
Abstract
Purpose of Review
The elevated adverse cardiovascular event rate among patients with low high-density lipoprotein cholesterol (HDL-C) formed the basis for the hypothesis that elevating HDL-C would reduce those events. Attempts to raise endogenous HDL-C levels, however, have consistently failed to show improvements in cardiovascular outcomes. However, steady-state HDL-C concentration does not reflect the function of this complex family of particles. Indeed, HDL functions correlate only weakly with serum HDL-C concentration. Thus, the field has pivoted from simply raising the quantity of HDL-C to a focus on improving the putative anti-atherosclerotic functions of HDL particles. Such functions include the ability of HDL to promote the efflux of cholesterol from cholesterol-laden macrophages. Apolipoprotein A-I (apoA-I), the signature apoprotein of HDL, may facilitate the removal of cholesterol from atherosclerotic plaque, reduce the lesional lipid content and might thus stabilize vulnerable plaques, thereby reducing the risk of cardiac events. Infusion of preparations of apoA-I may improve cholesterol efflux capacity (CEC). This review summarizes the development of apoA-I therapies, compares their structural and functional properties and discusses the findings of previous studies including their limitations, and how CSL112, currently being tested in a phase III trial, may overcome these challenges.
Recent Findings
Three major ApoA-I-based approaches (MDCO-216, CER-001, and CSL111/CSL112) have aimed to enhance reverse cholesterol transport. These three therapies differ considerably in both lipid and protein composition. MDCO-216 contains recombinant ApoA-I Milano, CER-001 contains recombinant wild-type human ApoA-I, and CSL111/CSL112 contains native ApoA-I isolated from human plasma. Two of the three agents studied to date (apoA-1 Milano and CER-001) have undergone evaluation by intravascular ultrasound imaging, a technique that gauges lesion volume well but does not assess other important variables that may relate to clinical outcomes. ApoA-1 Milano and CER-001 reduce lecithin-cholesterol acyltransferase (LCAT) activity, potentially impairing the function of HDL in reverse cholesterol transport. Furthermore, apoA-I Milano can compete with and alter the function of the recipient’s endogenous apoA-I. In contrast to these agents, CSL112, a particle formulated using human plasma apoA-I and phosphatidylcholine, increases LCAT activity and does not lead to the malfunction of endogenous apoA-I. CSL112 robustly increases cholesterol efflux, promotes reverse cholesterol transport, and now is being tested in a phase III clinical trial.
Summary
Phase II-b studies of MDCO-216 and CER-001 failed to produce a significant reduction in coronary plaque volume as assessed by IVUS. However, the investigation to determine whether the direct infusion of a reconstituted apoA-I reduces post-myocardial infarction coronary events is being tested using CSL112, which is dosed at a higher level than MDCO-216 and CER-001 and has more favorable pharmacodynamics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introductıon
Available therapies effectively reduce low-density lipoprotein cholesterol (LDL-C) levels. Yet, even with very well-controlled LDL-C concentrations, the residual burden of cardiovascular disease (CVD) remains substantial [1]. To reduce this residual risk of CVD, research has also focused on HDL-C as a potential therapeutic target. Several prospective epidemiologic studies, starting with the investigations of Gofman et al. [2] (1966) and Gordon et al. [3] (1977) in the Framingham Heart Study among many others, have demonstrated a consistent and robust inverse relationship between HDL-C levels and cardiovascular risk, irrespective of LDL-C levels or treatment with statins [4, 5, 6]. Furthermore, low HDL-C concentrations correlated with long-term adverse outcomes and mortality in patients with the acute coronary syndrome (ACS) [7, 8]. Meta-analyses have also demonstrated an inverse association of lower HDL-C with an increased cardiovascular (CV) risk among patients treated with statins [5, 9, 10].
These relationships gave rise to the “HDL hypothesis,” which states that “a reduction in plasma HDL may impair the efflux of cholesterol from the arterial wall and thereby accelerate the development of atherosclerosis.” [11, 12] Extensive research has explored various HDL-raising agents with the goal of further reducing cardiovascular risk. The “HDL hypothesis,” however, has encountered recent challenges based on the results of human genetic studies and the null results of several clinical trials that raised HDL-C by administration of fibrates, nicotinic acid (niacin), or various cholesteryl ester transfer protein (CETP) inhibitors.
Yet, strong experimental evidence indicates that HDL particles can mediate reverse cholesterol transport (RCT). The vasoprotective properties of HDL depend strongly on its protein composition, which undergoes substantial alterations and modifications in patients with coronary artery disease [13, 14]. ApoA-I is the principal protein component of HDL and mediates RCT and activation of LCAT, which can affect multiple steps in the RCT pathway. LCAT on HDL particles esterifies free cholesterol and both reduces free cholesterol on the particle surface and favors the accumulation of cholesteryl ester in the core. These actions enable the HDL to both acquire more free cholesterol and deliver more to the liver. Two previous infusible apoA-I compounds did not achieve a significant change in plaque volume in patients with coronary artery disease as assessed by intravascular ultrasound (IVUS). But these approaches had potential limitations mentioned below.
HDL-C as a Therapeutic Target
Fibrates
While fibrates increase HDL-C and lower triglyceride levels, their effectiveness has met with mixed results with respect to cardiovascular event reduction. Although gemfibrozil therapy raised HDL-C and lowered triglycerides in patients with CVD and low HDL-C, the reduction in events did not become apparent until approximately 2 years after randomization [15]. Moreover, the major studies with gemfibrozil (Helsinki Heart Study [16] and VA-HIT [15]) did not specify statins as background therapy. Clinically important drug interaction safety issues limit the use of gemfibrozil combined with statins [17]. In FIELD, fenofibrate did not demonstrate a significant reduction in the risk of coronary heart disease, death, or nonfatal myocardial infarction (MI) compared with placebo in patients with type 2 diabetes mellitus [18]. A subsequent study also demonstrated that the combination of fenofibrate and simvastatin did not reduce the rate of fatal cardiovascular events, nonfatal MI, or nonfatal stroke, as compared with simvastatin alone [19]. A large-scale outcome trial that evaluated a novel selective PPAR alpha modulator, pemafibrate, in patients with diabetes, hypertriglyceridemia (> 200 mg/dL), and low HDL (< 40 mg/dL), on a background of statin therapy. The study was recently halted for futility [20].
Niacin
Long-term niacin therapy decreases LDL-C and triglycerides and increases HDL-C levels. Niacin decreased the occurrence of MI in the long-term follow-up of the Coronary Drug Project conducted in the pre-statin era [21]. Outcome trials have not demonstrated a significant reduction in cardiovascular events with niacin-statin combinations despite significant increases in HDL-C levels, as compared with a statin-only approach [22, 23]. Furthermore, niacin therapy caused multiple adverse effects including elevated glucose levels among patients with diabetes [24], an increased risk of developing diabetes [25], peptic ulceration, myopathy, skin rash and ulceration, as well as excess bleeding events (mostly gastrointestinal and intracranial), and infections with combination niacin-laropiprant in patients receiving statins. [26]
CETP Inhibitors
CETP garnered great interest because of its role in transferring cholesteryl esters from HDL to apolipoprotein B-containing lipoproteins [27, 28]. The brisk rise in HDL-C induced by CETP inhibitors and the decrease in LDL-C levels with most agents offered considerable promise to prevent cardiovascular events. Yet, contrary to expectations, this therapy failed or only very modestly succeeded in clinical trials, either causing an increase in CV events and deaths (torcetrapib [29]) or lacking efficacy in reducing clinical outcomes despite the rise in HDL-C (dalcetrapib [30], evacetrapib [31]). Torcetrapib raises blood pressure, lowers potassium levels, and elevates sodium and bicarbonate levels due to an off-target mineralocorticoid agonist effect [32]. Anacetrapib treatment showed a modest beneficial effect but more likely due to LDL-C lowering than an elevation in HDL-C [33].
Inhibition of CETP activity increases HDL-C levels. However, the main atheroprotective property of HDL may derive from its ability to act as an acceptor of cholesterol from peripheral cells, specifically arterial wall macrophages. CETP inhibitors may interfere with this process by changing the cholesterol efflux capacity of donor cells and by modulating the uptake capacity of HDL particles [28]. Second, LDL, intermediate-density lipoprotein (IDL), and very-low-density lipoprotein (VLDL) contribute to RCT by CETP-mediated receipt of cholesteryl esters from HDL for the subsequent uptake into the liver. The blockage of this pathway may also contribute to the futility of CETP inhibitors. Furthermore, the concept that very high concentrations of HDL-C could actually deliver free cholesterol to macrophages might contribute to the lack of demonstrable cardiovascular benefit in the members of this class of drugs that elevate HDL concentrations substantially [34]. Taken together, the results of clinical trials indicate that raising HDL-C concentrations alone does not suffice to protect against atherosclerotic events in patients receiving statins.
Moving from Quantity to Quality
The measurement of the cholesterol in HDL alone cannot fully capture the putative antiatherogenic functions of HDL. HDL contains not only cholesterol but its major apolipoprotein (AI) and some > 50 other associated proteins in variable mixes [35, 36, 37, 38] giving rise to great heterogeneity in HDL particles. While increasing the quantity of HDL-C per se has not reduced CVD risk, altering other aspects of HDL particles may hold more promise [39, 40, 41]. Thus, some researchers have focused on validated atheroprotective functions of HDL like CEC rather than circulating HDL-C levels.
Cholesterol Efflux Capacity as a Therapeutic Target
Reverse cholesterol transport affects HDL-mediated transport of cholesterol from peripheral tissues to the liver. Cholesterol efflux (CE), cholesterol esterification, lipoprotein remodeling, and hepatic lipid uptake all contribute to this process (Fig. 1). CE from macrophages is the initial step and can be assessed ex vivo as a measure of the biological functionality of HDL particles [42]. CEC relates inversely with atherosclerosis burden, suggesting that CEC mediates atheroprotection by HDL [43]. Increased CEC also correlates strongly with a reduction in incident cardiovascular events, independent of HDL-C and LDL-C concentration [43, 44, 45]. Therefore, one newer path of investigation has focused on boosting HDL’s ability to efflux cholesterol. This shift reflects a refinement of the “HDL hypothesis” to a “cholesterol efflux hypothesis.”

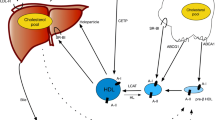
Schematic overview of reverse cholesterol transport. †Free cholesterol in peripheral tissues is effluxed by ABCA1 and ABCG1 transporters to lipid-poor ApoA-I (Preβ-HDL) and larger spherical HDL particles, respectively. The enzyme LCAT, carried on HDL particles, esterifies the free cholesterol molecules to form cholesteryl esters, which migrate to the core of the HDL particle to form mature HDL particles. Subsequently, mature HDL particles deliver the lipid cargo back to the liver through uptake mediated by the scavenger receptor SR-BI. Finally, cholesterol is converted to bile salts in the liver and secreted into the small intestine. CSL112 is apolipoprotein A-I purified from human plasma and reconstituted with phosphatidylcholine to form lipoprotein particles suitable for infusion. CSL112 fuses with HDL in plasma with subsequent release of lipid-poor apoA-I (pre-beta HDL). Abbreviations: ABCA1, ATP-binding cassette protein A1; ABCG1, ATP-binding cassette protein G1; ApoA-1, apolipoprotein A-I; HDL, high-density lipoprotein; LCAT, lecithin cholesterol acyltransferase; SR-BI, scavenger receptor class-B, type I; FC, free cholesterol, CE, cholesteryl ester, †Created with BioRender.com
Plasmas with similar HDL-C levels may differ in their ability to promote cholesterol efflux [46], indicating that the cholesterol content of HDL does not fully reflect antiatherogenic HDL properties. Specifically, a high HDL-C concentration may coexist with a low CEC. Therefore, qualitative changes to HDL function, particularly the ability to increase CEC, may relate directly to an improved CVD outcome [47].
Apolipoprotein A-I (ApoA-I)
ApoA-I, the major protein within HDL, governs in part its size and shape, removes cholesterol from peripheral cells, activates LCAT, and delivers cholesteryl esters to the liver. Evidence from animal and human studies suggests an important role of apoA-I in many of the antiatherogenic effects attributed to HDL [48, 49, 50]. Various peptides, imitating the amphipathic helices in apoA-I, have undergone testing as therapeutic agents including full-length apoA-I, mutated variants of apoA-I, and apoA-I mimetics such as 4F, 6F, FX-5A, ATI-5261, and ETC-642/MDCO-216 [51]. Phase II/III clinical trials in humans have not evaluated these apoA-I mimetic peptides.
ApoA-I-Based Infusion Therapies
Direct infusion of HDL-like particles or transgenic expression of its major proteins may reduce lesion burden in experimental atherosclerosis as well as favorably modify plaque functional features thought to relate to clinical benefit [52, 53, 54, 55]. Such findings in animals led to the hypothesis that infusion of apoA-I containing particles, such as lipid-poor pre-β HDL (the nascent lipid-poor precursor of mature HDL), can render plaques less likely to disrupt and regress atherosclerosis. Subsequently, small human studies have reported favorable effects of HDL-like particle infusion on endothelial vasodilator function and atherosclerosis [56, 57, 58, 59]. The effects of infusing purified forms of HDL on lipid transporting factors, endothelial functions, and changing atheroma volume [60] suggest that administering nascent HDL-like particles is functionally active in promoting cholesterol efflux as opposed to raising HDL-C concentration per se, and may have more relevance to reducing cardiovascular risk [61, 62]. Therefore, engineered HDL particles prepared in vitro from human plasma-derived or recombinant apoA-I and phospholipids merit testing to assess whether their infusion could reduce the burden of coronary atherosclerosis and yield clinical benefit. To date, three infusible HDL mimetics have proceeded from preclinical testing to clinical trials in humans (Fig. 2).

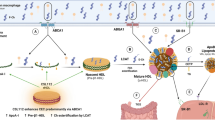
Timeline of human studies on apolipoprotein A-I infusion therapies. ApoA1 Milano (Nissen et al.) [67]; ApoA1 Milano (Kempen et al.) [98]; MILANO PILOT study (ApoA1 Milano, Nicholls et al.) 68.••; CER 001 (Keyserling et al.) [72]; CHI-SQUARE study (CER 001, Tardif et al.) [70]; CER 001 (Zheng et al.) [69]; CARAT study (CER 001, Nicholls et al.) 74.••; ERASE study (CSL111, Tardif et al.) [60]; CSL112 (Easton et al.) [81]; CSL112 (Gille et al.) 80, 82.••••; CSL112 (Tricoci et al.) [104]; AEGIS-I trial (CSL112, Gibson et al.) [83.••••]; AEGIS-II trial design (Gibson et al.) [84]. Abbreviations: AEGIS-I, ApoA-I Event Reducing in Ischemic Syndromes I; AEGIS-II, ApoA-I Event Reducing in Ischemic Syndromes-II; ApoA-I, apolipoprotein A-I; CARAT, CER-001 Atherosclerosis Regression Acute Coronary Syndrome Trial; ERASE, Effect of HDL on Atherosclerosis-Safety and Efficacy study
ApoA-I Milano
In 1974, University of Milan researchers first described apoA-I Milano. It contains an Arg to Cys mutation at residue 173 and is associated with low HDL-C concentration and modest hypertriglyceridemia but with no apparent elevation of cardiovascular risk [63, 64]. Recombinant apoA-I Milano produced in bacteria and complexed with phospholipids was synthesized and studied with the hypothesis that it would modulate atheroma.
Preclinical studies [65, 66] predicted a potential therapeutic scenario that contemplated an infusion following ACS to augment cholesterol efflux and stabilize plaque, while ongoing conventional lipid-modulating therapy would provide long-term clinical benefits. A phase II clinical trial evaluated 57 subjects following ACS. The subjects received weekly infusions of apoA-I Milano dimer complexed with palmitoyl-oleoylphosphatidylcholine (ETC-216, Esperion Therapeutics) at a dose of 15 or 45 mg/kg for 5 weeks. The investigators used intravascular ultrasonography (IVUS) to compare baseline coronary atherosclerosis to that measured 2 weeks after the final infusion of ETC-216. Plaque volume fell by 4.2% relative to the baseline, but the difference was not significant compared to the placebo arm [67]. Nonetheless, these findings offered promise regarding a novel strategy for the management of ACS.
MILANO-PILOT, a double-blind, randomized, placebo-controlled, multicenter trial evaluated the next iteration of an apoA-I Milano therapeutic, MDCO-216 [68.••]. Post-ACS patients (n = 122) on contemporary high-intensity statin therapy received MDCO-216 (20 mg/kg) for 5 weeks. HDL-C levels fell in the MDCO-216 group but not in the placebo group. IVUS showed no significant difference between the treatment and placebo arms in percent atheroma volume, total atheroma volume, and atheroma volume in the most diseased segment.
CER 001
CER-001 is an engineered negatively charged lipoprotein particle that contains recombinant human apoA-I derived from cultured cells and a combination of two phospholipids, sphingomyelin, and dipalmitoylphosphatidylglycerol. CER-001 has shown the ability to target plaque regions experimentally and clinically and elevate plasma-free (unesterified) cholesterol (FC) [69]. The “Can HDL Infusions Significantly Quicken Atherosclerosis Regression” (CHI-SQUARE) study [70] investigated the effects of six weekly CER-001 infusions at doses of 3, 6, or 12 mg/kg or placebo in 507 patients with ACS. IVUS and quantitative coronary angiography (QCA) evaluated coronary atherosclerosis at baseline and 3 weeks after the final infusion. CER-001 infusion induced a dose-related increase in blood FC. Yet, compared to placebo, no dose of CER-001 tested produced a statistically significant reduction in either the nominal or the percent total atheroma volume.
Subsequently, a post hoc analysis focusing on anatomically matched arterial segments demonstrated that infusions of CER-001 at a dose of 3 mg/kg provided the greatest atheroma regression in patients with a baseline percent atheroma volume of more than 30% but not with other concentrations of CER-001 [71]. The authors speculated that CER-001 had a U-shaped dose–response curve based on the imaging efficacy results, suggesting higher doses of CER-001 less effectively slowed the progression of atherosclerotic plaque. In this regard, CER-001 dose-dependently increases FC and plasma apo-A1 concentrations [70, 72], with a linear rather than a U-shaped relationship. These IVUS-based results led to the selection of 2–5 mg/kg of CER-001 as the optimal dose [71]. Building on this finding, the “Effect of Serial Infusions of CER-001, a Pre-β High-Density Lipoprotein Mimetic, on Coronary Atherosclerosis in Patients Following Acute Coronary Syndromes in the CER-001 Atherosclerosis Regression Acute Coronary Syndrome” (CARAT) study evaluated the effect of a 3 mg/kg CER-001 dose in 301 post ACS patients with a baseline percent atheroma volume > 30% in the proximal 10 mm in a target vessel [73, 74.••]. Subjects received randomly allocated weekly infusions of either CER-001 or placebo for 10 weeks. Compared to placebo, ten weekly infusions of 3 mg/kg CER-001 did not significantly affect coronary disease progression (percent atheroma volume and total atheroma volume) as assessed by IVUS. The negative results of infusing CER-001 in the setting of contemporary statin therapy in patients with recent ACS did not encourage the further development of this therapy.
CSL111 and CSL112
CSL111 combines apoA-I isolated from human plasma with phosphatidylcholine derived from soybean. The randomized, double-blind, placebo-controlled “Effect of HDL on Atherosclerosis-Safety and Efficacy” (ERASE) study used imaging to test CSL111’s actions [60]. One hundred and eighty-three patients received 4 weekly infusions of either placebo or 40 mg/kg of CSL111, starting within 2 weeks of ACS. The investigators performed IVUS and a quantitative coronary angiography (QCA) before randomization and 2 weeks after the last infusion. Atheroma volume fell significantly compared to baseline, similar to the ApoA-I Milano Trial. The therapy did not however promote a significant overall reduction in plaque volume when compared with a placebo.
Nevertheless, CSL111 had favorable effects on plaque characterization index on IVUS and coronary score on QCA [60]. Plaque characterization index is calculated using a detailed analysis of plaque composition for each IVUS cross-section chosen for the analysis at both baselines and after treatment. Every chosen cross-section was divided into 5 regions according to the type of plaque consisting calcific, fibrotic, fibrohypoechoic, hypoechoic, and normal. Plaque characterization scores including arc, area, inner perimeter, and outer perimeter were calculated by means of weighting factors. The summation of the characterization scores from each chosen cross-section was divided by the number of cross-sections analyzed [75, 76]. From a safety perspective, the highest dose of CSL111 was associated with transaminase elevations, which led to the early discontinuation of the 80 mg/kg study group. The hepatic side effect of CSL111 may have arisen from the high phosphatidylcholine content of the particle [77].
CSL112, a second-generation formulation, aims to optimize cholesterol efflux via ATP-binding cassette transporter A1 (ABCA1), a cholesterol transporter induced by excess cellular cholesterol [78] and present in atherosclerotic plaque [79]. CSL112 consists of purified human apoA-I combined with a lower amount of phosphatidylcholine than CSL111. CSL112 first underwent evaluation in a single ascending dose (SAD) study [80] in 57 healthy individuals and was well tolerated over the dose range of 5 to 135 mg/kg. Subsequently, a multiple ascending dose (MAD) study examined 36 healthy subjects within four treatment groups [81]. Groups 1 and 2 received 4 weekly infusions of either low (3.4 g) or high-dose (6.8 g) CSL112, group 3 received the low dose of CSL112 twice weekly for 4 weeks, and group 4 received a placebo. The infusion of 6.8 g rapidly produced a 17-fold increase in pre-β HDL levels [82.••••]. This immediate rise correlated with incremental ABCA1-mediated cholesterol efflux measured in vitro. The multiple infusions of CSL112 were well tolerated and did not cause clinically relevant elevations of liver function indices or evidence of anti-drug antibodies.
The AEGIS-I trial [83.••••] (ApoA-I Event Reducing in Ischemic Syndromes I), a multicenter, randomized, placebo-controlled, dose-ranging phase 2b clinical trial, examined the safety and tolerability of CSL112 compared with placebo. The study included 1258 patients following an acute MI. Four weekly administrations of CSL112 at both low (2 g) and high dose (6 g) were well tolerated and did not lead to significant alterations in the liver or kidney function or other safety concerns. Furthermore, this study confirmed the ability of CSL112 treatment to enhance cholesterol efflux capacity in vitro (3.67-fold for the 2 g dose, 4.30-fold for the 6 g dose).
The ApoA-I Event Reducing in Ischemic Syndromes-II (AEGIS-II) trial (NCT03473223) is investigating the potential of CSL112 to reduce major adverse cardiovascular events among patients with recent AMI. This phase III, a multicenter, double-blind, randomized, placebo-controlled, parallel-group trial is comparing 4 weekly infusions of 6 g CSL112 versus placebo in approximately 17,400 subjects [84].
Discussion
Although population-based epidemiological studies have consistently demonstrated an inverse relationship between HDL-C concentration and cardiovascular events, clinical trials have reported no or modest improvements in clinical outcomes from therapies that raise endogenous HDL-C. Following the disappointing results associated with HDL-C raising therapies [15, 17, 19, 21, 22, 23, 24, 25, 26, 27, 28, 29], which did not or only modestly improved CEC, the field moved on to assess apoA-I infusion strategies guided by the observation that these intravenous agents may in contrast improve CEC, a strong predictor for cardiovascular events, independent of blood HDL-C concentrations [39, 40].
IVUS-Based Surrogate Endpoints in Clinical Trials of ApoA-I Infusion Therapies
Although apoA-I infusion studies have suggested reductions in plaque burden assessed by IVUS when compared to baseline, these studies did not demonstrate a significant difference compared to placebo [60, 67, 69, 70, 74.••]. However, these negative imaging studies do not necessarily indicate that apoA-I-based therapies cannot contribute to cardiovascular event reduction. The evaluation of short-term atherosclerotic volume regression by IVUS may not exclude a reduction in cardiovascular events following apoA-I infusion, since these studies only measured plaque volumes within 6–9 weeks after the index event and plaque volume alone may not reflect numerous functional characteristics of plaques implicated in their propensity to provoke clinical events [85]. Statin trials assessed IVUS outcomes later than the HDL infusion studies, at a minimum of 6 months and generally up to 24 months [86, 87]. Indeed, when evaluated between 8 and 12 weeks after initiation of therapy, the statin IVUS trials did not reveal any significant changes in total atheroma volume or percentage of atheroma volume [88.••].
Elevation in CEC associates with an increase in fibrous cap thickness visualized using optical coherence tomography (OCT) [88.••], a crucial morphological change compatible with rendering plaques less susceptible to rupture. Although the concept of “plaque stabilization” generally considers cap structure and lipid pool size [89, 90], it should also encompass many other plaque properties such as the local inflammatory status, the functions of vascular smooth muscle cells, which synthesize structurally important interstitial collagens that confer fibrous cap strength, and activity of matrix-degrading proteinases, each of which may influence the biomechanical properties of the cap. [85, 91, 92] Intravascular ultrasound does not interrogate these important aspects of atheroma. Experimentally, apoA-I rapidly promotes reverse cholesterol transport in vivo and reduces macrophage number and cholesterol content, as well as increases collagen without any significant change in plaque volume [93]. These features may render plaques less likely to rupture.
The strategy of apoA-I-based infusion therapies has derived only modest support from surrogate endpoint data and has suffered from uncertainty with respect to incompletely validated biomarkers that may or may not associate with improved clinical outcomes. The modest sample size and surrogate endpoints of these studies may lead to false-negative or false-positive conclusions. An adequately powered phase III clinical outcomes trial furnishes the only strategy to assess definitively the efficacy of HDL infusion therapies in patients post myocardial infarction by monitoring outcomes including CV death, MI, and stroke. Statins generally do not manifest clinical benefit in the first year following initiation, and apoA-I infusions might offer a way to provide more immediate prevention of events during the high-risk period post-MI and awaiting the long-term actions of statins.
Comparison of Structural and Functional Properties of ApoA-1 Infusion Strategies
The major apoA-I-based infusion agents (MDCO 216, CER-001, CSL111, CSL112) differ substantially in composition, dosing, timing, frequency, pharmacokinetics, and pharmacodynamics, and these differences may account for differences in their effects (Table 1).
The components of these agents include variable proteins and lipid moieties. MDCO-216 contains recombinant apoA-I Milano, CER-001 contains recombinant apoA-I, CSL111, and CSL112 contains human plasma-derived apoA-I. Human apoA-I Milano often associates with low apoA-I levels [94]. Individuals heterozygous for the apoA-I Milano mutation have very low plasma apoA-I and HDL cholesterol levels as well as moderately elevated triglycerides [63]. Low levels of apoA-I Milano in plasma result at least in part from reduced LCAT activation [95], which reduces the transfer of cholesterol to the core of HDL particles reducing their size and increasing the clearance rate [96, 97]. Consistent with prior observations, the MILANO-PILOT trial [68.••] demonstrated that 5 weekly infusions of MDCO-216 caused a significant reduction in native apoA-I compared to placebo (p = 0.003). The mechanism of this reduction could result from either hypercatabolism similar to the pathway in heterozygous apoA-I Milano mutation or from a decrease in endogenous production of apoA-I.
The loss of native apoA-I due to apoA-I Milano administration may have adverse consequences. Furthermore, a single ascending dose study [98] reported that infusion of MDCO-216 caused a time and dose-dependent decrease in cholesterol esterification rate (CER) likely due to the reduced activity of LCAT. The proposed mechanism for this effect implicates the mutation in apoA-I Milano that affects the domain on apoA-I, which normally binds to and allosterically activates LCAT [99]. Patients receiving MDCO-216 exhibited a rapid increase in plasma FC, while cholesterol levels remained stable in the placebo group. The increase in FC may have resulted from incremental MDCO216-induced scavenger receptor class B, type I (SR-BI)-mediated efflux of cholesterol from tissues into plasma. This effect could also result from hampered esterification of FC by LCAT, since FC significantly correlated inversely with a decrease in cholesterol esterification rate. CER-001 may also inhibit LCAT activity since infusion leads to elevation of plasma FC with no elevation or even a reduction in cholesteryl ester [100]. CER-001 contains sphingomyelin, a lipid that can inhibit LCAT [101]. In contrast to the other HDL infusion therapies, however, infusion of CSL112 increases cholesterol esterification and maintains stable triglyceride levels [82.••••].
Comparison of Timing
The timing of administering these apoA-I therapies may prove critical because most recurrent events post ACS occur in the early days and weeks following initial presentation. Despite optimal medical therapy, acute MI survivors have a high risk of experiencing another CV event, especially in the first month. Recurrent CV events occur in 12% of these patients within 1 year, and half of those events occur in the first 30 days [102]. Therefore, an optimal intervention would achieve plaque stabilization very early after the initial presentation during a period before statins have achieved their expected clinical benefit. Participants in the CHI-SQUARE, MILANO, and CARAT studies received the first HDL therapy infusion within 14 days post ACS, and the infusions were repeated weekly on 6, 5, and 10 occasions, respectively. To address the high incidence of early recurrent CV events following ACS, AEGIS-II targets an earlier initiation and completion of treatment compared to previous studies. The first infusion of CSL112 will occur within 5 days after the index MI, and the total duration of once-weekly infusions is 4 weeks with an assessment of the primary endpoint at 90 days and follow-up through 1 year to assess the durability of the effect.
Comparison of Dosing
The dosing of these 3 agents differs over a 30-fold range, indicating the uncertainty in the optimal target dose and the potential for underdosing. The pivotal trials used CER-001 (3 mg/kg) and MDCO-216 (20 mg/kg) based on pilot imaging efficacy outcomes [71, 103]. CSL112 is dosed at a higher total dose of 6 g, corresponding to approximately 80 mg/kg based on its pharmacodynamic response. The higher dose of CSL112 yields a marked increase in plasma apoA-I concentrations and ABCA1-mediated CEC [80, 81, 104]. As a result, CEC differs substantially from these other three therapies. ABCA1-mediated CEC, which might be a relevant CEC measure for apoA-I therapies, increased by 242% in the AEGIS-I trial [83.••••] of CSL112, compared with the 80 to 90% increase with MDCO-216 in the MILANO-PILOT trial [68.••] and the minimal rise of 14% for CER-001 in an earlier study [69]. CARAT did not report CEC levels [74.••].
Conclusion
Although previous imaging studies of apoA-I infusion therapies did not demonstrate significant regression in coronary atherosclerotic plaque, post MI infusion of CSL112 differs in its properties from the other infusion strategies tested. CSL112 contains a human plasma-derived apoA-I, unlike the recombinant MDCO-216 and CER-001 proteins. Furthermore, CSL112 is not a mutant apoA-I like MDCO-216. CSL112 augments LCAT function in contrast to MDCO-216 and CER-001, which both lower LCAT function. CSL112 does not reduce endogenous apoA-I concentrations as does the MDCO-216 molecule.
Impaired CEC associates with worse clinical outcomes, and CSL112 substantially raises CEC. The effect of CSL112 to elevate concentrations of apoA-I and pre-β HDL results in a marked increase in CEC, as reported in phase I and validated in phase II clinical trials in healthy volunteers [81], patients with stable atherosclerotic coronary vascular disease [104], and after acute myocardial infarction [83.••••]. The ability of CSL112 to elevate strongly CEC suggests that it might rapidly remove plaque cholesterol and reduce the risk of early recurrent cardiovascular events. CSL112 has so far raised no safety concerns. The large phase III AEGIS-II trial will test the potential of early administration of CSL112 to reduce major adverse cardiovascular events in the 90-day high-risk period post-myocardial infarction. This study should provide insight into whether enhancing cholesterol efflux can improve cardiovascular outcomes.
Abbreviations
- ABCA1:
-
ATP-binding cassette transporter protein A1
- ACS:
-
Acute coronary syndrome
- ApoA-I:
-
Apolipoprotein A-I
- CE:
-
Cholesterol efflux
- CEC:
-
Cholesterol efflux capacity
- CETP:
-
Cholesteryl ester transfer protein
- CV:
-
Cardiovascular
- CVD:
-
Cardiovascular disease
- FC:
-
Free cholesterol
- HDL-C:
-
High-density lipoprotein cholesterol
- IDL:
-
Intermediate-density lipoprotein
- IVUS:
-
Intravascular ultrasonography
- LCAT:
-
Lecithin cholesterol acyltransferase
- LDL-C:
-
Low-density lipoprotein cholesterol
- MI:
-
Myocardial infarction
- MACE:
-
Major adverse cardiac events
- MAD:
-
Multiple ascending dose
- Pre-β HDL:
-
Lipid poor precursor of mature HDL
- QCA:
-
Quantitative coronary analysis
- RCT:
-
Reverse cholesterol transport
- SAD:
-
Single ascending dose
- SR-BI:
-
Scavenger receptor class B type I
- VLDL:
-
Very low-density lipoprotein
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Libby P. The forgotten majority: unfinished business in cardiovascular risk reduction. J Am Coll Cardiol. 2005;46:1225–8.
Gofman JW, Young W, Tandy R. Ischemic heart disease, atherosclerosis, and longevity. Circulation. 1966;34:679–97.
Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study Am J Med. 1977;62:707–14.
Gordon DJ, Probstfield JL, Garrison RJ, et al. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies Circulation. 1989;79:8–15.
Barter P, Gotto AM, LaRosa JC, et al. Treating to New Targets. Investigators HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med. 2007;357:1301–10.
Stone NJ, Robinson JG, Lichtenstein AH, A College of Cardiology, American Heart Association Task Force on Practice Guidelines, et al. ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;2014(129):1–45.
Rader DJ, Tall AR. The not-so-simple HDL story: is it time to revise the HDL cholesterol hypothesis? Nat Med. 2012;18:1344–6.
Kontush A, Therond P, Zerrad A, et al. Preferential sphingosine-1-phosphate enrichment and sphingomyelin depletion are key features of small dense HDL3 particles: relevance to antiapoptotic and antioxidative activities. Arterioscler Thromb Vasc Biol. 2007;27:1843–9.
Boekholdt SM, Hovingh GK, Mora S, et al. Very low levels of atherogenic lipoproteins and the risk for cardiovascular events: a meta-analysis of statin trials. J Am Coll Cardiol. 2014;64:485–94.
Reiner Z. Managing the residual cardiovascular disease risk associated with HDL-cholesterol and triglycerides in statin-treated patients: a clinical update. Nutr Metab Cardiovasc Dis. 2013;23:799–807.
Miller GJ, Miller NE. Plasma-high-density-lipoprotein concentration and development of ischaemic heart-disease. Lancet. 1975;1:16–9.
Glomset JA. The plasma lecithins: cholesterol acyltransferase reaction. J Lipid Res. 1968;9:155–67.
Besler C, Heinrich K, Rohrer L, Doerries C, et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J Clin Invest. 2011;121:2693–708.
Riwanto M, Rohrer L, Roschitzki B, et al. Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of high-density lipoprotein-proteome remodeling. Circulation. 2013;127:891–904.
Rubins HB, Robins SJ, Collins D, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med. 1999;341:410–8.
Frick MH, Elo O, Haapa K, et al. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med. 1987;317:1237–45.
Corsini A, Bellosta S, Davidson MH. Pharmacokinetic interactions between statins and fibrates. Am J Cardiol. 2005;96(44–49):34–5.
Keech A, Simes RJ, Barter P, et al. FIELD study investigators. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;36:1849–61.
Ginsberg HN, Elam MB, Lovato LC, ACCORD Study Group, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–74.
Pradhan AD, Paynter NP, Everett BM, et al. Rationale and design of the Pemafibrate to Reduce Cardiovascular Outcomes by Reducing Triglycerides in Patients with Diabetes (PROMINENT) study. Am Heart J. 2018;206:80–93.
Canner PL, Berge KG, Wenger NK, et al. Fifteen-year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol. 1986;8:1245–55.
Boden WE, Probstfield JL, Anderson T, AIM-HIGH Investigators, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67.
Landray MJ, Haynes R, Hopewell JC, HPS2-THRIVE Collaborative Group, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371:203–12.
Goldberg RB, Jacobson TA. Effects of niacin on glucose control in patients with dyslipidemia. Mayo Clin Proc. 2008;83:470–8.
Sazonov V, Maccubbin D, Sisk CM, Canner PL. Effects of niacin on the incidence of new onset diabetes and cardiovascular events in patients with normoglycaemia and impaired fasting glucose. Int J Clin Pract. 2013;67:297–302.
HPS2-THRIVE Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J. 2013;34: 1279–1291.
Shah PK. Inhibition of CETP as a novel therapeutic strategy for reducing the risk of atherosclerotic disease. Eur Heart J. 2007;28:5–12.
Xiang AS, Kingwell BA. Rethinking good cholesterol: a clinicians’ guide to understanding HDL. Lancet Diabetes Endocrinol. 2019;7(7):575–82. https://doi.org/10.1016/S2213-8587(19)30003-8.
Barter PJ, Caulfield M, Eriksson M, et al. ILLUMINATE Investigators. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–22.
Schwartz GG, Olsson AG, Abt, M, dal-OUTCOMES Investigators, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–99.
Lincoff AM, Nicholls SJ, Riesmeyer JS, et al. ACCELERATE Investigators. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N Engl J Med. 2017;376:1933–42.
Vergeer M, Bots ML, van Leuven SI, et al. Cholesteryl ester transfer protein inhibitor torcetrapib and off-target toxicity: a pooled analysis of the rating atherosclerotic disease change by imaging with a new CETP inhibitor (RADIANCE) trials. Circulation. 2008;118:2515–22.
Bowman L, Hopewell JC, Chen F, HPS3/TIMI55–REVEAL Collaborative Group, et al. Effects of anacetrapib in patients with atherosclerotic vascular disease. N Engl J Med. 2017;377:1217–27.
Pownall HJ, Rosales C, Gillard BK, Gotto AM Jr. High-density lipoproteins, reverse cholesterol transport and atherogenesis [published online ahead of print, 2021 Apr 8]. Nat Rev Cardiol. 2021;https://doi.org/10.1038/s41569-021-00538-z.
Vaisar T, Pennathur S, Green PS, et al. Shotgun proteomics implicates protease inhibition and complement activation in the anti-inflammatory properties of HDL. J Clin Invest. 2007;117:746–56.
Heinecke JW. The HDL proteome: a marker–and perhaps mediator–of coronary artery disease. J Lipid Res. 2009;50:167–71.
Davidson WS, Silva RA, Chantepie S, Lagor WR, Chapman MJ, Kontush A. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler Thromb Vasc Biol. 2009;29(6):870–6. https://doi.org/10.1161/ATVBAHA.109.186031.
Meikle PJ, Formosa MF, Mellett NA, Jayawardana KS, Giles C, Bertovic DA, Jennings GL, Childs W, Reddy M, Carey AL, Baradi A, Nanayakkara S, Wilson AM, Duffy SJ, Kingwell BA. HDL phospholipids, but not cholesterol distinguish acute coronary syndrome from stable coronary artery disease. J Am Heart Assoc. 2019;8(11): e011792. https://doi.org/10.1161/JAHA.118.011792.
Pirillo A, Norata GD, Catapano AL. Treating high density lipoprotein cholesterol (HDL C): quantity versus quality. Curr Pharm Des. 2013;19:3841–57.
Santos-Gallego CG. HDL: quality or quantity? Atherosclerosis. 2015;243:121–3.
Nazir S, Jankowski V, Bender G, et al. Interaction between high-density lipoproteins and inflammation: function matters more than concentration! Adv Drug Deliv Rev. 2020;S0169-409X:30143–5.
Anastasius M, Kockx M, Jessup W, et al. Cholesterol efflux capacity: an introduction for clinicians. Am Heart J. 2016;180:54–63.
Khera AV, Cuchel M, de la Llera-Moya M, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–35.
Rohatgi A, Khera A, Berry JD, et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371:2383–93.
Saleheen D, Scott R, Javad S, et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: a prospective case-control study. Lancet Diabetes Endocrinol. 2015;3:507–13.
de la Llera-Moya M, Drazul-Schrader D, Asztalos BF, Cuchel M, Rader DJ, Rothblat GH. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler Thromb Vasc Biol. 2010;30:796–801.
Rader DJ. New therapeutic approaches to the treatment of dyslipidemia. Cell Metab. 2016;23:405–12.
Moore RE, Kawashiri MA, Kitajima K, et al. Apolipoprotein A-I deficiency results in markedly increased atherosclerosis in mice lacking the LDL receptor. Arterioscler Thromb Vasc Biol. 2003;23:1914–20.
Voyiaziakis E, Goldberg IJ, Plump AS, et al. ApoA-I deficiency causes both hypertriglyceridemia and increased atherosclerosis in human apoB transgenic mice. J Lipid Res. 1998;39:313–21.
Hovingh GK, Brownlie A, Bisoendial RJ, et al. A novel apoA-I mutation (L178P) leads to endothelial dysfunction, increased arterial wall thickness, and premature coronary artery disease. J Am Coll Cardiol. 2004;44:1429–35.
Stoekenbroek RM, Stroes ES, Hovingh GK. ApoA-I mimetics. Handb Exp Pharmacol. 2015;224:631–48.
Nicholls SJ, Cutri B, Worthley SG, et al. Impact of short-term administration of high-density lipoproteins and atorvastatin on atherosclerosis in rabbits. Arterioscler Thromb Vasc Biol. 2005;25:2416–21.
Badimon JJ, Badimon L, Fuster V. Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. J Clin Invest. 1990;85:1234–41.
Badimon JJ, Badimon L, Galvez A, Dische R, Fuster V. High density lipoprotein plasma fractions inhibit aortic fatty streaks in cholesterol-fed rabbits. Lab Invest. 1989;60:455–61.
Ouimet M, Barrett TJ, Fisher EA. HDL and reverse cholesterol transport. Circ Res. 2019;124:1505–18.
Bisoendial RJ, Hovingh GK, Levels JH, et al. Restoration of endothelial function by increasing high-density lipoprotein in subjects with isolated low high-density lipoprotein. Circulation. 2003;107:2944–8.
Spieker LE, Sudano I, Hürlimann D, et al. High-density lipoprotein restores endothelial function in hypercholesterolemic men. Circulation. 2002;105:1399–402.
Nanjee MN, Cooke CJ, Garvin R, et al. Intravenous apoA-I/lecithin discs increase pre-beta-HDL concentration in tissue fluid and stimulate reverse cholesterol transport in humans. J Lipid Res. 2001;42:1586–93.
Weibel GL, Alexander ET, Joshi MR, et al. Wild-type ApoA-I and the Milano variant have similar abilities to stimulate cellular lipid mobilization and efflux. Arterioscler Thromb Vasc Biol. 2007;27:2022–9.
Tardif JC, Grégoire J, L’Allier PL, et al. Effect of rHDL on Atherosclerosis-Safety and Efficacy (ERASE) Investigators. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA. 2007;297:1675–82.
Mora S, Glynn RJ, Ridker PM. High-density lipoprotein cholesterol, size, particle number, and residual vascular risk after potent statin therapy. Circulation. 2013;128:1189–97.
Kingwell BA, Chapman MJ. Future of high-density lipoprotein infusion therapies: potential for clinical management of vascular disease. Circulation. 2013;128:1112–21.
Sirtori CR, Calabresi L, Franceschini G, et al. Cardiovascular status of carriers of the apolipoprotein A-I(Milano) mutant: the Limone sul Garda study. Circulation. 2001;103:1949–54.
Franceschini G, Sirtori CR, Bosisio E, et al. Relationship of the phenotypic expression of the A-I Milano apoprotein with plasma lipid and lipoprotein patterns. Atherosclerosis. 1985;58:159–74.
Shah PK, Yano J, Reyes O, et al. High-dose recombinant apolipoprotein A-I(Milano) mobilizes tissue cholesterol and rapidly reduces plaque lipid and macrophage content in apolipoprotein e-deficient mice. Potential implications for acute plaque stabilization. Circulation. 2001;103:3047–50.
Ameli S, Hultgardh-Nilsson A, Cercek B, et al. Recombinant apolipoprotein A-I Milano reduces intimal thickening after balloon injury in hypercholesterolemic rabbits. Circulation. 1994;90:1935–41.
Nissen SE, Tsunoda T, Tuzcu EM, et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290:2292–300.
Nicholls SJ, Puri R, Ballantyne CM, et al. Effect of infusion of high-density lipoprotein mimetic containing recombinant apolipoprotein A-I Milano on coronary disease in patients with an acute coronary syndrome in the MILANO-PILOT trial: a randomized clinical trial. JAMA Cardiol. 2018;3:806–14. The authors reported that infusion of MDCO-216 did not provide a significant plaque volume regression in statin-treated patients with acute coronary syndrome.
Zheng KH, van der Valk FM, Smits LP, et al. HDL mimetic CER-001 targets atherosclerotic plaques in patients. Atherosclerosis. 2016;251:381–8.
Tardif JC, Ballantyne CM, Barter P, et al. Can HDL Infusions Significantly Quicken Atherosclerosis REgression (CHI-SQUARE) Investigators. Effects of the high-density lipoprotein mimetic agent CER-001 on coronary atherosclerosis in patients with acute coronary syndromes: a randomized trial. Eur Heart J. 2014;35:3277–86.
Kataoka Y, Andrews J, Duong M, et al. Regression of coronary atherosclerosis with infusions of the high-density lipoprotein mimetic CER-001 in patients with more extensive plaque burden. Cardiovasc Diagn Ther. 2017;7:252–63.
Keyserling CH, Hunt TL, Klepp HM, et al. CER-001, a synthetic HDL-mimetic, safely mobilizes cholesterol in healthy dyslipidemic volunteers. Circulation. 2011;124:A15525 ((abstract)).
Andrews J, Janssan A, Nguyen T, et al. Effect of serial infusions of reconstituted high-density lipoprotein (CER-001) on coronary atherosclerosis: rationale and design of the CARAT study. Cardiovasc Diagn Ther. 2017;7:45–51.
Nicholls SJ, Andrews J, Kastelein JJP, et al. Effect of serial infusions of CER-001, a pre-β High-Density Lipoprotein Mimetic, on coronary atherosclerosis in patients following acute coronary syndromes in the CER-001 Atherosclerosis Regression Acute Coronary Syndrome Trial: a randomized clinical trial. JAMA Cardiol. 2018;3:815–22. Findings of this study demonstrate that infusion of CER-001 does not promote plaque regression in statin-treated patients following acute coronary syndrome.
Tardif JC, Grégoire J, Lespérance J, et al. Design features of the Avasimibe and Progression of coronary Lesions assessed by intravascular UltraSound (A-PLUS) clinical trial. Am Heart J. 2002;144:589–96.
Lee HS, Tardif JC, Harel F, et al. Effects of plaque composition on vascular remodelling after angioplasty in the MultiVitamins and Probucol (MVP) trial. Can J Cardiol. 2002;18:271–5.
Herzog E, Pragst I, Waelchli M, et al. Reconstituted high-density lipoprotein can elevate plasma alanine aminotransferase by transient depletion of hepatic cholesterol: role of the phospholipid component. J Appl Toxicol. 2015;36:1038–47.
Rosenson RS, Brewer HB Jr, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–19.
Puig O, Yuan J, Stepaniants S, et al. A gene expression signature that classifies human atherosclerotic plaque by relative inflammation status. Circ Cardiovasc Genet. 2011;4:595–604.
Gille A, Easton R, D’Andrea D, Wright SD, Shear CL. CSL112 enhances biomarkers of reverse cholesterol transport after single and multiple infusions in healthy subjects. Arterioscler Thromb Vasc Biol. 2014;34:2106–14.
Easton R, Gille A, D’Andrea D, et al. A multiple ascending dose study of CSL112, an infused formulation of ApoA-I. J Clin Pharmacol. 2014;54:301–10.
Gille A, D’Andrea D, Tortorici MA, Hartel G, Wright SD. CSL112 (apolipoprotein A-I [human]) enhances cholesterol efflux similarly in healthy individuals and stable atherosclerotic disease patients. Arterioscler Thromb Vasc Biol. 2018;38:953–63. This study provides compelling evidence of the ability of CSL112 to strongly elevate CEC despite underlying cardiovascular disease. The findings of this study suggest that CSL112 is a promising novel therapy for lowering the burden of atherosclerosis and reducing the risk of recurrent cardiovascular events.
Michael Gibson C, Korjian S, Tricoci P, et al. Safety and tolerability of CSL112, a reconstituted, infusible, plasma-derived apolipoprotein A-I, after acute myocardial infarction: the AEGIS-I Trial (ApoA-I Event Reducing in Ischemic Syndromes I). Circulation. 2016;134:1918–30. This phase-II study tested CSL112 for safety and tolerability. CSL112 was found to be well-tolerated, capable of increasing cholesterol efflux and did not significantly alter liver or kidney functions.
Gibson CM, Kastelein JJP, Phillips AT, et al. Rationale and design of ApoA-I Event Reducing in Ischemic Syndromes II (AEGIS-II): a phase 3, multicenter, double-blind, randomized, placebo-controlled, parallel-group study to investigate the efficacy and safety of CSL112 in subjects after acute myocardial infarction. Am Heart J. 2021;231:121–7.
Libby P. Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med. 2013;368:2004–13.
Lima JA, Desai MY, Steen H, et al. Statin-induced cholesterol lowering and plaque regression after 6 months of magnetic resonance imaging-monitored therapy. Circulation. 2004;110:2336–41.
Nissen SE, Nicholls SJ, Sipahi I, et al. ASTEROID Investigators. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295:1556–65.
Kini AS, Vengrenyuk Y, Shameer K, et al. Intracoronary imaging, cholesterol efflux, and transcriptomes after intensive statin treatment: the YELLOW II study. J Am Coll Cardiol. 2017;69:628–40. This study demonstrates a strong and independent association between improved CEC and fibrous cap thickening suggesting that cholesterol efflux may contribute to plaque stabilization.
Brown BG, Zhao XQ, Sacco DE, Albers JJ. Lipid lowering and plaque regression. New insights into prevention of plaque disruption and clinical events in coronary disease. Circulation. 1993;87:1781–91.
Libby P, Schoenbeck U, Mach F, Selwyn AP, Ganz P. Current concepts in cardiovascular pathology: the role of LDL cholesterol in plaque rupture and stabilization. Am J Med. 1998;104:14–8.
Newby AC, George SJ, Ismail Y, et al. Vulnerable atherosclerotic plaque metalloproteinases and foam cell phenotypes. Thromb Haemost. 2009;101:1006–11.
Dollery CM, Libby P. Atherosclerosis and proteinase activation. Cardiovasc Res. 2006;69:625–35.
Hewing B, Parathath S, Barrett T, et al. Effects of native and myeloperoxidase-modified apolipoprotein a-I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2014;34:779–89.
Sorci-Thomas MG, Thomas MJ. The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc Med. 2002;12:121–8.
Calabresi L, Franceschini G, Burkybile A, Jonas A. Activation of lecithin cholesterol acyltransferase by a disulfide-linked apolipoprotein A-I dimer. Biochem Biophys Res Commun. 1997;232:345–9.
Roma P, Gregg RE, Meng MS, Ronan R, Zech LA, et al. In vivo metabolism of a mutant form of apolipoprotein A-I, apo A-IMilano, associated with familial hypoalphalipoproteinemia. J Clin Invest. 1993;91:1445–52.
Perez-Mendez O, Bruckert E, Franceschini G, et al. Metabolism of apolipoproteins AI and AII in subjects carrying similar apoAI mutations, apoAI Milano and apoAI Paris. Atherosclerosis. 2000;148:317–25.
Kempen HJ, Gomaraschi M, Simonelli S, et al. Persistent changes in lipoprotein lipids after a single infusion of ascending doses of MDCO-216 (apoA-IMilano/POPC) in healthy volunteers and stable coronary artery disease patients. Atherosclerosis. 2016;255:17–24.
Alexander ET, Tanaka M, Kono M, et al. Structural and functional consequences of the Milano mutation (R173C) in human apolipoprotein A-I. J Lipid Res. 2009;50:1409–19.
Keyserling CH, Barbaras R, Benghozi R, Dasseux JL. Development of CER-001: preclinical dose selection through to phase i clinical findings. Clin Drug Investig. 2017;37:483–91.
Subbaiah PV, Liu M. Role of sphingomyelin in the regulation of cholesterol esterification in the plasma lipoproteins. Inhibition of lecithin-cholesterol acyltransferase reaction. J Biol Chem. 1993;268:20156–63.
Wallentin L, Becker RC, Budaj A, Freij A, Thorsén M, PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045–57.
Parolini C, Marchesi M, Lorenzon P, et al. Dose-related effects of repeated ETC-216 (recombinant apolipoprotein A-I Milano/1-palmitoyl-2-oleoyl phosphatidylcholine complexes) administrations on rabbit lipid-rich soft plaques: in vivo assessment by intravascular ultrasound and magnetic resonance imaging. J Am Coll Cardiol. 2008;51:1098–103.
Tricoci P, D’Andrea DM, Gurbel PA, et al. Infusion of reconstituted high-density lipoprotein, CSL112, in patients with atherosclerosis: safety and pharmacokinetic results from a phase 2a randomized clinical trial. J Am Heart Assoc. 2015;4: e002171.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Competing Interests
Dr. Gibson receives consultant fees from Portola Pharmaceuticals and reports grants from Angel Medical Corporation and CSL Behring; grants and other support from Bayer Corporation; grants and personal fees from Janssen, Johnson & Johnson, and Portola Pharmaceuticals; and personal fees from The Medicines Company, Boston Clinical Research Institute, Cardiovascular Research Foundation, Eli Lilly, Gilead Sciences Inc., Novo Nordisk, Pfizer, Web MD, UpToDate in Cardiovascular Medicine, Amarin Pharma, Amgen, Arena Pharmaceuticals, Bayer Corporation, Boehringer Ingelheim, Chiesi, Merck & Co., PharmaMar, Sanofi, Somahlution, St. Francis Hospital, and Verreseon Corporation. Dr. Libby is an unpaid consultant to, or involved in clinical trials for Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Norvo Nordisk, Novartis, Pfizer, and Sanofi-Regeneron. Dr. Libby is a member of the scientific advisory board for Amgen, Caristo, Cartesian, CSL Behring, DalCor Pharmaceuticals, Dewpoint, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, PlaqueTec, and XBiotech, Inc. Dr. Libby’s laboratory has received research funding in the last 2 years from Novartis. Dr. Libby is on the Board of Directors of XBiotech, Inc. Dr. Peter Libby has a financial interest in TenSixteen Bio, a company targeting somatic mosaicism and clonal hematopoiesis of indeterminate potential (CHIP) to discover and develop novel therapeutics to treat age-related diseases. Dr. Libby has a financial interest in Xbiotech, a company developing therapeutic human antibodies. Dr. Libby’s interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict-of-interest policies. Dr. Libby receives funding support from the National Heart, Lung, and Blood Institute (1R01HL134892), the American Heart Association (18CSA34080399), the RRM Charitable Fund, and the Simard Fund. Dr. Libby receives funding support from the National Heart, Lung, and Blood Institute (1R01HL134892), the American Heart Association (18CSA34080399), the RRM Charitable Fund, and the Simard Fund. Dr. Wright, Dr. Kingwell, Dr. Tricoci, Dr. Shaunik, Dr. Berman, and Dr. Duffy are employed by CSL Behring. All remaining authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Reviews and Scientific Meetings and New Research Implications
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kalayci, A., Gibson, C.M., Ridker, P.M. et al. ApoA-I Infusion Therapies Following Acute Coronary Syndrome: Past, Present, and Future. Curr Atheroscler Rep 24, 585–597 (2022). https://doi.org/10.1007/s11883-022-01025-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11883-022-01025-7