
Abstract
Purpose of Review
Lipoprotein(a) is an independent risk factor for cardiovascular disease. We review the ongoing shifts in consensus guidelines for the testing and management of Lp(a) and provide insight into whether current evidence suggests that awareness and testing of Lp(a) is clinically actionable.
Recent Findings
GWAS and Mendelian randomization studies have established causal links between elevated Lp(a) and forms of CVD, including CAD and calcific aortic valve disease. Testing of Lp(a) identifies patients with similar risk to that of heterozygous FH, enhances risk stratification in patients with borderline/intermediate risk as determined through traditional factors, and facilitates the assessment of inherited CVD risk through cascade screening in patients with known family history of elevated Lp(a). Reductions in Lp(a) through non-targeted therapies including PCSK9 inhibition and lipoprotein apheresis have demonstrated reductions in ASCVD risk that are likely attributable to lowering Lp(a). Targeted therapies to potently lower Lp(a) are in clinical development.
Summary
Lp(a) is actionable, and can be used to identify high risk patients for primary prevention and their family members through cascade screening, and to guide intensification of therapy in primary and secondary prevention of ASCVD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lipoprotein(a) [Lp(a)] is a plasma lipoprotein comprised of an apoB100 molecule bound to the glycoprotein apolipoprotein(a) [apo(a)]. Elevated Lp(a) levels have been identified as an independent risk factor for cardiovascular disease (CVD) via mechanisms of increased atherogenesis, thrombosis, and inflammation [1,2,3,4]. The emergence of RNA-based therapeutics aimed at potently reducing Lp(a) levels has identified Lp(a) as a key residual risk factor to focus on in the effort to combat lipid-driven atherosclerotic cardiovascular disease (ASCVD) risk. This review aims to provide an examination of the current knowledge of Lp(a) and ongoing shifts in consensus guidelines for the testing and management of Lp(a) and explore the question of whether “Lp(a) is actionable” in the absence of currently available targeted Lp(a)-lowering therapies.
Lipoprotein(a) and Evidence of Causality in Cardiovascular Disease
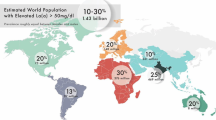
Lp(a) is composed of an LDL-like particle, which incorporates an apo(a) molecule via disulfide linkage with the apoB-100 protein on LDL. Apo(a) consists of multiple components, including a protease domain, ten distinct kringle IV units, and one kringle V unit. The length of the apo(a) tail is determined by the number of kringle IV type 2 (KIV2) repeats, which can range from 11 to more than 50 copies [5]. It is noteworthy that the plasma concentration of Lp(a) exhibits an inverse relationship with the size of the apo(a) particle [6]. Elevated Lp(a) is present in about 20% of the population, with higher prevalence seen in African Americans and South Asians as compared with White or East Asian individuals [7,8,9,10]. However, it should be noted that the lack of uniformity with which these measurements of Lp(a) were conducted across various studies prevents direct comparison between different ethnic groups. Regardless, Lp(a) plasma levels are estimated to be 70–90% genetically determined through codominant expression of the LPA gene on chromosome 6q26-27, leading to the expression of circulating isoforms [11]. Various single nucleotide polymorphisms have been identified to be strongly associated with increased levels of Lp(a) lipoprotein, owing largely to reduced LPA copy number and small Lp(a) isoforms [12].
The relationship between elevated Lp(a) and increased CVD risk is well established and understood to be independent of traditional risk factors such as LDL cholesterol (LDL-C) levels. In addition, the putative heritability of Lp(a) serum concentration have led to Mendelian randomization and large genome wide association studies (GWAS) supporting the association between Lp(a) levels and myocardial infarction [12, 13], ischemic stroke [14, 15], peripheral arterial disease [14, 16], and calcific aortic valve stenosis [17,18,19]. Additionally, large observational epidemiologic studies have established a link between coronary artery disease (CAD) and Lp(a) [1, 20, 21].
Lp(a) Measurement, Challenges Now and Beyond
Ongoing challenges exist given the lack of uniformity of Lp(a) measurement. Lp(a) serum levels are largely determined using immunoassays with apo(a) specific antibodies. However, given the widely variable size of apo(a) due to differential numbers of KIV2 motif repeats, the accuracy of ELISA-based methods is dependent on the binding sites of the apo(a) specific monoclonal antibodies used, as those specific to KIV2 motif repeats can yield significant variability in the measurement of Lp(a) levels [22]. Furthermore, there are two dominant units for reporting Lp(a) levels, with the first method reporting Lp(a) mass in milligrams per deciliter. ELISA-based methods, calibrated in nanomoles per liter of apo(a), account for the variability in Lp(a) size and therefore report measurements in Lp(a) serum molar concentrations; this method has been recommended by the National Heart, Lung and Blood Institute and likely offers the most accurate quantification method thus far [23].
Furthermore, it is important to note that clinically available laboratory reported LDL-C values (using Friedewald, Martin-Hopkins formula, or direct LDL-C measurement) are limited in that they include Lp(a) cholesterol [Lp(a)-C] within reported LDL-C. Patients with very elevated Lp(a) will carry a greater contribution of Lp(a)-C to LDL-C. While existing methods attempt to estimate LDL-C independent of Lp(a)-C include the Dahlen formula, assuming that Lp(a)-C is a fixed 30% of Lp(a) mass, this was demonstrated to overestimate Lp(a)-C and under-estimate true LDL-C (corrected LDL-C) in patients with elevated Lp(a), and therefore its use is no longer recommended [24]. Alternative methods to directly measure Lp(a)-C and therefore corrected LDL-C have been proposed and offer more accurate estimation of risk reduction attributable to Lp(a) reduction, particularly with forthcoming Lp(a)-specific therapies [25].
The establishment of Lp(a) risk thresholds has been important for facilitating ASCVD risk assessment and therapeutic guidance, with the most common thresholds being 50 mg/dL or ≥ 100–125 nmol/L, since the first threshold was introduced by the European Atherosclerosis Society (EAS) in 2010 [26]. However, more recent recommendations in 2022 now advocate for broader thresholds for ruling in or out Lp(a)-associated CVD risk, with “grey zones” encompassing intermediate values within the range of 30–50 mg/dL or 75–125 nmol/L, likely reflecting the understanding of the continuous, linear risk between Lp(a) and cardiovascular outcomes [27••].
Due to the understanding of significant variations in average Lp(a) values among various racial and ethnic groups, both the National Lipid Association (NLA) and HEART UK associations offered recommendations in their 2019 guidelines acknowledging this racial/ethnic heterogeneity while also noting the limited quality of current evidence supporting the use of population-specific thresholds, and as such recommended a universal cut-point with the caveat that it holds greater clinical relevance in predominantly White populations [28, 29]. In contrast, the American Heart Association (AHA) 2021 statement acknowledged the challenges posed by differences in Lp(a) levels among population ancestries and refrained from providing a specific Lp(a) risk threshold in their guidance [30].
Recent guidelines place emphasis on interpreting Lp(a) elevations in the context of a patient’s global ASCVD risk, with two major studies motivating this shift. The EPIC-Norfolk study stratified 14,051 patients by a composite cardiovascular health metric based on traditional cardiovascular risk factors and found that patients with elevated Lp(a) and metric scores in the healthiest category had only ~ 1/3 of the incident CVD risk compared to those with similar Lp(a) elevations but metric scores in the unhealthiest category [31]. The importance of overall CVD risk in contextualizing Lp(a)-specific risk was redemonstrated in a study of 415,274 individuals of European ancestry in the UK Biobank. In this analysis, patients were stratified by their baseline estimated lifetime CVD risk, and the absolute increase in ASCVD risk associated with elevated Lp(a) was found to be proportional to a patient’s baseline absolute risk [21]. Considering these findings, the EAS in its 2022 statement recommended that the interpretation of Lp(a) levels and subsequent therapeutic decisions be made in the context of a patients’ global ASCVD health after accounting for Lp(a), rather than the Lp(a) levels alone [27••].
Guidance on Lp(a) Testing from Professional Societies
Prior to 2019, consensus guidelines offered recommendations for testing only in select individuals with elevated risk profiles based on personal or family history of premature ASCVD, heterozygous familial hypercholesterolemia (HeFH), borderline ASCVD risk in the setting of primary prevention, or progressive ASCVD or refractory LDL-C despite optimal therapy. In response to the accumulating evidence linking Lp(a) to CVD, several professional societies have recently issued or updated their clinical guidance regarding Lp(a) with a trend among recent guideline publications towards advocating for universal Lp(a) screening among all adults (2019 European Society of Cardiology/European Atherosclerosis Society (ESC/EAS), 2021 Canadian Cardiovascular Society (CCS) dyslipidemia guidelines, and 2022 EAS Lp(a) consensus statement) [27••, 32, 33] (Table 1).
Proponents of universal screening cite the high epidemiologic burden of elevated Lp(a) and increasing evidence linking Lp(a) to ASCVD, as well as the ability to enhance ASCVD risk assessment. In contrast, the 2019 NLA scientific statement recommended against testing in the general population due to the lack of currently available targeted Lp(a)-lowering therapies and insufficient evidence linking Lp(a)-specific treatments to improved outcomes [28]. Nevertheless, some societal guidelines highlight that universal screening carries likely little harm and identification of patients with extremely high Lp(a) levels ≥ 180 mg/dL (≥ 430 nmoL/L) is important as these individuals have lifetime ASCVD risk similar to patients with HeFH [33]. Recommendations may continue to shift toward universal screening for Lp(a) in the general population should targeted Lp(a) lowering therapies demonstrate benefit in lowering CVD risk.
Furthermore, patients in the highest tertiles of Lp(a) and oxidized phospholipids on apoB (OxPL-apoB) exhibit more rapid hemodynamic deterioration and need for aortic valve replacement [34,35,36]. Several societies have proposed that Lp(a) examination may inform the frequency of valve surveillance in those patients with established calcific aortic valve disease (CAVD) [28, 29, 37].
Mitigation of Lp(a)-Driven CVD Risk
There are no currently approved Lp(a) lowering therapeutics available for clinical use, and thus the consensus recommendation among professional societies for Lp(a) management centers primarily on tighter control of other traditional ASCVD risk factors, including lowering LDL-C, blood pressure, blood glucose, and promotion of improved dietary/lifestyle changes. Intensification of such changes, irrespective of their effect on Lp(a) levels, can mitigate some ASCVD risk as those with elevated Lp(a) in ideal cardiovascular health had lower risk than those not following healthy lifestyle [31].
As new data have emerged on how Lp(a) and other risk factors impact overall ASCVD risk, recent guidelines have started providing more detailed recommendations. The 2022 EAS guideline, for instance, has outlined targets for LDL-C reduction required to mitigate Lp(a)-associated risk based on different Lp(a) levels and the age at which LDL-C-lowering therapy is initiated [27••]. While these recommendations suggest the potential for some patients to mitigate their Lp(a)-related risk through aggressive management of other factors, this approach has important limitations and may be inadequate for individuals whose primary ASCVD risk stems disproportionately from elevated Lp(a). For example, work by Trinder et al. [38] suggests that apoB is insufficient to explain Lp(a)-driven risk for CAD, and therefore it is unclear whether aggressive LDL-C and/or apoB lowering can fully offset risk from Lp(a). Importantly, statin therapy may lead to increases in Lp(a) [6, 39]. In a large meta-analysis of seven, randomized placebo-controlled statin outcomes trials, ASCVD risk persisted in a linear relationship with Lp(a) despite statin treatment [40]. However, despite the potential for statins to increase Lp(a) levels, statin therapy remains a cornerstone of ASCVD prevention and risk reduction and statins remain first-line lipid-lowering therapy for ASCVD risk reduction including for patients with elevated Lp(a).
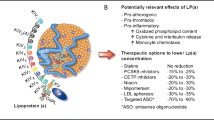
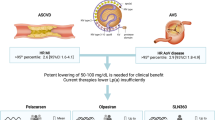
Beyond the general recommendation for traditional risk factor control, the use of PCSK9 inhibitor (PCSK9i) monoclonal antibodies (mAbs) for managing elevated Lp(a) has been shown to reduce Lp(a) levels by up to 15–30% [41, 42]. Post hoc analyses of the FOURIER and ODYSSEY OUTCOMES clinical trials showed that patients with elevated Lp(a) (defined as Lp(a) > 50 mg/dL in the post-hoc FOURIER study, Lp(a) > 60 mg/dL in the post-hoc ODYSSEY OUTCOMES study) derived greater absolute risk reduction from PCSK9i mAb treatment compared to those with Lp(a) levels below the threshold. Further analysis of the ODYSSEY OUTOMES study suggested that the Lp(a)-lowering effect of alirocumab contributed independently to ASCVD risk reduction [43•]. In keeping with these findings, the NLA dyslipidemia guideline suggested consideration of PCSK9 mAb for very high-risk patients taking a maximally tolerated statin and ezetimibe, with an Lp(a) of ≥ 50 mg/dL (or ≥ 100 nmol/L) and LDL-C ≥ 70 mg/dL (or non–HDL-C ≥ 100 mg/dL) [28]. Similarly, the CCS in 2021 recommended consideration of PCSK9i mAb for secondary prevention in high-risk patients with Lp(a) > 60 mg/dL [32].
Lipoprotein apheresis has also been examined as a therapeutic option in a number of single arm studies, with reductions in Lp(a) levels by up to 75% (time averaged reduction of 30–35%), with some studies suggesting associated reductions in CVD events [44,45,46]. However, apheresis remains an invasive and expensive procedure, thereby limiting its widespread use. Nevertheless, guidelines have suggested utility for this modality in select patients with progressive coronary artery disease and Lp(a) greater than 150 nmol/L (> 60 mg/dL) and whose LDL-C remains greater than 125 mg/dL despite maximally tolerated lipid lowering therapy [27••, 29]. In the USA and Germany, a current indication for lipoprotein apheresis is HeFH with LDL-C ≥ 100 mg/dL and Lp(a) ≥ 60 mg/dL and either CAD or peripheral artery disease (PAD) [47, 48].
Niacin has been shown to reduce Lp(a) levels by up to 30%, though concerns over tolerability as well as the unknown cardiovascular benefit have limited recommendations for its use [49, 50].
Finally, antisense oligonucleotides (pelacarsen) targeting apo(a) and investigational small interfering RNA molecules targeting apo(a) RNA are in clinical trials (e.g., olpasiran and SLN360) [51,52,53]. These investigational, targeted therapies have achieved potent serum Lp(a) lowering by up to 80–100% at the highest tested doses, in contrast with more modest Lp(a) reductions demonstrated with currently available therapies, such as PCSK9i.
Is Lp(a) Clinically Actionable?
Our perspective is that Lp(a) has important clinical implications that are actionable with the current literature on multiple levels. Despite the lack of targeted therapies for Lp(a)-driven CVD risk reduction in clinical practice, the expansion of the knowledge base surrounding Lp(a) and its conferred risks for various CVD profiles in recent years offers new insights into how more widespread testing of Lp(a) is of greater utility than previously realized. Shifting consensus among various societal guidelines has also reflected the growing body of evidence that understanding Lp(a)-driven risk in the broader sense is of utility in a number of clinical scenarios [27••, 32, 33].
Primary Prevention of ASCVD
The utility of Lp(a) screening in primary prevention is multifold, and our perspective is that universal testing should be considered in all adults at least once, as suggested in several recent statements from professional societies [27••, 32, 33]. Lp(a) testing provides a practical method in identifying these high CVD risk patients for aggressive lipid lowering therapy, particularly as evaluation of traditional risk factors may not capture these patients. Furthermore, certain racial/ethnic backgrounds demonstrate higher prevalence for elevated Lp(a), including those of African or South Asian heritage, and the identification of elevated Lp(a) in patients of South Asian or Latin descent may support maximizing lifestyle modifications to reduce CVD if demonstrated to have Lp(a) > 50 mg/dL, particularly as these populations may have the highest Lp(a) attributable risk for myocardial infarction (MI), independent of other traditional risk factors [54]. Finally, both the 2018 Multi-Society Cholesterol Guideline and the 2019 American College of Cardiology (ACC)/American Heart Association (AHA) Primary Prevention Guideline recommend the use of Lp(a) as a CVD “risk enhancer” among those with borderline and intermediate risk as determined by the 10-year Pooled Cohort Equation. Together with additional evidence-based tools for risk stratification (e.g., coronary artery calcium scoring), elevated Lp(a) may inform discussions with patients on diet/lifestyle counseling and consideration of the initiation and/or intensification of statin therapy and non-statin lipid-lowering therapy [55, 56]. In primary prevention, the use of aspirin has also been associated with potential benefit in patients with elevated Lp(a)-associated genotypes as demonstrated in secondary analyses of the ASPREE trial and Women’s Health Study [57, 58]. Further investigation with randomized trials will help define the role of aspirin in primary prevention of ASCVD in patients with elevated Lp(a).
Secondary Prevention of ASCVD
While post hoc analyses of FOURIER and ODYSSEY OUTCOMES suggest a potential benefit of Lp(a) lowering in the secondary prevention of ASCVD, data from randomized, controlled cardiovascular outcomes trials with potent Lp(a)-lowering therapies (HORIZON NCT04023552 and OCEAN(a) NCT05581303) are poised to better examine the ability of Lp(a)-lowering to reduce ASCVD risk. Until such data are available, however, the examination of Lp(a) levels may be particularly beneficial in two populations: 1) patients with recurrent ASCVD events despite aggressive lipid lowering therapy and 2) patients with less than expected LDL-C lowering on evidence-based lipid lowering therapy [28, 37]. Identification of patients with elevated Lp(a) in this context may facilitate practicing clinicians to pursue aggressive CVD risk factor management, consideration of the lowest guideline recommended LDL-C targets, and consideration of the use of PCSK9 inhibitors or lipoprotein apheresis in select cases. Finally, in patients with established CAVD, Lp(a) may help identify patients who may stand to benefit from more aggressive aortic valve surveillance, given the association of more rapid valve deterioration and need for replacement in those at the highest tertiles of Lp(a) levels [29, 37].
Furthermore, given that Lp(a) levels are largely determined by genetic factors, with little influence from lifestyle modifications, cascade testing in patients with either premature ASCVD and/or known elevated Lp(a) may help prognosticate the CVD risk profiles of offspring and other family members and help direct lifetime risk reduction and primary prevention [27••, 30]. These recommendations are summarized in the proposed “Lp(a) action plan” for clinicians (Fig. 1).

Proposed lipoprotein(a) action plan for practicing clinicians
Conclusions
In summary, despite the current lack of targeted therapies for Lp(a) lowering, Lp(a) is clearly actionable today. Recommendations for more widespread Lp(a) testing continue to gain traction with the goal of helping to refine our ability to estimate CVD risk in patients and families, and guide treatment decisions in primary and secondary prevention of ASCVD.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Emerging Risk Factors C, Erqou S, Kaptoge S, Perry PL, Di Angelantonio E, Thompson A, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412–23.
Kiechl S, Willeit J. The mysteries of lipoprotein(a) and cardiovascular disease revisited. J Am Coll Cardiol. 2010;55(19):2168–70.
Steinberg D, Witztum JL. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30(12):2311–6.
Saleheen D, Haycock PC, Zhao W, Rasheed A, Taleb A, Imran A, et al. Apolipoprotein(a) isoform size, lipoprotein(a) concentration, and coronary artery disease: a mendelian randomisation analysis. Lancet Diabetes Endocrinol. 2017;5(7):524–33.
Brunner C, Lobentanz EM, Petho-Schramm A, Ernst A, Kang C, Dieplinger H, et al. The number of identical kringle IV repeats in apolipoprotein(a) affects its processing and secretion by HepG2 cells. J Biol Chem. 1996;271(50):32403–10.
Tsimikas S. A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69(6):692–711.
Virani SS, Brautbar A, Davis BC, Nambi V, Hoogeveen RC, Sharrett AR, et al. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2012;125(2):241–9.
Steffen BT, Thanassoulis G, Duprez D, Stein JH, Karger AB, Tattersall MC, et al. Race-based differences in lipoprotein(a)-associated risk of carotid atherosclerosis. Arterioscler Thromb Vasc Biol. 2019;39(3):523–9.
Bhatnagar D, Anand IS, Durrington PN, Patel DJ, Wander GS, Mackness MI, et al. Coronary risk factors in people from the Indian subcontinent living in west London and their siblings in India. Lancet. 1995;345(8947):405–9.
Enkhmaa B, Anuurad E, Zhang W, Kim K, Berglund L. Heritability of apolipoprotein (a) traits in two-generational African-American and Caucasian families. J Lipid Res. 2019;60(9):1603–9.
Kronenberg F. Human Genetics and the Causal Role of Lipoprotein(a) for Various Diseases. Cardiovasc Drugs Ther. 2016;30(1):87–100.
Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518–28.
Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331–9.
Nordestgaard BG, Langsted A. Lipoprotein (a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res. 2016;57(11):1953–75.
Pan Y, Li H, Wang Y, Meng X, Wang Y. Causal effect of Lp(a) [Lipoprotein(a)] Level on ischemic stroke and Alzheimer disease: a Mendelian randomization study. Stroke. 2019;50(12):3532–9.
Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Genetic evidence that lipoprotein(a) associates with atherosclerotic stenosis rather than venous thrombosis. Arterioscler Thromb Vasc Biol. 2012;32(7):1732–41.
Cairns BJ, Coffey S, Travis RC, Prendergast B, Green J, Engert JC, et al. A replicated, genome-wide significant association of aortic stenosis with a genetic variant for lipoprotein(a): meta-analysis of published and novel data. Circulation. 2017;135(12):1181–3.
Arsenault BJ, Boekholdt SM, Dube MP, Rheaume E, Wareham NJ, Khaw KT, et al. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: a prospective Mendelian randomization study and replication in a case-control cohort. Circ Cardiovasc Genet. 2014;7(3):304–10.
Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368(6):503–12.
Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117(2):176–84.
Patel AP, Wang M, Pirruccello JP, Ellinor PT, Ng K, Kathiresan S, et al. Lp(a) (lipoprotein[a]) concentrations and incident atherosclerotic cardiovascular disease: new insights from a large national biobank. Arterioscler Thromb Vasc Biol. 2021;41(1):465–74.
Marcovina SM, Albers JJ, Gabel B, Koschinsky ML, Gaur VP. Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a). Clin Chem. 1995;41(2):246–55.
Tsimikas S, Fazio S, Ferdinand KC, Ginsberg HN, Koschinsky ML, Marcovina SM, et al. NHLBI working group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol. 2018;71(2):177–92.
Yeang C, Karwatowska-Prokopczuk E, Su F, Dinh B, Xia S, Witztum JL, Tsimikas S. Effect of pelacarsen on lipoprotein(a) cholesterol and corrected low-density lipoprotein cholesterol. J Am Coll Cardiol. 2022;79(11):1035–46.
Yeang C, Witztum JL, Tsimikas S. Novel method for quantification of lipoprotein(a)-cholesterol: implications for improving accuracy of LDL-C measurements. J Lipid Res. 2021;62: 100053.
Nordestgaard BG, Chapman MJ, Ray K, Boren J, Andreotti F, Watts GF, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844–53.
•• Kronenberg F, Mora S, Stroes ESG, Ference BA, Arsenault BJ, Berglund L, et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: a European Atherosclerosis Society consensus statement. Eur Heart J. 2022;43(39):3925–46. This document represents the first consensus recommendation by a major professional society for universal testing of Lp(a) on the basis of identifying patients with very high Lp(a) ≥180 mg/dL or ≥430 nmoL/L given its association with high lifetime ASCVD risk.
Wilson DP, Jacobson TA, Jones PH, Koschinsky ML, McNeal CJ, Nordestgaard BG, et al. Use of Lipoprotein(a) in clinical practice: a biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol. 2019;13(3):374–92.
Cegla J, Neely RDG, France M, Ferns G, Byrne CD, Halcox J, et al. HEART UK consensus statement on Lipoprotein(a): a call to action. Atherosclerosis. 2019;291:62–70.
Reyes-Soffer G, Ginsberg HN, Berglund L, Duell PB, Heffron SP, Kamstrup PR, et al. Lipoprotein(a): a genetically determined, causal, and prevalent risk factor for atherosclerotic cardiovascular disease: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2022;42(1):e48–60.
Perrot N, Verbeek R, Sandhu M, Boekholdt SM, Hovingh GK, Wareham NJ, et al. Ideal cardiovascular health influences cardiovascular disease risk associated with high lipoprotein(a) levels and genotype: The EPIC-Norfolk prospective population study. Atherosclerosis. 2017;256:47–52.
Pearson GJ, Thanassoulis G, Anderson TJ, Barry AR, Couture P, Dayan N, et al. 2021 Canadian Cardiovascular Society guidelines for the management of dyslipidemia for the prevention of cardiovascular disease in adults. Can J Cardiol. 2021;37(8):1129–50.
Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111–88.
Capoulade R, Chan KL, Yeang C, Mathieu P, Bosse Y, Dumesnil JG, et al. Oxidized phospholipids, lipoprotein(a), and progression of calcific aortic valve stenosis. J Am Coll Cardiol. 2015;66(11):1236–46.
Zheng KH, Tsimikas S, Pawade T, Kroon J, Jenkins WSA, Doris MK, et al. Lipoprotein(a) and oxidized phospholipids promote valve calcification in patients with aortic stenosis. J Am Coll Cardiol. 2019;73(17):2150–62.
Chan KL, Teo K, Dumesnil JG, Ni A, Tam J, Investigators A. Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation. 2010;121(2):306–14.
Handelsman Y, Jellinger PS, Guerin CK, Bloomgarden ZT, Brinton EA, Budoff MJ, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the management of dyslipidemia and prevention of cardiovascular disease algorithm - 2020 executive summary. Endocr Pract. 2020;26(10):1196–224.
Trinder M, Zekavat SM, Uddin MM, Pampana A, Natarajan P. Apolipoprotein B is an insufficient explanation for the risk of coronary disease associated with lipoprotein(a). Cardiovasc Res. 2021;117(5):1245–7.
Tsimikas S, Gordts P, Nora C, Yeang C, Witztum JL. Statin therapy increases lipoprotein(a) levels. Eur Heart J. 2020;41(24):2275–84.
Willeit P, Ridker PM, Nestel PJ, Simes J, Tonkin AM, Pedersen TR, et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: individual patient-data meta-analysis of statin outcome trials. Lancet. 2018;392(10155):1311–20.
O’Donoghue ML, Fazio S, Giugliano RP, Stroes ESG, Kanevsky E, Gouni-Berthold I, et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation. 2019;139(12):1483–92.
Bittner VA, Szarek M, Aylward PE, Bhatt DL, Diaz R, Edelberg JM, et al. Effect of alirocumab on lipoprotein(a) and cardiovascular risk after acute coronary syndrome. J Am Coll Cardiol. 2020;75(2):133–44.
• Szarek M, Bittner VA, Aylward P, Baccara-Dinet M, Bhatt DL, Diaz R, et al. Lipoprotein(a) lowering by alirocumab reduces the total burden of cardiovascular events independent of low-density lipoprotein cholesterol lowering: ODYSSEY OUTCOMES trial. Eur Heart J. 2020;41(44):4245–55. This post-hoc analysis of the ODYSSEY OUTCOMES study demonstrated that baseline Lp(a) was linearly associated with ASCVD in post-ACS patients and that Lp(a) lowering via PCSK9 inhibition contributed independently to ASCVD risk reduction.
Roeseler E, Julius U, Heigl F, Spitthoever R, Heutling D, Breitenberger P, et al. Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of follow-up and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol. 2016;36(9):2019–27.
Leebmann J, Roeseler E, Julius U, Heigl F, Spitthoever R, Heutling D, et al. Lipoprotein apheresis in patients with maximally tolerated lipid-lowering therapy, lipoprotein(a)-hyperlipoproteinemia, and progressive cardiovascular disease: prospective observational multicenter study. Circulation. 2013;128(24):2567–76.
Rosada A, Kassner U, Vogt A, Willhauck M, Parhofer K, Steinhagen-Thiessen E. Does regular lipid apheresis in patients with isolated elevated lipoprotein(a) levels reduce the incidence of cardiovascular events? Artif Organs. 2014;38(2):135–41.
Khan TZ, Hsu LY, Arai AE, Rhodes S, Pottle A, Wage R, et al. Apheresis as novel treatment for refractory angina with raised lipoprotein(a): a randomized controlled cross-over trial. Eur Heart J. 2017;38(20):1561–9.
Schettler VJJ, Neumann CL, Peter C, Zimmermann T, Julius U, Roeseler E, et al. The German Lipoprotein Apheresis Registry (GLAR) - almost 5 years on. Clin Res Cardiol Suppl. 2017;12(Suppl 1):44–9.
Investigators A-H, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255–67.
Group HTC, Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371(3):203–12.
O’Donoghue ML, Rosenson RS, Gencer B, Lopez JAG, Lepor NE, Baum SJ, et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N Engl J Med. 2022;387(20):1855–64.
Rider DA, Eisermann M, Loffler K, Aleku M, Swerdlow DI, Dames S, et al. Pre-clinical assessment of SLN360, a novel siRNA targeting LPA, developed to address elevated lipoprotein (a) in cardiovascular disease. Atherosclerosis. 2022;349:240–7.
Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, Tardif JC, Baum SJ, Steinhagen-Thiessen E, et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med. 2020;382(3):244–55.
Pare G, Caku A, McQueen M, Anand SS, Enas E, Clarke R, et al. Lipoprotein(a) Levels and the Risk of Myocardial Infarction Among 7 Ethnic Groups. Circulation. 2019;139(12):1472–82.
Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596–646.
Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139(25):e1082–143.
Chasman DI, Shiffman D, Zee RY, Louie JZ, Luke MM, Rowland CM, et al. Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis. 2009;203(2):371–6.
Lacaze P, Bakshi A, Riaz M, Polekhina G, Owen A, Bhatia HS, et al. Aspirin for primary prevention of cardiovascular events in relation to lipoprotein(a) genotypes. J Am Coll Cardiol. 2022;80(14):1287–98.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
GSM and TTC have no relevant conflicts of interest to disclose. MJW is a consultant to Amarin, Regeneron, and The Kinetix Group, reports advisory board fees from Novartis and speaker fees from Regeneron, and has received grant support from Amgen (investigator-initiated study) and the National Institutes of Health, grant KL2TR001444. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, G.S., Chiou, T.T. & Wilkinson, M.J. Is Lipoprotein(a) Clinically Actionable with Today’s Evidence? The Answer is Yes. Curr Cardiol Rep 25, 1175–1187 (2023). https://doi.org/10.1007/s11886-023-01937-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11886-023-01937-z