
Abstract
The solution of elements from metallic alloys is analyzed, notably the initial stage characterized by solution in proportion to the alloy composition and subsequent selective leaching of alloying elements. For the latter stage of the process, characteristic features of the originating depletion zone are derived for different formation mechanisms. The results are compared with observations for steels and nickel-based alloys after exposure to lead-based liquid alloys or liquid tin, and, where possible, the prevailing mechanism is identified. Furthermore, the influence of dissolved oxygen and formation of intermetallic compounds are addressed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Application of lead (Pb)-based liquid alloys or liquid tin (Sn) to thermal energy conversion or storage opens the avenue to compact design, highly efficient components in the high-temperature section of respective plants, however, at the cost of increased corrosion of metallic materials of construction, namely nickel (Ni)-containing steels or Ni-based alloys. Experimental studies and corresponding theoretical work identify selective leaching of constituent parts, especially Ni, as an intermediate stage of complete dissolution,1 with the near-surface depletion zone originating in the solid alloys being dependent on the alloy composition,2,3 the liquid metal3 and temperature.1,2,3 If the oxygen content in the liquid allows, formation of solid oxides is likely to interfere with the leaching process or even changes the corrosion mode to oxidation. A similar role intermetallic compounds may play.
As to fundamental interactions, the focus is on the transfer to and transport in the liquid metal of dissimilar metals.4 The dissimilarity primarily refers to the solubility or maximum enrichment the elements may achieve if dissolving from the pure solid, and this solid constitutes the thermodynamically stable solid modification of the element under consideration. However, when metals dissolve from an alloy, concentrations in the liquid metal cannot reach solubility, except for the element that would remain at the solid/liquid interface after complete leaching of the other elements. Of major alloying elements in steels or Ni-based alloys, the possible enrichment of Ni in liquid metals typically is clearly higher than for iron (Fe) or chromium (Cr), which is illustrated in Fig. 1 for liquid lead-bismuth eutectic (LBE).5,6,7

Solubility of Fe,5 Cr6 and Ni6,7 in LBE as a function of temperature. The solid section of the data plots indicates the temperature range in which experimental data are actually available. It should be noted that at temperature less than about 450°C, the thermodynamically stable Ni-containing solid is intermetallic NiBi rather than pure nickel.2,7 Reprinted from C. Schroer, TMS2021 150th Annual Meeting and Exhibition Supplemental Proceedings (The Minerals, Metals and Materials Society, Pittsburgh PA: Springer 2021) p. 642-656.
After analyzing the elemental steps that result in the transition of alloying elements to the bulk of the liquid, while others stay behind, the theoretical implications are compared to experimental observations. The influence of oxygen dissolved in the liquid metal as well as the formation of intermetallic compounds is discussed. The observations evaluated with respect to dissimilar metal solution stem from experiments in Pb-based alloys1, 3, 8,9,10,11,12,13,14,15,16,17,18,19 and liquid Sn20,21,22,23 at 400–750 and 500–1000°C, respectively.
Fundamental Aspects of the Solution of Metal Alloys
The solution of a solid material, a metallic alloy in this case, may be understood as the gradual transition of its constituent parts to the bulk volume of a liquid. Initially, the alloying elements are likely to dissolve in proportion to their concentration in the alloy (general or proportionate solution), but the situation will change if one of the alloying elements reaches a thermodynamic limit of further solution. This is expressed as the equality of thermodynamic activity of the respective element in the solid and the liquid or
f denotes the activity coefficient that corresponds to the mole fraction y in the solid alloy, whereas x is the mole fraction in the liquid. As a first approximation, the inverse of the solubility xs of the element under consideration replaces the activity coefficient in the liquid, implying an ideal diluted solution with negligible interaction between the different solutes. Inserting Eq. 1 into the element mass balance and re-arranging gives
for the moles of alloy having dissolved (i) in j moles of liquid solvent at the time the equality of activities is achieved. x0 considers the initial concentration in the liquid. An equation of this type applies to each element of the solid alloy separately, and the element that will be the first to impose a limit on proportionate solution obviously is the one for which the dissolved moles of alloy are smallest (smallest i at given j). At negligible initial concentrations in the liquid, this is the element with smallest product of activity coefficient in the solid and solubility in the liquid (0 < f × xs < 1), and its concentration in the liquid at the time the conditions for solution change is f × y × xs. The criterion for negligible initial concentration in the liquid is x0 << f × y × xs. For solid Cr–Fe–Ni alloys, indicative values of activity coefficients are < 5,2, 24 so that, at negligible initial concentrations in the liquid, the alloying element with lowest solubility determines the end of the stage of proportionate solution if differences in solubility exceed about half an order of magnitude. Equation (2) implies that a highly soluble alloying element is limiting for proportionate solution especially if its initial concentration in the liquid approaches the solubility and the concentration in the alloy is low.
Subdividing solution into elemental steps (subprocesses) such as mass transfer to and transport in the liquid, above considerations may also be applied to the transfer of the alloying elements across the solid/liquid interface, if the elements have approximately the same diffusivity in the liquid. For same diffusivity and negligible initial concentrations in the liquid, concentration gradients in the liquid rise in proportion to the transfer across the solid/liquid interface, so that also the transport of alloying elements away from this interface occurs in proportion to the alloy composition, at least initially. The element exhibiting the smallest f × xs again reaches the threshold concentration first, however, at smaller effective volume of liquid, i.e., earlier than for infinitely fast dispersion of the transferring elements in the liquid, which was tacitly presumed above. The hydrodynamic approach to diffusion (Stokes-Einstein) predicts the same diffusivity for particles of the same size, which approximately applies to Cr–Fe–Ni alloys dissolving in the form of atoms. Experimental evidence is available for the diffusion coefficients of Fe and Ni in LBE.25 Disproportionate element transport tends to change the element limiting proportionate transfer, which possibly occurs if the element with lowest solubility has a comparatively low initial concentration in the liquid or the concentration of a highly soluble element already approaches the solubility before solution starts.
Solution not in proportion to the alloy composition, typically named dealloying or selective leaching in the technical literature, necessarily involves disproportionate transport away from the solid/liquid interface but does not necessarily mean disproportionate transfer across this interface. However, should proportionate transfer remain, an alternative to dispersion in the bulk of the liquid must exist for elements that would otherwise oversaturate the liquid at the solid/liquid interface. Such an alternative is reprecipitation after some short-range diffusion,1 but the elements in question must locally achieve a thermodynamic activity in the liquid that is higher than in the newly formed solid phase, which is equivalent to the original alloy being a metastable modification for the primarily settled elements such as austenite for Fe at temperature less than about 900°C. If reprecipitation at a minimum of diffusion is an option especially for elements that are only sparingly soluble in the liquid, they do not restrain proportional transfer across the solid/liquid interface of the original alloy. It should be noted that, as to the qualitative effect on element transfer across the instantaneous surface of the original alloy, the formation of a compound with constituent parts of the liquid is comparable to reprecipitation in the form of a more stable allotrope as long as the compound layer that establishes is permeable to the other, especially the highly soluble elements in the original alloy.
In the following, the term selective leaching is used for solution (transition of elements to the bulk of the liquid) of a metal alloy that is not in proportion to the alloy composition.
Development of a Depletion Zone
Selective leaching from metallic alloys typically features a layer on the alloy surface that is depleted in those alloying elements that preferentially change over to the bulk of the liquid (Fig. 2). The layer is respectively enriched in elements that rather stay behind. Unless the selectively removed element occupies only interstitial sites in the alloy crystallographic lattice, the depletion zone that develops tends to be porous, generally allowing the liquid to penetrate this zone. Elementary processes specifically involved in the development of the depletion zone may be lattice and surface diffusion or reprecipitation.26 Parameters determining the evolution of the depletion zone26,27 especially are the concentration of the preferentially removed elements and the homologous temperature, i.e., the ratio of the prevailing temperature and the solidus temperature of the alloy (both in Kelvin).

Schematic illustration of depletion and penetration of liquid as expected for (a) disproportionate element transfer and dominant lattice diffusion in solid phases; (b) disproportionate element transfer above the percolation limit; (c) proportionate element transfer followed by reprecipitation of sparingly soluble elements.
If the original (parent) alloy is a substitutional solid solution, disproportionate element transfer across the solid/liquid interface creates vacancies in the solid, at this interface. These vacancies may partly be refilled by atoms from the bulk of the parent alloy, of the elements that preferentially transfer to the liquid, or annihilated alternatively through diffusion in the opposite direction, of the elements that transfer to the liquid to a lesser extent. The concentration gradients required for sustaining this counter-diffusion across the depletion zone must develop. With the enrichment at the depletion zone/liquid interface, the element that once has stopped proportionate transfer from the parent alloy regains some driving force for transition to the liquid phase, which, along with mass transport toward the bulk of the parent alloy, contributes to the reduction of the near-surface vacancy concentration at the cost of a recession of the depletion zone surface. Local accumulation of vacancies may lead to near-surface pores, just as porosity originates at the parent alloy/depletion zone interface if counter-diffusion across the depletion zone is non-balanced in favor of the resupply of the preferentially transferring elements from the parent alloy (Kirkendall effect).28 The penetration of liquid via such pores, deep into the depletion zone, requires a continuous network, which naturally must stretch across the whole depletion zone if the liquid should reach the parent alloy. Disregarding for a moment the role grain boundaries may play in respect of penetration of the liquid, an alternative are paths that open in consequence of anisotropy in interfacial energy between the liquid and the crystal lattice planes in the depletion zone if lattice diffusion in the depletion zone is rate-limiting.26 These paths are likely to exhibit a nodular through dendritic shape,26 but are unlikely to reach the parent alloy/depletion zone interface (Fig. 2a).
While lattice diffusion needs a comparatively high homologous temperature for its contribution to become significant, a depletion zone evolves without lattice diffusion if the concentration of elements that preferentially transfer to the liquid exceeds a certain threshold concentration (percolation limit, Fig. 2b). At or above this concentration, the respective elements may be regarded as forming chains in the parent alloy, with a width that allows the liquid to enter the voids that would otherwise originate from these elements transferring to the liquid phase.26,27 The elements that stay behind form a so-called bi-continuous solid structure. The ligaments of the solid coarsen especially by surface diffusion.27 Even below the percolation limit, surface diffusion may effect a decomposition of superficially depleted and liquid-penetrated parent alloy, which can be understood to be of spinodal type,29,30 into domains of liquid and grains of depleted alloy. The latter naturally form immediately on the parent alloy surface, and it seems very likely that depleted solid is connected locally to the parent alloy, which also applies to the lattice diffusion and percolation mechanism. At homologous temperature too low for significant surface diffusion, disproportionate element transfer to the liquid will stop shortly after it has begun. Grain boundaries in the parent alloy provide paths of locally enhanced element transfer and intergranular penetration of the liquid. Depletion of the parent alloy grains proceeds from the grain boundaries into the adjacent grains. The characteristic patterns of depleted solid and liquid that result from the particular mechanism of depletion are found in the interior of the former parent alloy grains, separated by the former grain boundaries that are clearly dominated by penetrated liquid,27 so that the gross microstructure of the parent alloy is conserved (Fig. 2a, b).
As to grain boundaries being the sites where element transfer from solid to liquid is likely to start, there is no principal difference between this transfer becoming selective at some point in time and continuous transfer in proportion to the parent alloy composition. Reprecipitation to maintain proportionate element transfer to the liquid is expected to occur first in the grain boundaries, too (Fig. 2c). Such particles of the newly formed phase are an obstacle to necessary element transport in the liquid volume that has penetrated the grain boundaries, especially of the highly soluble alloying elements; consequently, solution of and reprecipitation on the grain faces gain in importance. The precipitates above the interior of the grains are likely to be large compared to particles forming inside the limited space along grain boundaries, so that the parent alloy microstructure is traced through small between large precipitates. Growth of large at the cost of small particles (Ostwald ripening) may clear the former grain boundaries from precipitates, by which the original gross microstructure becomes even more apparent in the depletion zone. In contrast to depletion by disproportionate transfer to the liquid, there is no diffusion of the elements that remain in the depletion zone toward the bulk of the parent alloy, because the activity of these elements in the precipitating solid is always less than in the parent alloy. Accordingly, for the surface of the depletion zone to recede, repeated transfer to and dispersion in the liquid are required.1 It should be noted that the presence of a newly formed phase is not distinctive for the reprecipitation mechanism. Missing depletion below the deepest penetration of the liquid excludes disproportionate element transfer coupled to lattice diffusion in the solid phase(s), whereas a continuous film of liquid separating the depletion zone from the parent alloy is an indicator of proportionate element transfer followed by reprecipitation from the liquid. Especially in the initial stage of depletion zone development, reprecipitation may possibly occur immediately on the parent alloy surface, but in the long run, such particles are likely to be undercut by the liquid, and growth of existing particles is more favorable.
Observations from Experiments
Most of the experimental studies evaluated in the following were performed on steels. As illustrated in Fig. 1 for LBE, significantly different solubility primarily occurs for austenitic grades because of the high solubility of Ni. At < 900°C, ferrite is the generally more stable modification of the parent element (Fe) in the alloy, so that disproportionate element transfer and proportionate transfer followed by reprecipitation of Fe are equally likely to occur, especially because the solubility of Fe in liquid metals is relatively low but not insignificant.
Figure 3 shows an early stage of solution for austenitic 15–15 Ti steel 1.4970 after exposure to flowing LBE at 400°C, found on the flat, polished surface of the material, in both the solution annealed and cold-worked (40%) state.11 In the particular experiment, local solution of the steel co-occurred with surface oxidation at a concentration of 10–7% (by mass) oxygen carried by the LBE. Numerous small Fe crystallites (ferrite) along grain boundaries in the steel microstructure as well as larger ones found on the grain faces imply reprecipitation of Fe. While it may not be excluded that part of the accumulated Fe deposited from the flowing liquid metal, because other materials such as ferritic/martensitic steel and technically pure Fe were tested in the same experimental run,12 the interaction of the austenitic steel and the liquid metal is obvious from cross-section analyses, especially for grain and subgrain boundaries.11 The reprecipitation of Fe seems even more required for maintaining proportionate element transfer from the steel if the Fe concentration in the liquid is already high.

Surface of 15-15 Ti steel 1.4970 (40 % cold work, flat surface, polished) after exposure for about 5000 h to flowing LBE at 400°C and 10–7% dissolved oxygen.11 Reprinted from C. Schroer, TMS2021 150th Annual Meeting and Exhibition Supplemental Proceedings (The Minerals, Metals and Materials Society, Pittsburgh PA: Springer 2021) p. 642-656.
Preferential interaction at grain and subgrain boundaries, in which the austenitic steel does not differ, e.g., from technically pure Fe,12 is also evident from the top view on the surface in Fig. 3. Penetration of the constituents of the liquid metal along and adsorption onto the opposing grain faces31 may support accelerated element transfer, especially at high-angle grain boundaries, where crystallographic mismatch between grains is largest. However, the penetration of liquid metal may be more apparent at subgrain boundaries such as between the deformation twins in cold-worked austenitic steel after exposure to LBE at 45013 or 500°C,14 suggesting a special affinity of the twinning planes to the liquid metal. Where the LBE has penetrated, electron microscopy reveals ferrite with reduced Ni and Cr content, always enclosed by solidified LBE,13 whereas element depletion on the side remote from the ferrite is not apparent from the presented elemental maps and other micro-analyses, which is a clear indicator of element transfer in proportion to the steel composition and subsequent reprecipitation of ferrite. The presented idea that a narrow austenite twin widens through selective leaching of elements, which opens the space for further ingress of liquid metal,13 may apply analogously for a single twin boundary, considering the loss of material in connection with the removal of atoms via the penetrating liquid. In an advanced stage of selective leaching in austenitic steel, such as observed after exposure to flowing oxygen-containing LBE at 550°C (Fig. 4), liquid metal that penetrated the depletion zone seems to trace at least part of the grain boundaries of the original steel microstructure. Progress at the depletion zone/steel interface is largely transgranular.15 The particular shape of this interface, however, depends on temperature,1 concentration and orientation of deformation twins in the material14 or other special features of the steel microstructure.16 The results from energy-dispersive x-ray spectroscopy (EDS) that are presented in Fig. 4 indicate the film of solidified LBE at the transition to the unaffected steel and missing depletion beyond that are characteristic for the reprecipitation mechanism. The EDS analysis presented in Fig. 4 furthermore suggests that a small part of Cr removed from the steel has oxidized on the depletion zone surface alternatively to transfer to the bulk of the LBE.

Cross section of a depletion zone that has formed in austenitic steel 1.4571 during exposure for 5012 h to oxygen-containing flowing LBE at 550°C.15 Reprinted from C. Schroer, TMS2021 150th Annual Meeting and Exhibition Supplemental Proceedings (The Minerals, Metals and Materials Society, Pittsburgh PA: Springer 2021) p. 642-656.
The interaction between austenitic steel and liquid Sn20 is, at < 650°C, dominated by the formation of Fe–Sn intermetallic phases. Ferrite first occurs above this temperature, and absorption of Sn atoms stabilizes the ferritic phase, which is still observed at > 1050°C. At > 840°C, where ferrite exclusively forms, a relatively high share of lattice and especially surface diffusion in the development of the depletion zone can be expected, and the presumably spinodal-type decomposition29,30 of depleted and Sn-penetrated steel into ferrite and liquid seems rather probable. The habitus of ferrite changes from globular to columnar under conditions hampering the removal of Ni, i.e., after pre-saturation of the liquid with Ni, for a comparatively small volume of liquid or decreasing temperature.20 At > 1050°C, the depletion zone shows prominent surface recession, and the ferrite tends to recrystallize into a continuous layer. Additionally, Sn diffuses into the austenite grain boundaries, with a preference for twins in the austenitic structure, triggering local austenite/ferrite transitions inside the steel.20
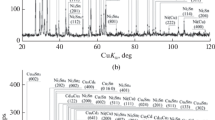
In the case of Ni-based alloys, the highly soluble constituents include the parent element of the alloy (Ni), suggesting that the percolation mechanism26,27 is likely to operate. This is reflected by the innermost part of the depletion zone that forms in Alloy 2.4663 (Ni–22Cr–12Co–9Mo) during the exposure to liquid Sn at 700°C (Fig. 5a). Cr and molybdenum (Mo) are the major alloying elements of low solubility in liquid Sn, and what forms is a fine-structured conglomerate of Sn–Ni and Cr–Mo phases (Fig. 5b). The Sn–Ni component is at least partially liquid at 700°C and also traces the former grain boundaries in the original alloy microstructure.22 The fine dispersion coarsens with time, so that, in the older, outer portion of the depletion zone, Cr–Mo has clearly separated from Sn–Ni and accumulated in the domains that appear dark in the backscatter electron (BSE) micrograph depicted in Fig. 5a. At 1000°C, the inner part of the depletion zone is columnar Cr–Mo interspersed with Sn–Ni (labeled A in Fig. 5c). In the direction toward the bulk of the liquid, a transition zone (labeled B) follows, which shows a high percentage of Sn–Ni along with small particles that seem to have separated from the Cr–Mo columns in the subzone A. Such a pinch-off of particles is typical for aging of rod-type structures.32,33,34 The particles in the outermost portion (labeled C) are comparatively large and have gained in size partly through absorption of Fe from the liquid Sn, which is a circumstance of the particular experimental setup.22 The columnar Cr–Mo is reminiscent of the ferrite structure attributed to relatively low velocity of Ni transfer to the bulk of the liquid metal.20 Obviously, at temperature higher by 300°C, improved lattice and surface diffusion renders interconnection of the atoms of the highly soluble elements in the alloy less important for selective leaching to proceed at the maximum possible rate. Additionally, the capacity of the liquid for taking up Ni as well as other alloying elements is naturally higher.

Selective leaching in Ni-based alloy 2.4663 (Ni–22Cr–12Co–9Mo) after contact with static liquid Sn (a, b) for 25 h at 700°C and (c) for 50 h at 1000°C.22 Figure 5b is a detail of the innermost part of the depletion zone depicted in Fig. 5a. A–C in Fig. 5c marks distinguishable subzones. Reprinted from C. Schroer, TMS2021 150th Annual Meeting and Exhibition Supplemental Proceedings (The Minerals, Metals and Materials Society, Pittsburgh PA: Springer 2021) p. 642-656.
Influence of Dissolved Oxygen
The constituents of a solid metal alloy may not only differ in solubility or diffusivity but also in their affinity to oxygen dissolved in the liquid. Surface oxides that form may effectively protect the solid from solution; however, defects accumulate along with the growth of an oxide film, so that the oxide becomes prone to local failure,35,36 and solution in the liquid is only delayed rather than irreversibly avoided. If and where solid/liquid element transfer remains possible, the loss in oxygen-affine elements increases with increasing oxygen activity, as observed, e.g., for selective leaching in austenitic steel caused by liquid Pb–lithium (Li) with different oxygen concentrations.17
When alloying elements form solid oxide only after transfer to the liquid, the site where this happens depends on the resupply of oxygen from the bulk of the liquid. Since oxygen tends to deplete at alloy/liquid interfaces, solid oxide typically precipitates primarily in some distance from the site of element transfer to the liquid, as, e.g., observed for austenitic steel at 450°C in the presence of static LBE containing 10–7% oxygen.18 At the site of oxide precipitation, the local concentration of the respective alloying elements is always low, which keeps up the concentration gradients required for transport in the liquid. This enhances the removal of these elements from the liquid at the alloy/liquid interface and contributes to maintaining element transfer in proportion to the original alloy composition, especially if the sparingly soluble elements of the alloy are involved.1 Accordingly, oxidation of Fe and Cr suggests itself as an explanation for different ferrite content in depletion zones formed in austenitic steel at 450 and 550°C in flowing LBE with 10–6% dissolved oxygen in either case.1 The precipitation of oxides may even reverse the situation regarding element depletion in the solid alloy, exemplified in Fig. 6, by the exposure of binary Ni–Cr, especially Ni with 35% Cr, to static liquid Pb at 750°C and 10–6% dissolved oxygen.19 The presented EDS linescan indicates near-surface Cr depletion in the alloy dspite the solubility of Ni in Pb being clearly higher than the solubility of Cr.37 Cr has reprecipitated to form Cr-rich oxide.

Ni–35Cr model alloy after exposure for 120 h to oxygen-containing static Pb at 750°C: BSE micrograph and results of qualitative EDS analyses performed along the indicated line.19 Reprinted from C. Schroer, TMS2021 150th Annual Meeting and Exhibition Supplemental Proceedings (The Minerals, Metals and Materials Society, Pittsburgh PA: Springer 2021) p. 642-656.
Interference of solid oxide formation with element solution is also possible for steels and Ni-based alloys in contact with liquid tin.23
Formation of Intermetallics
As mentioned earlier, the formation of intermetallic compounds is particularly important in the Fe–Sn system. In the context of dissimilar metal solution, the establishment of an intermetallic phase, in the form of a layer that separates alloy and liquid, involves an additional element transfer to and transport across this layer. Formally, it seems useful to regard an intermetallic that grows into the solid alloy as a constituent part (possibly the only constituent) of a depletion zone, alternative to the penetration of liquid. Then, considerations as to solid/solid element transfer, subsequent transport, re-ordering or reprecipitation are largely equivalent to direct transfer to the liquid, except for the alloying element that becomes a principal constituent of the intermetallic. As a first approach, the transfer of this element to the intermetallic phase may be regarded as dependent on the supply of its counterpart in the liquid (e.g., Sn) across the growing layer or the space available for such growth into the original alloy. Growth at the intermetallic/liquid interface means that such a depletion zone may possibly go beyond the original position of the alloy surface.
When typical austenitic steel is being exposed to liquid Sn at 500°C, FeSn2 and, to a lesser extent, FeSn form a bi-layer intermetallic zone on the steel surface, whereas Ni primarily permeates these intermetallics and changes over to the liquid metal. Element transfer from the steel to the intermetallic layers is largely in proportion to the original steel composition.21,22 Under conditions that result in local initiation of enhanced degradation, pits formed in the steel surface exhibit FeSn2 dispersed in a network of ferrite or another phase that is rich in Cr and Mo,23 suggesting that the percolation mechanism was active, at least temporarily. In the center of the pits, it rather is Sn that penetrates this network, unless FeSn2 formation is not particularly favored, i.e., for liquid Sn not pre-saturated with Fe.23 At 700°C, FeSn gains in importance for the intermetallic layers that now form along with columnar ferrite.21,22 From this ferrite, particles pinch off, similar to the situation depicted in Fig. 5c. Such particles, enclosed by FeSn, are rich in Cr and Mo, with Fe content and size decreasing with increasing distance from the steel.22 Locally enhanced interaction between austenitic steel and liquid Sn again results in Fe–Sn intermetallics dispersed in a network of Ni- and Fe-depleted steel.23
Due to the high solubility of Ni in liquid Sn, Ni–Sn intermetallics tend to form in a later stage of interaction with the liquid metal than, e.g., Fe-containing intermetallics. Accordingly, they are likely to influence aging rather than early development of a depletion zone that results from dissimilar metal solution in the form of selective leaching. This especially applies to Ni-based alloy 2.4663 when being exposed to liquid Sn at 700°C,22,23 however, only if the formation of, in this case, Ni3Sn4 is not enforced by pre-saturating the liquid with Ni. Pre-saturation with Ni generally produces a thin, heterogeneous zone, all in all, depleted in Ni, with similar constituent parts but clearly different in structure compared to the situation without pre-saturating with Ni. Ni3Sn4 forms on top and is likely to also be the Sn–Ni component in the inner part of the depletion zone23 (cf. Fig. 5b). Where locally enhanced interaction occurred, the largely two-phase layer with Ni3Sn4 on top is found in the bottom and on the edges of pits in the alloy surface, whereas the aged structure in the center of the pit shows coarser, needle-shaped Cr–Mo interspersed with Sn.23 The original alloy grain structure is lost, in contrast to the depletion zones observed if conditions do not particularly favor formation of Ni3Sn4.
Conclusion
Regarding the transition of dissimilar metals from a solid alloy to the bulk volume of a liquid, the conditions for solution in proportion to the alloy composition will change, if, at the solid/liquid interface, the thermodynamic activity of alloying elements in the liquid reaches the same value as in the solid. As a first approximation, this happens first to the element with the smallest product of solubility in the liquid and activity coefficient in the solid. In the following stage of selective leaching, different mechanisms may apply, with characteristic features of the originating depletion zone, which may be utilized for identifying the mechanism for specific couples of solid alloy and liquid.
The particular mechanism that involves proportionate element transfer to the liquid and reprecipitation of sparingly soluble elements is indicated by a film of liquid that separates the still unaffected solid alloy from the originating depletion zone. At constant temperature, reprecipitation is viable especially for elements for which the alloy is not the thermodynamically most stable modification under the prevailing conditions, such as austenite for Fe at temperature below about 900°C. The implications of proportionate element transfer followed by reprecipitation of alloying elements of low solubility (in the form of ferrite) are clearest in the results from high-resolution microscopy on twin grain boundaries in austenitic steel after exposure to LBE at 450°C.
Selective leaching caused by liquid Sn without influence of the formation of intermetallic phases is to be expected only at temperatures high enough for remarkable lattice or surface diffusion, so that one of the main arguments against disproportionate element transfer to the liquid loses validity. The same mechanism of selective element leaching from austenitic steel as in liquid Pb alloys is not a matter of course, also because Sn becomes a constituent part of the ferrite that forms. The habitus of the latter changes with the conditions for Ni removal. Ni-based alloys are predestined to deplete through the percolation mechanism (700°C), which changes with increasing temperature when lattice and surface diffusion gain in importance (1000°C).
Oxygen dissolved in the liquid supports leaching especially of oxide-forming elements and may even reverse the situation as to the elements preferentially removed from the alloy.
The influence of intermetallic phases on dissimilar metal solution or selective leaching naturally is largest the earlier these intermetallics form. Depletion zones may primarily consist of intermetallics such as for austenitic steel after exposure to liquid tin at 500°C. In heterogeneous parts that are reminiscent of depletion zones observed for direct element transfer to the liquid, intermetallics may be considered to assume the role of the penetrating liquid. Ferrite or another steel phase formed from austenitic steel at 700°C is rich in Cr and Mo rather than Fe, which is the steel element abundant in the intermetallic (FeSn). In the case of Ni-based alloys at 700°C, a prominent influence on the structure of observed depletion zones, especially the heterogeneous part, is evident for liquid tin pre-saturated with Ni. In general, a solid intermetallic instead of liquid inside the depletion zone is likely to reduce degradation of the solid alloy.
References
C. Schroer, O. Wedemeyer, J. Novotny, A. Skrypnik, and J. Konys, Corros. Sci. 84, 113. (2014).
S. Gossé, J. Nucl. Mater. 449, 122. (2014).
E. Yamaki, K. Ginestar, and L. Martinelli, Corros. Sci. 53, 3075. (2011).
W.D. Manly, Corrosion 12, 336t. (1956).
J.R. Weeks, and A.J. Romano, Corrosion 25, 131. (1969).
G. Rosenblatt and J. R. Wilson, in Corrosion by liquid metals: Proceedings of the sessions on corrosion by liquid metals of the 1969 Fall meeting of the Metallurgical Society of AIME, October 13–16, 1969, Philadelphia, Pennsylvania, eds. by J. E. Draley and J. R. Weeks (Plenum Press, New York, London, 1970), p. 469.
L. Martinelli, F. Vanneroy, J. C. Diaz Rosado, D. L’Hermite and M. Tabarant, J. Nucl. Mater. 400, 232 (2010).
R.C. Asher, D. Davies, and S.A. Beetham, Corros. Sci. 17, 545. (1977).
V. Tsisar, C. Schroer, O. Wedemeyer, A. Skrypnik, and J. Konys, J. Nucl. Mater. 454, 332. (2014).
V. Tsisar, C. Schroer, O. Wedemeyer, A. Skrypnik, and J. Konys, J. Nucl. Mater. 468, 305. (2016).
V. Tsisar, C. Schroer, O. Wedemeyer, A. Skrypnik, and J. Konys, J. Nucl. Eng. Radiat. Sci. 4, 41001. (2018).
C. Schroer, V. Tsisar, A. Durand, O. Wedemeyer, A. Skrypnik, and J. Konys, J. Nucl. Eng. Radiat. Sci. 5, 11006. (2019).
P. Hosemann, D. Frazer, E. Stergar, and K. Lambrinou, Scr. Mater. 118, 37. (2016).
O. Klok, K. Lambrinou, S. Gavrilov, E. Stergar, J. Lim, T. van der Donck, W. van Renterghem, and I. de Graeve, J. Nucl. Mater. 510, 556. (2018).
C. Schroer, O. Wedemeyer, J. Novotny, A. Skrypnik, and J. Konys, J. Nucl. Mater. 418, 8. (2011).
K. Lambrinou, E. Charalampopoulou, T. van der Donck, R. Delville, and D. Schryvers, J. Nucl. Mater. 490, 9. (2017).
M.G. Barker, V. Coen, H. Kolbe, J.A. Lees, L. Orecchia, and T. Sample, J. Nucl. Mater. 155–157, 732. (1988).
O. Klok, K. Lambrinou, S. Gavrilov, J. Lim, and I. de Graeve, J. Nucl. Eng. Radiat. Sci. 4, 31019. (2018).
O. Picho, Beständigkeit von Nickel-Chrom-Legierung und Eisenaluminidschichten in sauerstoffhaltigen flüssigen Bleilegierungen, Karlsruher Institut für Technologie (KIT), Karlsruhe, Germany (2014). https://doi.org/10.5445/IR/1000043553.
D. Duc, D. Marchive, D. Treheux, and P. Guiraldenq, J. Cryst. Growth 24–25, 559. (1974).
A. Heinzel, A. Weisenburger, and G. Müller, Mater. Corros. 68, 831. (2017).
T. Emmerich, and C. Schroer, Corros. Sci. 120, 171. (2017).
T. Emmerich, A. Durand, V. Tsisar, and C. Schroer, Mater. Corros. 69, 832. (2018).
F.N. Mazandarany, and R.D. Pehlke, Metall. Trans. 4, 2067. (1973).
Y. Gao, M. Takahashi, and M. Nomura, Mech. Eng. J. 2, 15–00149. (2015).
Q. Chen, and K. Sieradzki, J. Electrochem. Soc. 160, C226. (2013).
I. McCue, B. Gaskey, P.-A. Geslin, A. Karma, and J. Erlebacher, Acta Mat. 115, 10. (2016).
K. Geng, and K. Sieradzki, J. Electrochem. Soc. 164, C330. (2017).
P.-A. Geslin, I. McCue, B. Gaskey, J. Erlebacher, and A. Karma, Nat. Commun. 6, 8887. (2015).
J. Erlebacher, M.J. Aziz, A. Karma, N. Dimitrov, and K. Sieradzski, Nature 410, 450. (2001).
J. Luo, H. Cheng, K.M. Asl, C.J. Kiely, and M.P. Harmer, Science 333, 1730. (2011).
V. Gorshkov, and V. Privman, J. Appl. Phys.122, 204301. (2017).
L.K. Aagesen, A.E. Johnson, J.L. Fife, P.W. Voorhees, M.J. Miksis, S.O. Poulsen, E.M. Lauridsen, F. Marone, and M. Stampanoni, Nat. Phys. 6, 796. (2010).
A.J. Bernoff, A.L. Bertozzi, and T.P. Witelski, J. Stat. Phys. 93, 725. (1998).
L.F. Lin, C.Y. Chao, and D.D. Macdonald, J. Electrochem. Soc. 128, 1194. (1981).
C.Y. Chao, L.F. Lin, and D.D. Macdonald, J. Electrochem. Soc. 128, 1187. (1981).
T. Alden, D.A. Stevenson, and J. Wulff, Trans. Metall. Soc. AIME 212(2), 15. (1958).
Acknowledgements
An early version of this paper was presented on the occasion of TMS2021 Virtual, March 15–18, 2021, and published in TMS2021 150th Annual Meeting and Exhibition Supplemental Proceedings (The Minerals, Metals and Materials Society, Pittsburgh PA: Springer 2021) p. 642–656.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schroer, C. Dissimilar Metal Solution from Solid Alloys as Observed for Steels and Nickel‐Based Alloys in the Presence of Lead‐Based Liquid Alloys or Liquid Tin. JOM 73, 4000–4008 (2021). https://doi.org/10.1007/s11837-021-04948-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11837-021-04948-9