
Abstract
The material extrusion 3d printing technique has proved to be reliable and low cost for obtaining metal parts. Therefore, in the present work, its suitability for obtaining pieces of a material composed of iron/iron oxide from an industrial waste was studied. Once the powder was obtained by chemical partial reduction, filaments were manufactured with a polymer matrix of TPE/PP/SA in a polymer–powder proportion of 13–87 wt.%. The influence of temperature and printing speed on the geometry of the printed and sintered parts, as well as on their mechanical and microstructural properties, was evaluated. The results showed that the printing speed had a significant impact on the print geometry of the parts, while the printing temperature had no effect. During thermal debinding and sintering, an additional chemical reduction due to the production of reducing agents during the decomposition of the polymer matrix was observed. This shows that the selection of the polymer matrix affects the microstructure of the sintered parts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Two of the pillars of Industry 4.0 are the circular economy and additive manufacturing. The circular economy is based on reusing, repairing, and recycling materials and products resulting from the production process, in order to reduce waste to a minimum.1 Regarding material recycling, during the processes of obtaining a product, leftover industrial material is generated which, under the concept of a linear economy, or “take–make–dispose”, was generally considered as waste. On the other hand, under the concept of a circular economy, the aim is to reintegrate this waste into the production chain as raw material through other manufacturing processes, which can help reduce the use of non-renewable resources and decrease the carbon footprint. In this regard, for example, Furlani et al.2 investigated the addition of calamine in the manufacture of mortars with different proportions, seeking to replace the fine aggregate. Esguerra-Arce et al.3 evaluated the obtaining of an aluminum compound and iron oxides from industrial waste through compaction, varying the percentage of calamine addition in the pieces. In the first of these two studies, pieces were not manufactured with 100% calamine waste; however, Tirado González et al.4 used calamine at 100% to obtain metal pieces reinforced with iron oxide through the compaction process. While pieces could be obtained in this way, geometric freedom was compromised. Therefore, it is important to seek new processes that can use waste as raw material with a high percentage of participation, in addition to allowing the obtaining of complex geometries. One example of such processes is additive manufacturing, which makes it possible to obtain different geometries that would be impossible to obtain through conventional processes.
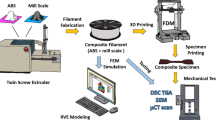
Additive manufacturing refers to the creation of pieces layer-by-layer using computer assisted design. This technique facilitates the creation of prototypes,5 highly customized products with minimal waste of material, in addition to optimizing the parameters6 of the produced parts, such as weight, geometry, and mechanical properties, among others.
The additive manufacturing techniques used to print metal parts include selective laser melting (SLM),7 selective laser sintering (SLS),8 and the binder jetting technique (BJT)9 These techniques make use of metal powders as starting material. SLM and SLS involve the use of high-energy lasers and controlled atmospheres, making them expensive. BJT, on the other hand, allows the obtaining of metal parts in green for subsequent sintering, but the machine used for the process is expensive. A more economical alternative is metal material extrusion (MEX), in which the filament is composed of metal particles and polymer and, after printing, the piece is submitted to a thermal cycle in which the polymer is removed, and the metal particles are sintered.10,11 At present, the mechanical properties of parts obtained by MEX are comparable to the properties of the parts obtained by metal injection molding; however, MEX has some advantages such as design freedom and the fact that no metal molds are required.5 As reported in several investigations,12,13,14 porosity between 7% and 8% has been found for pieces obtained by this method.
There are some reports of research projects regarding iron manufacturing by MEX. Hwang et al.15 reported the manufacture of polymer/metal composite filaments using ABS with iron particles (smaller than 43 μm in size), in which they evaluated mechanical properties such as tensile strength; however, they did not attempt to sinter the parts. Zhang et al.16 explored the production of a sponge-shaped iron composite filament with an average particle size of 25 μm. It was observed that channels formed between the irregularities of the particles emerging to the surface. The above demonstrates the need for further study on the obtaining of reinforced Fe pieces with a second phase, as well as the evaluation of printing parameters that affect the final properties of the piece.
Conventional techniques for the production of metal powders for powder metallurgy including MEX, such as gas or water atomization and electrochemical methods, are complicated and expensive.17 As an alternative, an industrial waste generated during the hot-rolling process of low carbon steels, called mill scale, has been studied to obtain iron with a oxide core by partial chemical reduction. This residue is composed of three different oxides: the outer layer consists of hematite (Fe2O3), the intermediate layer of magnetite (Fe3O4), and the inner layer of wüstite (FeO).18 After being subjected to grinding, this powder was subjected to partial reduction with H2 and can be used to manufacture parts by pressing and sintering, exhibiting mechanical properties similar to those of a low-carbon steel and magnetic properties typical of a hard ferromagnetic.4
Therefore, this work focuses on evaluating the processability of this recovered waste as iron/iron oxide composite powder by metal MEX, evaluating the influence of printing speed and temperature on the geometry of the printed samples, taking into account the volumetric dimensional tolerance. This has been carried out considering that a better processability in 3D printing is related to smaller variation in the dimensions.
Materials and Methods
Powder
The mill scale was obtained from Gerdau Diaco, a steel-making factory, and was turned into powder using a ball mill with an alumina jar of 5 L and steel grinding bodies. The mill scale to grinding bodies ratio was 400 ml:600 ml, the grinding rotation speed was 50 rpm, and the grinding time was 131 h. For the particle size distribution, morphological characterization, chemical composition, and microstructure analysis, the obtained powder was characterized by laser granulometry (Hydro 2000MU (A)) using water as dispersant, scanning electron microscopy (SEM; Phenom XL) coupled with energy dispersive spectroscopy (EDS) and X-ray diffraction (XRD; PANalytical model X’PERT PRO MPD), with a Cu anode (λ = 1.54 Ǻ).
Following the procedures described above, the powder was chemically reduced in a tubular chamber made of AISI 304 stainless steel at 700°C in a furnace (Nabertherm) using a H2 atmosphere (5.0 SFCH) as reductant agent and Ar (5.0 SFCH) as carrier gas. The reduction was carried out for 2 h with a prior homogenization of 1 h under Ar. The chamber used had dimensions of 128 cm length and 6.6 cm diameter, with an internal gas dispersion perforated tube 42 cm long and 1.3 cm in diameter. Then, the powder was sieved with mesh number 10 (150 μm). After that, the powder was characterized by laser granulometry, SEM/EDS, and XRD.
Binder and Filament
To obtain the filament to be used in MEX, the metallic powder (IRONOX) was mixed with a thermoplastic elastomer (TPE; 30A shore hardness), polypropylene (PP; 95A shore hardness), and stearic acid (SA; Sigma Aldrich™), in the proportions shown in supplementary Table S-I (refer to online supplementary material). The matrix materials were determined in a previous investigation by our research group.11 The nominal temperature of 140°C was graduated in the mixer, and when the temperature controller showed this temperature, the polymer pellets were introduced into the mixer and, after softening, the IRONOX powder was gradually added and repeatedly mixed.
Once the mixture was well homogenized and cooled to room temperature, it was cut to obtain pellets. These pellets were put into a Filastruder™ extruder, with a screw speed of 7 rpm. The appropriate temperature to obtain a non-defect filament with a diameter of 1.74 mm was 162°C. Then, the filament was characterized by measuring its tensile properties and thermogravimetry (TGA) using an atmosphere of argon at 0.05 L/min and a heating rate of 0.5°C/min (NETZSCH STA 449C) from room temperature to 1200°C. TGA measurements of the pure polymers and blend were also carried out for comparison purposes. Finally, a microscopy characterization of the filament was carried out by SEM after cryo-fracturing in liquid nitrogen.
Printing
Cylindrical samples were designed for printing using a Fusion 360 by Autodesk™, with nominal dimensions of 8.0 mm height and 16.0 mm diameter. Then, Ultimaker Cura™ software was used to fix the printing parameters, as shown in supplementary Table S-II. Layer height, build plate temperature, and flow values were selected by a previous test. A 23 experimental design was carried out and an ANOVA was carried out to analyze the influence of temperature and printing speed. Printing temperatures were fixed at 210, 220, and 230°C, and printing speeds at 7, 9, and 12 mm/s, respectively. These parameters were selected as a starting point, taking into account previously investigated values in the literature.11,19,20 The codes of the combination of parameters are T230-S7 for a printing temperature of 230°C and a printing speed of 7 mm/s, and so on. This process was carried out for a total of nine treatments with three repetitions each.
An Ender 3 Pro™ printer was used with a direct drive extruder. The nozzle used was bronze with a diameter of 6.0 mm. After printing, the samples were characterized by measuring their dimensions and mass, and by XRD (Rigaku SmartLab) with a Cu anode (λ = 1.54 Ǻ). An ANOVA analysis of variance was performed considering the affect of both factors (temperature and speed) separately, as well as the interaction between them. Posing as null hypotheses, hoA = there are no significant differences between nozzle temperatures at 210°C, 220°C, and 230°C, hoB = there are no significant differences between speeds at 7 mm/s, 9 mm/s, and 12 mm/s, hoC = there are no significant interactions between nozzle temperatures and print speeds; and, as alternative hypotheses, h1A = there are significant differences between nozzle temperatures at 210°C, 220°C, and 230°C, h1B = there are significant differences between speeds at 7 mm/s, 9 mm/s, and 12 mm/s, and h1C = there is a significant interaction between temperatures and print speeds. This was used to obtain an analysis against the factors that affect the volume of the printed specimens. To accept or reject null hypotheses, a comparison was made between the probability value of 0.05 against the obtained results, taking into account that, if the probability is less than this value, the null hypotheses are rejected.
Three of the nine treatments were selected for debinding and sintering considering the lowest variation of the dimensions with respect to the nominal dimensions, taking into account the results of the ANOVA.
Debinding and Sintering
The selected samples were subjected to solvent debinding at 50°C in cyclohexane (purity ≥ 99.9%; Sigma Aldrich™) for 24 h using a magnetic stirrer at 500 rpm. These parameters were selected based on previous research12,16 and preliminary testing. The samples were dried at 50°C for 12 h and the mass was measured. Taking into account that a PP remains after solvent debinding, thermal debinding was carried out before sintering. The thermal debinding and sintering temperatures were selected taking into account the TGA results. The furnace used was tubular (Carbolite™ Gero) using an inert atmosphere of argon with a flux of 0.8 L/min. The samples were placed on a refractory recipient with alumina powder (100–365 μm; Sigma Aldrich™). After sintering, the samples were characterized by measuring their density in a balance (Sartorius LA 230S). They were then polished to a mirror finish with diamond particles (0.25 μm) to measure the hardness (LEICA VMHT MOT), analyze the morphology by SEM/EDS (Quanta 250 ESEM; FEI) and analyze the microstructure by XRD. For each sintered sample, considering the percentage of phases assessed by Rietveld refinement and their densities (Fe = 7.87 g/cm3 and FeO = 5.75 g/cm3), the bulk density was calculated with the mixing rule shown in Eq. 1, where ρ is the density and f is the volumetric fraction. With the respective values, the relative density was calculated. Porosity of the samples was measured using images from SEM and ImageJ™ software.
Results and Discussion
Powder
Grinding
The particle size distribution of the mill scale after grinding is shown in Fig. 1a. As can be seen, the mean particle size is 7.765 μm, the maximum is 208.930 μm, and the minimum is 0.550 μm. The curve is bimodal. The morphology shown in Fig. 1b is angular with an irregular shape, which is typical of ceramic particles obtained by grinding.21 The EDS analysis showed aluminum in addition to iron and oxygen, which could be a contaminant of the grinding process, because the jar used for grinding the mill scale is made of Al2O3 and is also used to grind aluminum chips. Manganese was also found with 0.68 wt.%, which is a common contaminant of mill scale.22 As can be corroborated in the XRD pattern in Fig. 1c, the mill scale under study is composed of the three iron oxides, FeO, Fe3O4, and Fe2O3, without the presence of metallic iron. After Rietveld refinement, 64.5% of FeO, 35.1% Fe3O4, and 0.4% Fe2O3 by mass were found. It should be noted that there is no presence of crystalline phases containing aluminum according to XRD.

(a) Particle size distribution, (b) SEM/EDS image and (c) XRD pattern of ground mill scale.
Chemical Reduction
The particle size distribution of the chemically reduced and meshed particles is shown in Fig. 2a. As can be seen, the mean particle size is 12.192 μm, the maximum is 39.811 μm, and the minimum is 0.209 μm. Comparing the average particle size between the reduced mill scale and the ground mill scale, it can be seen that the mean size increases as a result of the reduction process, meaning that agglomerations could have formed between the particles due to the formation of iron in the outer layer.23 The morphology of the particles is angular and spongy, as shown in Fig. 2b. When the chemical reduction of phases occurs, that is, from hematite to magnetite and from magnetite to wüstite, stress is caused within the particles, generating porosities and cracks during the reduction process.24 In addition to this, Matthew et al.25 explain that there are three mechanisms by which porous iron growth occurs during reduction: (1) a continuous reaction involving pore and iron growth, (2) a dendritic growth mechanism, in which porosity is formed before the iron, and (3) a mechanism in which layers of iron are formed on the oxide surface. On the other hand, in terms of the elemental composition measured by EDS, the presence of iron and oxygen is evident; also, aluminum appears. However, as can be seen in Fig. 2c, aluminum phases do not appear, which indicates that Al is in very low proportion or that it exhibits an amorphous structure. After Rietveld refinement, 89% of iron α, 7% of magnetite, and 4% of wüstite by mass were found. Comparing these findings with those for the unreduced mill scale XRD shown in Fig. 1c, it can be noted that the hematite was totally reduced to magnetite, the magnetite partially reduced to wüstite, and the wüstite was partially reduced to iron. A zoom of the peaks between 34 and 38 2θ (in degrees) of magnetite and wüstite is shown in Fig. 2d. The purpose of this is to enable a further reduction process to be explained later.

(a) Particle size distribution, (b) SEM/EDS, (c) XRD pattern, and (d) representative peaks of magnetite and wüstite after deconvolution of mill scale after chemical reduction.
Binder and Filament
The filament obtained after extrusion is shown in Fig. 3a. The tensile strength was 2.27 MPa, as can be seen in Fig. 3b, which is within the values reported by Kukla et al.26 for filaments manufactured with Nd. The elastic modulus was 372.62 MPa. The filament did not present problems when rolling and was able to be passed through the rollers of the printing machine, indicating that the tensile strength and elastic modulus were appropriate for printing. The cross-section of the filament can be seen in Fig. 3c and d, where the presence of porosities inside the IRONOX particles can be seen. Two benefits have been attributed to these porosities: (1) these channels act as paths through which gases are eliminated during thermal debinding, thus avoiding possible swellings; and (2) solvent debinding times are reduced.16 On the other hand, it can be observed that there is a uniform distribution of IRONOX particles in the polymer mixture developed.

(a) Filament, (b) stress–strain curve of the filament. Cross-section SEM images of filament: (c) ×1000, (d) ×3000.
Figure 4a shows the TGA curves obtained for the pure polymers under study, namely PP, TPE, and SA, as well as for the mixture of these. Part (b) of the figure shows the TGA curve obtained for the polymer mixture together with the IRONOX powder in the proportions mentioned in supplementary Table S-I. Total degradation of pure polypropylene (backbone), TPE, and the polymer mixture occurs in all three cases at a temperature of approximately 425°C, with the percentages of material remaining at 1200°C of 0.44% for PP, 1.22% for TPE, and 6.20% for the mixture. This last value is explained by the sum of the remaining material of PP and TPE in their proportions. The mixture of the three polymers with the IRONOX powder has the highest mass loss up to 460°C, indicating that the polymer mixture became more stable at 35°C above the original degradation temperature through its interaction with the metal powder. This could be explained by the interaction between the polar and nonpolar organic molecules with metal surfaces. In the case of the polar molecules, van der Waals forces are generated by the imbalance of charges in the atoms, which generates non-permanent dipoles that interact with the metal surface.27 In the case of the nonpolar molecules, since electrons are not evenly distributed around the atomic nucleus, permanent dipoles are generated, which also lead to the formation of van der Waals forces with the metal surface.28,29

TGA feedstock of binder: (a) pure components and blend of polymers and (b) blend with the powder of IRONOX.
However, the result of this study is contrary to that found by Tafti et al.,30 where the polymer mixture, consisting of EVA, AS, and paraffin wax, degraded to a lower temperature when mixed with the metal powder. This could indicate that the morphology and porosity of the IRONOX particles also affects the thermal stability of the polymer matrix through polymer/metal surface interaction. Finally, the fact that the degradation temperature is higher is beneficial for the parts obtained by this process, since the shape of the parts is maintained for as long as possible until the sintering temperature is reached.
Printability
The resulting printed samples are shown in Fig. 5, according to the design of experiments. In the front views, it can be seen that, for all three temperatures and at a speed of 7 mm/s, there is an overlap of the layers. On the other hand, at a speed of 9 mm/s, a low adhesion between the layers is observed (red arrows), especially at temperatures of 220°C and 230°C. Finally, at a speed of 12 mm/s, there is evidence of good adhesion between the layers; however, with the outer layer surrounding the rectilinear filling, there is poor adhesion in some areas (orange arrows). Taking this into account, it can be observed that there is a better adhesion between the layers at 7 mm/s printing speed, indicating that there is more time for the deposition of the layers.

Printed samples; one of the three test specimens printed per treatment is shown (Color figure online).
In the side view, in general, good quality printing, without discontinuities, can be observed. Small accumulations of material can be observed on the external surface of the wall (white circles), which occurred in the nozzle at the time of the growth of the layers.
Sample volume measurements compared with the nominal volume are shown in Fig. 6. It can be seen that, at a printing speed of 7 mm/s, there is less variation compared to the nominal value for the three temperature values. On the other hand, taking into account the printing temperatures, it can be observed that there is a lower dispersion of the data with respect to the nominal value at 230°C. Therefore, at this temperature, better behavior is generated against the design values.

Volume of the printed samples compared with the nominal volume.
With respect to Table I, which depicts the ANOVA analysis for the volume of the printed samples, it is found that the null hypothesis of the velocity factor must be rejected, because the p value is less than 0.05, which indicates that there are significant differences between the printing speeds under study. On the other hand, both the temperature factor and the interaction between speed and temperature have not been significantly differentiated, so alternative hypotheses are rejected in these cases.
Supplementary Fig. S-1 shows the XRD patterns for each combination of parameters. In general, the peaks of iron α, wüstite, and magnetite are observed, and are consistent with the starting powders shown in Fig. 2c. For the combination of T210-S9, a different peak is observed, which is attributed to zirconium oxide, indicating contamination by the grinding process; although this did not appear in the characterization of the powders.
According to these results, it was decided to continue working with the printed parts at a temperature of 230°C, evaluating the influence of printing speeds on physical properties (density, porosity, and shrinkage) and mechanical properties (hardness) after sintering.
Solvent Debinding
Supplementary Table S-III shows the percentage of weight loss for each of the specimens, as well as the polymeric residual percentage inside the specimens after solvent debinding. As cyclohexane dilutes TPE in the printed samples, and considering that the percentage by weight of the developed filament has a contribution of 7.79 wt.% of TPE (supplementary Table S-I), the TPE remainder in supplementary Table S-III was calculated by subtracting the mass loss from 7.79%. In this way, an elimination of approximately 82.80% of this component was obtained after 24 h.
Thermal Debinding and Sintering
According to the TGA shown in Fig. 4b, and taking into account that the dissolution of the TPE was not complete, the two degradation temperatures present in the thermogram were considered, the first from 200 to 350°C and the second from 350 to 460°C. Based on these temperatures, the thermal cycle shown in supplementary Fig. S-2a was developed. The heating rates were established according to Gonzalez-Gutierrez et al.31 and Thompson,12 who found that the use of low rates during this process is appropriate in order to avoid defects such as bloating. The pieces obtained after this thermal cycle are shown in supplementary Fig. S-2b. The S before the name means sintered. As can be seen, no blistering or surface cracking was generated after the debinding and sintering processes. Therefore, both the debinding process and the thermal cycle were successful.
Figure 7 shows the microstructures of the sintered specimens in the plane X–Y. For the S-T230-S7 parameters combination, it can be seen that there was a good adhesion between the particles, meaning that there were no printing defects present. On the other hand, for the S-T230-S9 combination, there were printing defects in terms of the union of the layers, which were not well adhered from the printing, as evidenced in Fig. 5. These defects cannot be eliminated during sintering, since the distance by which they are divided becomes considerable, preventing cohesion between the particles at these points.32 In the case of S-T230-S-12, the same problem occurs in terms of print quality but only on the outer layer, which is also evident from the printing. Regarding the microstructure, the second phase (the dark phase) can be seen to be embedded in the metallic iron matrix (the clearest phase).

SEM images of sintered pieces: (a) and (b) S-T230-S7, (c) and (d) S-T230-S9, (e) and (f) S-T230-S12.
Figure 8a shows the microstructure in relation to the elemental composition obtained by EDS, which is representative of the three sintered samples. Supplementary Table S-IV shows the atomic and weight percentages for the different elements presented in spectra 1, 2, and 3 of Fig. 8b, c, and d, respectively. Spectrum 1 refers to the matrix of the sintered material. Mainly iron with a little manganese is observed, manganese being a common contaminant in mill scale due to the process of obtaining the steel from which it comes,22 as explained before. Spectra 2 and 3 show oxygen, iron, manganese, and aluminum, which means that the darker phases correspond to iron oxide (wüstite). As mentioned before, aluminum is present as a contaminant from grinding and could be dissolved in the material.

Chemical composition by EDS of the sintered sample S-T230-S7 (a) region and its spectra (b) 1, (c) 2, and (d) 3.
Figure 9 shows the XRD patterns for the three sintered specimens. In general, for all three patterns the presence of iron α and wüstite is evident. When analyzing the graphs corresponding to 2θ between 34 and 38 degrees, a disappearance of the peak corresponding to the magnetite is observed. This peak appeared in the starting powders (Fig. 2c) and in the printed specimens (Supplementary Fig. S-1). Figure 10 shows the percentages of phases obtained by Rietveld refinement for these sintered parts. For comparison purposes, the percentages obtained in ground mill scale and reduced powders are also shown.

XRD patterns of the sintered samples: (a) S-T230-S7, (b) S-T230-S9, and (c) S-T230-S12.

Phases percentage of the materials under study by Rietveld refinement.
The absence of magnetite is explained by the thermal debinding process. During the degradation process, polypropylene, hydrogen, and carbon are released (Eq. 2). The released hydrogen reacts with magnetite, reducing it to wüstite and producing water vapor as a by-product (Eq. 3). Moreover, carbon produced via the process shown in Eq. 2 reacts with the water vapor molecules produced in Eq. 3, producing carbon monoxide and hydrogen (Eq. 4), both reducing agents that further reduce magnetite to wüstite, as shown in Eq. 5.33 The reason why the reduction of magnetite to wüstite occurs preferably, rather than from wüstite to metallic iron, is because, above 570°C, the first reaction is favored more than the second, since the first requires a higher activation energy, as reported by Jozwiak et al.34 Again, aluminum phases do not appear, which indicates that Al is in very low proportion or that it exhibits an amorphous structure.
The densities of the printed specimens at speeds of 7, 9, and 12 mm/s were 6.7525 g/cm3, 6.6677 g/cm3, and 6.5123 g/cm3, respectively, finding overall that the lower the printing speed, the higher the density. The relative density values were 90.55%, 88.47%, and 86.10%, respectively, which are comparable to those reported by Mousapour et al.35 for 316L stainless steel parts sintered at a temperature of 1360°C. The porosity, measured using SEM images at 1000×, showed values of 1.65%, 3.23%, and 4.41% for the sintered samples S-T230-S-7, S-T230-S-9, and S-T230-S-12, respectively. Therefore, the density values shown before can be explained by the porosity of the samples; the higher the porosity, the lower the density.
The volumetric shrinkage of the sintered parts is shown in Fig. 11a. The contraction was greater at a lower printing speed, as the lower printing speed results in greater cohesion between the paths (Fig. 7), which in turn results in a smaller final volume of the piece. Therefore, the volumetric shrinkage of the part without the presence of defects is around 46.47%, obtained at a printing speed of 7 mm/s. Meanwhile, the hardness of the sintered parts is shown in Fig. 11b. As Vickers hardness must be explained in terms of microstructural aspects, such as dislocation density in an iron matrix and iron oxide quantity, the last phase is the second phase that minimizes the dislocation movement of the iron. Since the sintering of the specimens was carried out in the same furnace under the same conditions, the density of dislocations of the iron matrix should not vary in the three samples; furthermore, this dislocation density corresponds to an annealed metal.36 As can be seen in Fig. 10, the quantity of wüstite is highest in the specimen that was printed at 7 mm/s, and the amount of this oxide is lower in the specimens printed at speeds of 9 and 12 mm/s. The lower the amount of dispersed phase, the lower the hardness. However, microhardness can also be affected by the porosity that remains after sintering which, as already explained, increases with printing speed, explaining the lower hardness of the S-T230-S12 specimen and the higher hardness of the S-T230-S7 specimen.

(a) Volumetric shrinkage and (b) hardness of the sintered samples.
Currently, there has been no evaluation metric to judge how good these processes are in comparison to other techniques, such as die pressing. However, by analyzing previous research, we can make a comparison. In the case of copper powder processed by die pressing, the resultant hardness is 118 HV,37 while, when the material is processed by MEX, the resultant hardness is 54.8 ± 2.1 HV,38 or 46.44% of that achieved by die pressing. In the case of stainless steel 316L processed by die pressing, the resultant hardness was 355.2 HV,39 while, when the material was processed by MEX, it was 285 ± 5.5 HV,14 or 43.91% of that achieved by die pressing. In our case, the hardness of IRONOX processed by die pressing was 143 HV,4 while, when the material was processed by MEX, the resultant hardness was 129 HV, or 90.20% of that achieved by die pressing. Although the hardness percentages vary, in general the hardnesses attained by the MEX process are lower compared to die pressing. The differences are related to the manufacturing process, as long times at high temperatures are required for MEX/die pressing, causing an increase in grain size. Moreover, the printing process causes a higher presence of pores in MEX than in die pressing. The higher hardness in the case of IRONOX obtained by die pressing in comparison to MEX could be related to the nature of the second phase, which is an oxide and does not experience changes with temperature, unlike the metallic phase.
Conclusion
Parts were obtained by metal MEX of a non-stainless metal, originating from an industrial waste. This residue, mill scale, obtained from a hot-rolling process of low-carbon steel, was subjected to a process of grinding and partial reduction with hydrogen, obtaining iron particles with an iron oxide core (IRONOX). The filament was manufactured with a TPE/PP/AS matrix with 50 vol.% of metallic powder. The printing was carried out on an Ender™ 3 Pro desktop printer, varying the temperature and printing speed. Printed parts were submitted to chemical debinding, thermal debinding, and sintering. The following conclusions were made:
-
Based on the temperatures and printing speeds studied, namely 210, 220, and 230°C and 7, 9, and 12 mm/s, respectively, according to the design of experiments it was found that the printing speed has a significant effect on the volume of the printed parts, while printing temperature has no significant effect with respect to the volume.
-
Regarding the mixture developed with TPE/PP/AS and IRONOX, it was found that the IRONOX powder has a significant influence on the thermal stability of the polymer mixture, which was attributed to the van der Waals forces that can occur between the metal surface and the polymer mixture and/or to the morphology and porosity of the metal particles.
-
Regarding the selected printed parts, T230-S7, T230-S9, and T230-S12, no significant differences were found in the characterization by XRD with respect to the IRONOX powder. However, differences were found with respect to the sintered parts, due to an additional chemical reduction presented from magnetite to wüstite, which in turns resulted from the polymer’s decomposition during thermal debinding, which made the magnetite in the sintered parts disappear.
-
The parts printed at 7 mm/s and 230°C were successfully depolymerized, as they exhibited the highest contraction during sintering, producing samples with no defects of any kind.
References
T.M. Tavares, G.M.D. Ganga, M. Godinho Filho, and V.P. Rodrigues, Sustain. Prod. Consum. 37, 369 https://doi.org/10.1016/j.spc.2023.03.006 (2023).
E. Furlani and S. Maschio, Case Stud. Constr. Mater. https://doi.org/10.1016/j.cscm.2016.02.001 (2016).
A. Esguerra-Arce, G. Pinto-Arciniegas, S. Silva-Hurtado, R. Setchi, and J. Esguerra-Arce, JOM. https://doi.org/10.1007/s11837-023-05993-2 (2023).
J.G. Tirado González, B.T. Reyes Segura, J. Esguerra-Arce, A. Bermúdez Castañeda, Y. Aguilar, and A. Esguerra-Arce, J. Clean. Prod. https://doi.org/10.1016/j.jclepro.2020.122362 (2020).
C. Suwanpreecha and A. Manonukul, Metals https://doi.org/10.3390/met12030429 (2022).
R. Ranjan, Z. Chen, C. Ayas, M. Langelaar, and F. Van Keulen, Addit. Manuf. 61, 103339 https://doi.org/10.1016/j.addma.2022.103339 (2023).
Y. Li, S. Liu, H. Zhi, X. Yang, J. Zhang, and Y. Wang, J. Manuf. Process. 94, 374 https://doi.org/10.1016/j.jmapro.2023.03.064 (2023).
L.S. Santos, S.K. Gupta, and H.A. Bruck, Addit. Manuf. 23, 235 https://doi.org/10.1016/j.addma.2018.08.002 (2018).
C. Su, J. Wang, H. Li, Z. You, and J. Li, J. Manuf. Process. 84, 652 https://doi.org/10.1016/j.jmapro.2022.09.025 (2022).
Z. Lotfizarei, A. Mostafapour, A. Barari, A. Jalili, and A.E. Patterson, Addit. Manuf. 61, 103335 https://doi.org/10.1016/j.addma.2022.103335 (2023).
A.-F. Gil-Plazas, J.-D. Rubiano-Buitrago, L.-A. Boyacá-Mendivelso, L.-K. Herrera-Quintero, A. Gil-Plazas, J. Rubiano-Buitrago, and L. Boyacá-Mendivelso, Revista Facultad de Ingeniería 31, e13913 https://doi.org/10.19053/01211129.v31.n60.2022.13913 (2022).
Y. Thompson, J. Gonzalez-Gutierrez, C. Kukla, and P. Felfer, Addit. Manuf. 30, 100861 https://doi.org/10.1016/j.addma.2019.100861 (2019).
B. Liu, Y. Wang, Z. Lin, and T. Zhang, Mater. Lett. https://doi.org/10.1016/j.matlet.2019.127252 (2020).
M. Sadaf, M. Bragaglia, and F. Nanni, J. Manuf. Process. https://doi.org/10.1016/j.jmapro.2021.04.055 (2021).
S. Hwang, E.I. Reyes, K. Sik Moon, R.C. Rumpf, and N.S. Kim, J. Electron. Mater. 44, 771 https://doi.org/10.1007/BF02648403 (2015).
Y. Zhang, L. Poli, E. Garratt, S. Foster, and A. Roch, 3D Print Addit. Manuf. 7, 279 https://doi.org/10.1089/3dp.2020.0136 (2020).
K. Kassym and A. Perveen, in Mater Today Proc (2019). https://doi.org/10.1016/j.matpr.2020.02.364.
N. Birks, G.H. Meier, and F.S. Pettit, Introduction to the High Temperature Oxidation of Metals (Cambridge University Press, Cambridge, 2006), p84.
M.A. Wagner, A. Hadian, T. Sebastian, F. Clemens, T. Schweizer, M. Rodriguez-Arbaizar, E. Carreño-Morelli, and R. Spolenak, Addit. Manuf. 49, 102472 https://doi.org/10.1016/j.addma.2021.102472 (2022).
P. Singh, V.K. Balla, A. Tofangchi, S.V. Atre, and K.H. Kate, Int. J. Refract. Metals Hard Mater. 91, 105249 https://doi.org/10.1016/j.ijrmhm.2020.105249 (2020).
P. Sharma, S. Sharma, and D. Khanduja, Mater. Manuf. Process. 30, 1370 https://doi.org/10.1080/10426914.2015.1037904 (2015).
H. El-Shiekh, M.K. El-Fawakhry, M.E. Mitwally, and A.Y. Shash, Results Mater. 19, 100429 https://doi.org/10.1016/j.rinma.2023.100429 (2023).
M.I.A. Barustan and S.M. Jung, Met. Mater. Int. https://doi.org/10.1007/s12540-019-00259-6 (2019).
J. Chen, R. Zhang, T. Simmonds, and P.C. Hayes, Metall. Mater. Trans. B Process Metall. Mater. Process. Science 50, 66 https://doi.org/10.1007/s11663-019-01659-0 (2019).
S.P. Matthew, T.R. Cho, and P.C. Hayes, Metall. Trans. B. https://doi.org/10.1007/BF02654252 (1990).
C. Kukla, J. Gonzalez-Gutierrez, S. Cano, S. Hampel, C. Burkhardt, T. Moritz, and C. holzer, ACTAS DEL VI Congreso Nacional de Pulvimetalurgia y i congreso Iberoamericano de Pulvimetalurgia, ed. In G. Herranz, B. Ferrari, & J. M. Cabrera (Comité Español de Pulvimetalurgia, Spain, 2017), pp. 1-6.
H. Lgaz and H. Seung Lee, J. Mol. Liq. https://doi.org/10.1016/j.molliq.2021.118071 (2022).
H. Öström and A. Nilsson, Chemical Bonding of Hydrocarbons to Metal Surfaces 2004. diva2:190462.
P.O. Bedolla, G. Feldbauer, M. Wolloch, S.J. Eder, N. Dörr, P. Mohn, J. Redinger, and A. Vernes, J. Phys. Chem. C 118, 17608 https://doi.org/10.1021/jp503829c (2014).
A.A. Tafti, V. Demers, S.M. Majdi, G. Vachon, and V. Brailovski, Metals. https://doi.org/10.3390/met11020264 (2021).
J. Gonzalez-Gutierrez, Y. Thompson, D. Handl, S. Cano, S. Schuschnigg, P. Felfer, C. Kukla, C. Holzer, and C. Burkhardt, Mater. Lett. https://doi.org/10.1016/j.matlet.2020.128909 (2021).
M.K. Agarwala, V.R. Jamalabad, N.A. Langrana, A. Safari, P.J. Whalen, and S.C. Danforth, Rapid. Prototyp. J. https://doi.org/10.1108/13552549610732034 (1996).
T. Murakami and E. Kasai, ISIJ Int. https://doi.org/10.2355/isijinternational.51.9 (2011).
W.K. Jozwiak, E. Kaczmarek, T.P. Maniecki, W. Ignaczak, and W. Maniukiewicz, Appl. Catal. A Gen. https://doi.org/10.1016/j.apcata.2007.03.021 (2007).
M. Mousapour, M. Salmi, L. Klemettinen, and J. Partanen, J Manuf Process 67, (2021). https://doi.org/10.1016/j.jmapro.2021.05.021.
J. G. Tirado González, A. Esguerra-Arce, L. K. Herrera, and J. Esguerra-Arce, in Sustainable Design and Manufacturing. SDM 2022. Smart Innovation, Systems and Technologies, Vol 338., edited by S. G. Scholz, R. J. Howlett, and R. Setchi (Springer, Singapore, 2023), pp. 67-76. https://doi.org/10.1007/978-981-19-9205-6_7.
H. Ayub, L.A. Khan, E. McCarthy, I.U. Ahad, K. Fleischer, and D. Brabazon, J. Mater. Res. Technol. https://doi.org/10.1016/j.jmrt.2022.05.012 (2022).
A. Cañadilla, A. Romero, G.P. Rodríguez, M. Caminero, and Ó.J. Dura, Materials. https://doi.org/10.3390/ma15134644 (2022).
N. Kurgan, Mater. Des. https://doi.org/10.1016/j.matdes.2013.06.035 (2013).
Acknowledgements
The authors must express their gratitude to Professors Oliver Clemmens and Guido Schmitz for laboratory accommodation in Max Planck Institute, and many other facilities which have greatly assisted the present study. The authors also thank the Powder Metallurgy Laboratory of Universidad Nacional de Colombia - Bogotá, and the Centro de Materiales y Ensayos of the SENA. Grateful acknowledgment of financial assistance is made to Escuela Colombiana de Ingeniería Julio Garavito and the project 82305 with contingent recovery Contract 2021-1012.
Funding
Open Access funding provided by Colombia Consortium.
Author information
Authors and Affiliations
Contributions
J.G.T.-G.: Writing—original draft, Methodology, Conceptualization, Data curation. J.E.-A.: Writing—review & editing, Supervision, Resources, Methodology, Conceptualization. A.E.-A.: Review & editing, Resources, Supervision. L.K.H.-Q.: Review & editing, Supervision, Resources, Project administration, Methodology, Conceptualization.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tirado-González, J.G., Esguerra-Arce, J., Esguerra-Arce, A. et al. 3D Printing Iron/Iron Oxide Composites by Metal Material Extrusion from an Industrial Waste. JOM 76, 1924–1936 (2024). https://doi.org/10.1007/s11837-024-06371-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11837-024-06371-2