999, 277:G662–G670.
Ray JM, Squires PE, Curtis SB, et al.: Expression of the calcium-sensing receptor on human antral gastrin cells in culture. J Clin Invest 1997, 99:2328–2333.
•• Nakamura E, Hasumura M, Gabriel AS, et al.: Functional role of calcium-sensing receptor on somatostatin release from rat gastric mucosa [abstract]. Gastroenterology 2010, 138:S404. These investigators presented work on fractioned rat gastric cells that measured mRNA expression for CaSR on individual cell types, identifying CaSR expression to be predominantly found on D cells inducing activating response with somatostatin release. This action was verified with CaSR synthetic agonist calcitonin and antagonized with NPS2143.
•• Feng J, Lu M, Anthony S, et al.: The calcium-sensing receptor (CaSR) mediates meal stimulated gastrin secretion [abstract]. Gastroenterology 2007, 132:A101. This abstract provided the initial data supporting the hypothesis that the CaSR is the antral pH sensor.
Dufner MM, Kirchhoff P, Remy C, et al.: The calcium-sensing receptor acts as a modulator of gastric acid secretion in freshly isolated human gastric glands. Am J Physiol Gastrointest Liver Physiol 2005, 289:G1084–G1090.
Buchan AM, Squires PE, Ring M, Meloche RM: Mechanism of action of the calcium-sensing receptor in human antral gastrin cells. Gastroenterology 2001, 120:1128–1139.
• Ceglia L, Harris SS, Rasmussen HM, Dawson-Hughes B: Activation of the calcium sensing receptor stimulates gastrin and gastric acid secretion in healthy participants. Osteoporos Int 2009, 20:71–78. This double-blind, randomized, placebo-controlled study was the first in vivo human study to demonstrate the actions of of CaSR. Subjects on fixed metabolic diet who were given the CaSR agonist cinacalcet were shown to have increased serum gastrin and basal gastric acid output compared to placebo.
Isenberg JI, Maxwell V: Intravenous infusion of amino acids stimulates gastric acid secretion in man. N Engl J Med 1978, 298:27–29.
Busque SM, Kerstetter JE, Geibel JP, Insogna K: L-type amino acids stimulate gastric acid secretion by activation of the calcium-sensing receptor in parietal cells. Am J Physiol Gastrointest Liver Physiol 2005, 289:G664–G669.
Kirchhoff P, Dave MH, Remy C, et al.: An amino acid transporter involved in gastric acid secretion. Pflugers Arch 2006, 451:738–748.
Quinn SJ, Bai M, Brown EM: pH sensing by the calcium-sensing receptor. J Biol Chem 2004, 279:37241–37249.
Acknowledgments
The authors thank Coleen Palileo for her editorial and artistic support. This work was supported by Department of Veterans Affairs Merit Review funding, and National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases grant R01 543221.
Disclosure
No potential conflict of interest relevant to this article was reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Goo, T., Akiba, Y. & Kaunitz, J.D. Mechanisms of Intragastric pH Sensing. Curr Gastroenterol Rep 12, 465–470 (2010). https://doi.org/10.1007/s11894-010-0147-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11894-010-0147-7