
Abstract
The oxidative stability of mixtures of edible oils containing polyunsaturated fatty acids (PUFA) and microcrystalline cellulose (MCC) was investigated. The mixtures studied consisted of oils of either camelina (CAM), cod liver (CLO), or salmon (SO) mixed with either colloidal or powdered MCC. A 50:50 (w/w) ratio of oil:MCC resulted in an applicable mixture containing high levels of PUFA edible oil and dietary fiber. The oxidative stability of the formulated mixtures and the pure oils was investigated over a period of 28 days. The peroxide value (PV) was assessed as a parameter for primary oxidation products and dynamic headspace gas chromatography mass spectrometry (GC/MS) was used to analyze secondary volatile organic compounds (VOC). CAM and the respective mixtures were oxidatively stable at both 4 and 22 °C during the storage period. The marine oils and the respective mixtures were stable at 4 °C. At 22 °C, an increase in hydroperoxides was found, but no increase in VOC was detected during the time-frame investigated. At 42 °C, prominent increases in PV and VOC were found for all oils and mixtures. Hexanal, a common marker for the degradation of n-6 fatty acids, propanal and 2,4-heptadienal (E,E), common indicators for the degradation of n-3 fatty acids, were among the volatiles detected in the headspace of oils and mixtures. This study showed that a mixture containing a 50:50 ratio of oil:MCC can be obtained by a low-tech procedure that does not induce oxidation when stored at low temperatures during a period of 1 month.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Omega-3 polyunsaturated fatty acids (n-3 PUFA) in human nutrition have drawn considerable interest due to their potential health benefits [1, 2]. In particular, the effects of α-linolenic acid (ALA; C18:3n-3) common in vegetable oils and eicosapentaenoic acid (EPA; C20:5n-3) and docosahexaenoic acid (DHA; C22:6n-3) in marine oils are well documented [3–5]. Still, the consumption of fish is relatively low in many countries and the recommended daily intake difficult to attain, which has created an interest in fortifying food products with n-3 PUFA [6, 7]. However, this implementation is not without problems; the unsaturated nature of these compounds makes them susceptible to oxidation. The hydroperoxides that are primarily formed can degrade further into secondary oxidative products, such as aldehydes, alcohols, and ketones [8]. This deterioration causes quality changes in foods and is of major concern both nutritionally and from a safety viewpoint [9]. In this respect, the enhancement of food quality strategies regarding the control of lipid oxidation is of great importance.
To retard the development of oxidative deterioration in n-3 PUFA oils, antioxidants are added [6]. Strategies such as encapsulation in gelatin and microencapsulation have also been used to lower the oxidation rate. The process of microencapsulation transforms the oils into powdery solids that can be used for the supplementation of food products such as bread, milk products, and infant formulas, among others [6]. However, complete control regarding the oxidation rate might be difficult to foresee due to the high number of variables involved in such heterogeneous systems, e.g., matrix type, drying procedures, lipid distribution, and so forth [10]. Moreover, in microencapsulation, the amount of active ingredient may be low compared to the coating material, leading to difficulties regarding adequate dosing in foods. The encapsulation efficacy of oils are reported to vary between 300 and 400 g/kg considering the oil content [11], but the distribution of oil in the microcapsules might differ. Therefore, techniques yielding higher amounts of the bioactive ingredients and more cost-efficient processing might be more suited for satisfying these ends.
Microcrystalline cellulose (MCC) is a polysaccharide often used as an excipient, serving as a vehicle for active substances [12]. It is derived from cellulose by acid depolymerization of α-cellulose, yielding crystalline bundles which can be further processed. The drying of crystalline bundles result in powdered MCC, yielding aggregates of very porous particles often used as an inert fiber source and zero-calorie bulking agent. Co-processing the crystalline bundles with a soluble hydrocolloid gives colloidal MCC. The hydrocolloid functions as a barrier coating for particles and it facilitates water uptake and dispersion [13]. The application of MCC in the food industry is extensive, as it is inexpensive, non-toxic, and has the ability to act as a carrier, fat replacer, emulsifier, and stabilizer [14]. However, to our knowledge, the potential of MCC as a carrier of n-3 PUFA in foods has not yet been investigated.
The aims of the presented study were to: (1) formulate mixtures containing polyunsaturated edible oils and MCC; (2) investigate the oxidative stability of the mixtures by monitoring the formation of primary and secondary oxidation products at different temperatures during a period of 28 days. Selected oils were salmon and cod liver oil high in EPA and DHA, and camelina oil high in ALA, all of which are relevant alternatives for food fortification.
Materials and Methods
Chemicals and Materials
Isooctane, glacial acetic acid, starch, potassium iodide, sodium, and sodium thiosulfate solution, were obtained from Merck KGaA (Darmstadt, Germany). Sodium dodecyl sulfate (SDS), hexane, and methanol were from Sigma-Aldrich (Steinheim, Germany). Helium and nitrogen gas was supplied from Yara International ASA (Oslo, Norway). All chemicals and gases mentioned above were of analytical grade. Heptanoic acid ethyl ester (>98%) and hexanal (>97%) were obtained from Fluka (Buchs, Switzerland), Food Industry FAME Mix was supplied by Restek (Bellefonte, PA), and nonadecanoic acid methyl ester, C19:0 methyl ester from Larodan AB (Malmö, Sweden).
Crude, cold-pressed oil from camelina seeds (CAM; Camelina sativa) was provided by Bioforsk Øst (Apelsvoll, Norway). TINE EPADHA Oil 1200, refined and deodorized cod liver oil (CLO; Gadus morhua), was supplied by TINE SA (Oslo, Norway). Salmon oil (SO; Salmo salar) refined, deacidificated, bleached, and deodorized was obtained from Nofima Mat AS (Ås, Norway). The tocopherol content determined in the pure oils, CAM, CLO, and SO, amounted to ~800, ~950, and ~200 mg/kg, respectively, as stated by the suppliers. Miglyol 812 (Axo Industry SA, Wavre, Belgium), a blend of saturated medium-chain triglycerides (MCT), was used as a blank The oils were split into smaller aliquots, flushed with nitrogen, and stored at −80 °C until testing.
MCC powders Avicel GP 1030 (coprocessed with approximately 11% carboxymethyl cellulose) and Avicel PH 101 (pure MCC) were obtained from FMC Biopolymer (Philadelphia, PA). Throughout this paper, the notation “-c” (colloid) and “-p” (powdered) has been used, indicating Avicel GP 1030 and PH 101, respectively.
Preparation
The formulation of oil:MCC mixtures was established after several preliminary tests with different concentrations of oil and MCC. The ratios tested were 30:70, 40:60, 50:50, and 60:40 oil:MCC (w/w). Oil was added to the MCC powder to obtain a saturated and relatively dry mixture, with the possibility for usage in food fortification. The mixing procedure consisted of using a hand blender for 5 s, followed by 5 s of stirring with a spatula, before another 5 s with a blender until homogeneous.
Storage
Two grams of the 50:50 ratio of the homogenous mixture were weighed into 20 ml dark brown headspace vials (Agilent Technologies, Waldbronn, Germany). The vials were sealed with atmospheric oxygen, using tight-fitting screw caps, and stored at 4, 22, and 42 °C. Temperatures were chosen to reflect refrigerated storage, ambient, and accelerated conditions during storage. Pure oils were stored under the same conditions as the mixtures and used as references. In addition to these, blank samples were implemented throughout the experiment, consisting of MCT and the MCC powders alone. The combinations were the same as for the samples and they were implemented in the same manner throughout the experiment. Randomly chosen sample vials were analyzed initially and after storage for 3, 7, 14, 21, and 28 days. The storage periods were chosen in accordance with fish oil-enriched food products with short shelf life, which were considered to be the most comparable products in this case.
Characterization
Scanning Electron Microscopy
The surface appearances of MCC and oil:MCC mixtures were examined using a Zeiss EVO-50-EP (Carl Zeiss SMT Ltd., Cambridge, England) scanning electron microscopy (SEM). Samples were adhered to stubs using double-sided tape and sputter coated with gold using a Polaron Sputter Coater SC 7640 (Quorum Technologies Ltd., East Sussex, England), and examined at 5.0 kV.
Fatty Acid Composition
The fatty acid composition of the selected oils was determined as methyl esters analyzed by gas chromatography mass spectrometry (GC/MS). The transesterification of lipids to fatty acid methyl esters (FAME) were performed as described by Devle et al. [15], with minor modifications. Briefly, 200 μg oil was dissolved in 2.0 mL hexane and 1.5 mL of 3.33 mg/mL sodium methanolate solution was added, and placed on a shaker for 30 min, before the samples were left to settle for 10 min. Two hundred microliters of the top layer were transferred to a new vial, and internal standard (IS) nonadecanoic acid methyl ester at 50 μg/mL was added prior to dilution with hexane to a final volume of 1.0 mL. FAME were identified by comparing their retention times to those of a known 37-component standard FAME mix, as well as comparing the mass spectra with spectra available from the National Institute of Standards and Technology (NIST) database. For all samples, four replicates were analyzed.
Separation was performed using an Agilent 6890 Series GC (Agilent Technologies), equipped with a TRACE TR-FAME (50 m × 0.32 mm × 0.25 μm) capillary column (Thermo Fischer Scientific Inc., Waltham, MA). Helium was used as the carrier gas at a constant pressure set to 95 kPa. Injections were made in splitless mode at 250 °C with a volume of 1.0 mL. The purge time was 2 min at a flow rate of 10 mL/min. The temperature program was 50 °C for 5 min, then 10 °C/min to 175 °C with a 15-min hold-time, then 5 °C/min to 240 °C with a 10-min hold-time, and, finally, 90 °C/min to 250 °C followed by a 5-min hold-time. Detection was performed using an Autospec Ultima GC/MS (Micromass, Manchester, England). Electron ionization was set at 70 eV and the mass range was 45–700 m/z with a scan time of 1.0 s and interscan delay of 0.50 s. The ion source temperature was set to 250 °C and the transfer line was held at 250 °C. The software used was MassLynx version 4.0 (Waters, Milford, MA). NIST 08 (NIST, Gaithersburg, MD) was used as a reference library. The performance of the system was verified with hexane as blanks prior, during, and after analysis of the samples.
Stability
The oxidative stability was assessed directly through the lipid degradation parameters. Primary and secondary lipid oxidation products were investigated at six time-points up to almost 1 month at three different temperatures.
Primary Oxidation Products
Primary oxidation products were assessed by means of the peroxide value (PV) using the American Oil Chemists’ Society (AOCS) method Cd 8b-90 [16]. In brief, PV were measured directly on the oils and oil:MCC mixtures dissolved in acetic acid–isooctane solution and determined by iodometric titration. The results are expressed as meq/kg and samples were analyzed in triplicate.
Secondary Oxidation Products
Secondary oxidation was assessed monitoring the volatile compounds (VOC) using dynamic headspace GC/MS, according to Volden et al. [17]. The headspace volatile compounds were isolated using a Teledyne Tekmar HT3™ (Teledyne Tekmar, Mason, OH). In short, the conditions used were a preheat time of 5 min and a helium flow of 50 ml/min for 10 min, trapping the analytes on a 24-cm Tenax® GR 60/80 mesh size (Supelco Analytical, Bellefonte, PA) at 25 °C. Desorption was performed at 280 °C with a gas flow of 75 ml/min for 5 min and the analytes were transferred to the GC via a transfer line at 100 °C. The software version was HT3 Teklink ver. 1.2.1104 (Teledyne Tekmar, Mason, OH).
Separation of the volatile compounds was achieved using an Agilent gas chromatograph 6890 N equipped with a DB-WAXETR (Agilent Technologies) fused silica capillary column (30 m × 0.25 × 0.50 μm). The injector inlet temperature was 250 °C. Helium was used as a carrier gas with a flow rate of 1.0 ml/min. The temperature program was 30 °C for 10 min, then 1.0 °C/min to 40 °C, 3.0 °C/min to 70 ˚C, and 6.5 °C/min to 230 °C, followed by a 5-min hold-time at 230 °C. Detection was obtained using a 5975 Inert XL Mass Selective Detector (Agilent Technologies) in electron ionization mode with the ion source temperature at 200 °C scanning continuously within a mass range of 33–300 m/z. Ionization was done by electronic impact at 70 eV. MSD ChemStation D.02.00.275 software was used. Compounds were identified using the NIST 05 Mass Spectral Library ver. 2.0d (Agilent Technologies), as well as with single-reference compounds. The quantification was in accordance with Olsen et al. [18], with brief modifications. In short, the peak areas of standards were plotted and the response factor for hexanal was used for all analytes, regardless of individual response factors, yielding a semiquantitative approach. Heptanoic acid ethyl ester and hexanal were mixed into MCT and analyzed in the same manner as for the samples. The performance of the system was verified with N2 blanks prior, during, and after analysis of the samples, which were analyzed in triplicate.
Statistical Analysis
The statistical analysis was performed in R ver. 2.12.0, which is a free software environment maintained by the R Development Core Team (http://www.r-project.org/). Analysis of variance (ANOVA) was carried out on PV and total volatile compounds. The statistical models included the main effects of oils and oil:MCC mixtures, storage temperature, and storage time, plus all possible interactions between the main effects (all effects were considered to be fixed). A significance level of 0.05 was used when testing for effects in the ANOVA and in the accompanying Tukey’s honestly significant difference (HSD) test for pairwise differences of means.
Results and Discussion
Oil:MCC Mixture
In order to obtain a mixture that was neither too “oily” nor too “powdery”, and contained as much oil as possible, the ratio 50:50 (w/w) of oil:MCC was found to be the most suitable. Overall, the described process and system is a low-tech approach requiring no heating or drying steps. It is recognized that high temperatures applied during processing can induce oxidation in systems containing PUFA oils [11, 19]. The amount of added oil in the studied mixtures was 50%, which is higher than that reported from microencapsulation studies showing oil contents of around 40% [11]. As a consequence, the studied mixtures would make it easier to provide recommended amounts of n-3 PUFA. In addition, MCC is an excellent source of dietary fiber, which can have important implications in human health [20].
Scanning Electron Microscopy
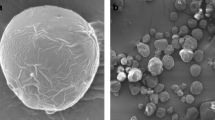
Figure 1 shows the structures of colloidal and powdered MCC and the respective oil:MCC mixtures. The particle sizes of the two MCC powders were slightly different; ~35 μm for colloidal and ~50 μm for powdered MCC, in agreement with Jumaa et al. [21]. In all cases, the oil covered the whole available outer surface area of the particles. The surface of mixtures with camelina oil, CAM-c and CAM-p, respectively, showed a dim appearance on the SEM images, which may be related to the darker yellow color of the camelina oil compared to the marine oils.

Scanning electron microscopy (SEM) images of colloidal (a) and powdered (b) microcrystalline cellulose (MCC). The magnification was 1,000×
Fatty Acid Composition
Table 1 presents the fatty acid composition of the oils investigated. Linoleic acid (18:2n-6) and ALA were the main PUFA found in CAM, whereas EPA and DHA were dominant in CLO and SO. This is in good agreement with previous reports on these oils [17, 18, 22, 23]. The MCT was comprised of the saturated fatty acids C8:0, C10:0, and C12:0 in the relative abundances 49.6 ± 0.1, 49.4 ± 0.1, and 0.7 ± 0.1%, respectively.
Stability
Primary Oxidation Products
Table 2 shows the PV in pure oils and oil:MCC mixtures. The initial PV of the oils were 0.8 ± 0.1 meq/kg for CAM and 1.1 ± 0.1 meq/kg for both CLO and SO, indicating that the fresh pure oils were unoxidized. MCT gave no PV response, which was to be expected, given its content of saturated fatty acids. According to Let et al. [24], the PV can be regarded as an even more important parameter when considering oxidation rates than the actual composition of fatty acids.
The pure oils CAM and SO showed no significant increase in PV at 4 °C, and were considered to be stable at this temperature. For CLO, a significant increase was shown at day 28 (Table 2). At 22 °C, only CAM was found to be stable. For CLO and SO, a significant change was found at day 21 and 7, respectively. Eidhin et al. [22] reported that camelina oil was more stable than fish oil at room temperature, which is in good agreement with the present results. Edible oil consists of mostly triacylglycerols, but also minor components and natural antioxidants such as tocopherols are present. In fresh CAM, the content of total tocopherols was approximately 800 mg/kg, comprising mainly of γ-tocopherol. The relatively high content was probably one of the reasons for the sustained oxidative stability at 4 and 22 °C, since tocopherols function by interrupting the autoxidation chain process.
A similar pattern was found at the accelerated storage temperature of 42 °C (Table 2). CAM was the most stable oil and a significant change in PV was detected first after 2 weeks. For CLO, the PV started to increase 1 week earlier, whereas SO oxidized rapidly with no induction period and had a high PV already at day 3. Abramovič et al. [25] reported that camelina oil stored for 27 days at 50 °C in the dark had a 40% reduction of γ-tocopherol content. This may be one of the reasons for the increase in primary oxidation products in CAM as a result of the chosen storage conditions. Also, the high content of ALA in the oil may influence its resistance to lipid oxidation. The results from SO were in accordance with studies investigating comparable oils at 40 °C [26]. The high content of PUFA like EPA and DHA, and their inherent susceptibility to oxidation, especially at elevated temperatures, were most likely the reasons for the increase in primary oxidation products in CLO and SO [8]. Antioxidants or a cocktail thereof are usually added to improve the oxidative stability of unsaturated oil, by extending the induction period or lowering the oxidation rate [27]. Considering the high levels of added antioxidants in CLO (~950 mg/kg), a prolonged induction period compared to SO (~200 mg/kg) with no exogenous antioxidants added was to be expected.
Considering the oil:MCC mixtures, the PV in CAM-c and CAM-p showed the same development as for the pure oil; i.e., no significant increases at 4 and 22 °C during 28 days of storage (Table 2). At 42 °C, CAM-c and CAM-p showed significantly lower PV than CAM at days 14 and 21, indicating the reduced production of peroxides in the mixtures compared with pure oil. It is assumed that the absorption and adsorption capacity of oil to MCC powder influences the oxidative stability of the oils. As for CLO-c and CLO-p, the development of PV showed the same pattern as for CLO at 4 and 22 °C. The PV was found to be significantly lower in CLO-c and CLO-p at days 21 and 28 at 22 °C and day 14 at 42 °C compared to the pure cod liver oil at the same time-points. At 22 °C SO-c and SO-p showed significantly lower PV than SO from day 7 and throughout the study. Under accelerated storage (i.e., at 42 °C), the development of peroxides, especially in SO-c and SO-p, increased at earlier time-points than for SO. A quality standard on omega-3 products has been proposed by the Council for Responsible Nutrition (CRN), recommending that the PV should be ≤5 meq/kg [25]. This criterion was adopted for the oils and oil:MCC mixtures investigated in this study. In that respect, CAM, CAM-c, and CAM-p stored at 4 and 22 °C were considered to be stable during the chosen time-frame. Mixtures with cod liver oil stored at 4 and 22 °C showed PV up to 5.3 meq/kg after 21 and 28 days for CLO-c and CLO-p at 22 °C, respectively, bordering on the desired threshold. As for the mixtures, CLO was stable at 4 °C, but above the threshold value at 21 days at 22 °C. SO, SO-c, and SO-p were stable at 4 °C, while at 22 °C, the respective mixtures were stable until day 7. SO exceeds the threshold value already at day 7 under ambient conditions. The mixtures containing marine oils, CLO, and SO, respectively, were oxidative-stable when stored at 4 °C during the investigated period. Camelina oil and the respective mixtures showed low PV when stored at 4 and even 22 °C. The storage time could possibly be increased by storing the samples in the absence of oxygen.
Secondary Oxidation Products
Tables 3, 4, and 5 show the development of the lipid-derived volatiles found in the headspace of the oils and oil:MCC mixtures during the 28 days of storage at the three selected temperatures. The main volatile degradation products were aldehydes, ketones, and alcohols. Overall, the aldehydes comprised of 3-methylbutanal, 2-butenal (E), hexanal, 2-hexenal (E), 2-pentenal (E), nonanal, 2-heptenal (Z), 2-octenal, 2,4-heptadienal (E,E), propanal, 2-propenal, butanal, pentanal, heptanal, and octanal. The ketones identified were 2-butanone and 1-penten-3-one. Only one alcohol was found, namely, 1-penten-3-ol. Propanal, 2-propenal, butanal, pentanal, heptanal, and octanal were not found in CAM or its respective MCC mixtures, while 3-methylbutanal was only found in CAM, CAM-c, and CAM-p. The detected volatiles are typical oxidation products for PUFA [8, 28]. Hexanal and pentanal are common markers of n-6 PUFA and their aromas are characterized as green, grassy, and pungent [6]. Hexanal was found in all oils and oil:MCC mixtures, while pentanal was found in the mixtures with CLO and SO. Propanal, 2-pentenal, 1-penten-3-one, 1-penten-3-ol, and 2,4-heptadienal were among the components which characterize the oxidative decomposition n-3 PUFA and have odor characteristics, like pungent, fishy, green, and oily [8, 17, 28, 29]. The oxidizability of fatty acids are related to the number of bis-allylic positions available [8], therefore, ALA, EPA, and DHA were expected to be precursors to the major volatile oxidation products found in CAM, CLO, and SO.
At day 0, the total volatile contents were 0.62 ± 0.03, 0.52 ± 0.03, and 0.54 ± 0.01 mg/g for CAM, CAM-c, and CAM-p, respectively, and 0.24 ± 0.01, 0.14 ± 0.01, and 0.15 ± 0.02 mg/g for CLO, CLO-c, and CLO-p, respectively. No volatile compounds were detected from salmon oil initially. For both CAM and CLO, the levels were significantly higher than the levels of the respective mixtures at day 0, indicating that the availability of VOC decreases in the presence of MCC. Low initial values of volatile compounds in CLO and SO are most likely related to the deodorization step applied in the refining process [6]. For SO, additional steps, including bleaching and de-acidification, were applied for the removal of impurities, including pro-oxidants, which may help explain the low content of VOC at 4 and 22 °C during the investigated time-frame. Deodorization removes volatile substances in order to obtain a product bland in flavor and odor. This was not applied to camelina oil and may explain the higher initial VOC content in comparison to the marine oils.
In Table 3, no significant increase (p < 0.05) in the total VOC was found in CAM, CAM-c, and CAM-p at 4 and 22 °C during 28 days of storage. This was in accordance with the PV for CAM at the same time and temperatures (Table 2). Lipid hydroperoxides have been reported to be relatively stable at room temperature [28] and are, therefore, not readily decomposed to volatile compounds in the absence of metals or elevated temperatures. However, under the accelerated storage conditions (i.e., 42 °C), an increase in the total VOC was found at day 28 for CAM. The emission of total volatiles was found to be higher in CAM compared with CAM-c and CAM-p at day 28. The total volatile alcohol content was higher in CAM at day 28 compared to CAM-c and CAM-p, where the aldehydes accounted for the highest contribution to the total VOC.
Figure 2 shows a more detailed description of the most prominent volatile compounds found in CAM, CAM-c, and CAM-p at 42 °C storage and their influence on the total VOC. Hexanal, 2-pentenal (E), and 1-penten-3-ol dominated the headspace volatiles for CAM, CAM-c, and CAM-p, and had a pronounced influence on the high level of total aldehydes and alcohols found at days 21 and 28.

Development of selected volatile organic compounds (VOC) from CAM (a), CAM-c (b), and CAM-p (c) stored at 42 °C for 28 days. The error bars indicate standard deviation
Table 4 shows the total volatile content as well as the contribution from aldehydes, alcohols, and ketones found in CLO, CLO-c, and CLO-p at the selected time-points and temperatures. As for PV in Table 2, CLO and the respective mixtures were shown to be stable at 4 °C during the storage time. An increase in PV was found for CLO, CLO-c, and CLO-p during storage at 22 °C for 28 days (Table 2). By contrast, no increase was found in the total VOC under the same storage conditions and time-points. At 42 °C, trends from PV that showed a significant increase from day 7 were reflected with an increase in total volatiles at day 14 for CLO and CLO-c. CLO-p showed an increase at day 7. The aldehydes accounted for a higher contribution to the total VOC levels than the alcohols and ketones at 42 °C. In Fig. 3, the contributions from some of the most prominent single VOC found in CLO, CLO-c, and CLO-p are shown. The most abundant compounds were typical degradation products from n-3 PUFA, namely, propanal and 1-penten-3-ol. 1-penten-3-ol was especially abundant regarding the high alcohol content found in the headspace of CLO-p at day 28 and, hence, was a major contributor to the high total VOC at the same time-point.

Development of selected VOC from CLO (a), CLO-c (b), and CLO-p (c) stored at 42 °C for 28 days. The error bars indicate standard deviation
In Table 5, the total volatile compounds found in SO, SO-c, and SO-p during storage are shown. At 4 °C, no increase in the total aldehyde, alcohol, and ketones were found, reflecting the stability observed for the PV, as shown in Table 2. Volatile compounds were expected to be more abundant at 22 °C, due to the large increase in PV throughout the storage interval. Significant increases on day 7 at 42 °C were found for SO and its MCC mixtures, which is in accordance with the high PV values detected from day 3 (Table 2). The effect of antioxidant addition to highly unsaturated fish oils were clearly shown by comparing the induction period in PV and volatile compounds for CLO and SO and the respective mixtures at 42 °C; the oxidation of SO, SO-c, and SO-p was initiated and reached a high production rate of primary and secondary products at an earlier time-point than that of CLO, CLO-c and CLO-p. Propanal, and 1-penten-3-ol were the most prominent single VOC detected in SO and the mixtures (Fig. 4), accountable for some of the contribution to the high concentrations of total VOC, especially alcohols in SO-p (Fig. 4).

Development of selected VOC from SO (a), SO-c (b), and SO-p (c) stored at 42 °C for 28 days. The error bars indicate standard deviation
Conclusion
This study demonstrated that it was fully feasible to mix polyunsaturated edible oil and microcrystalline cellulose (MCC). A mixture containing 50% oil can be obtained using a method that was simple, cost-efficient, and required no heating or drying steps. The stability investigated over a period of 28 days indicated that the mixtures might have an expedient potential if stored at low temperatures, which can form an appropriate basis for a commercial product.
References
World Health Organization/Food and Agriculture Organization of the United Nations (WHO/FAO) (2009) Fats and fatty acids in human nutrition. Report from a Joint WHO/FAO Expert Consulation. Ann Nutr Metab 55
International Society for the Study of Fatty Acids and Lipids (ISSFAL) ISSFAL Board Statement No.3. http://www.issfal.org.uk. Accessed October 2010
de Lorgeril M, Renaud S, Mamelle N, Salen P, Martin JL, Monjaud I, Guidollet J, Touboul P, Delaye J (1994) Mediterranean alpha-linolenic acid-rich diet in secondary prevention of coronary heart disease. Lancet 343:1454–1459
Calder PC (2006) n-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am J Clin Nutr 83:1505S–1519S
Zatonski W, Campos H, Willett W (2008) Rapid declines in coronary heart disease mortality in Eastern Europe are associated with increased consumption of oils rich in alpha-linolenic acid. Eur J Epidemiol 23:3–10
Kolanowski W, Laufenberg G (2006) Enrichment of food products with polyunsaturated fatty acids by fish oil addition. Eur Food Res Technol 222:472–477
Jacobsen C, Let MB, Nielsen NS, Meyer AS (2008) Antioxidant strategies for preventing oxidative flavour deterioration of foods enriched with n-3 polyunsaturated lipids: a comparative evaluation. Trends Food Sci Technol 19:76–93
Frankel EN (2005) Lipid oxidation, 2nd edn. The Oily Press, Bridgwater, England
Eriksson CE (1987) Oxidation of lipids in food systems. In: Chan HWS (ed) Autooxidation of unsaturated lipids. Academic Press, New York, pp 207–231
Velasco J, Dobarganes C, Márquez-Ruiz G (2003) Variables affecting lipid oxidation in dried microencapsulated oils. Grasas Aceites 54:304–314
Kolanowski W, Laufenberg G, Kunz B (2004) Fish oil stabilisation by microencapsulation with modified cellulose. Int J Food Sci Nutr 55:333–343
Fechner PM, Wartewig S, Füting M, Heilmann A, Neubert RHH, Kleinebudde P (2003) Properties of microcrystalline cellulose and powder cellulose after extrusion/spheronization as studied by Fourier transform Raman spectroscopy and environmental scanning electron microscopy. AAPS PharmSci 5:77–89
Tuason DC, Krawczyk GR, Buliga G (2010) Microcrystalline cellulose. In: Imeson A (ed) Food stabilisers, thickeners and gelling agents. Blackwell Publishing Ltd., Oxford, pp 218–238
Humphreys WM (1996) Fiber-based fat mimetics: microcrystalline cellulose. In Roller S, Jones SA (eds) Handbook of fat replacers. CRC Press, Boca Raton, FL, pp 131–144
Devle H, Rukke EO, Naess-Andresen CF, Ekeberg D (2009) A GC-magnetic sector MS method for identification and quantification of fatty acids in ewe milk by different acquisition modes. J Sep Sci 32:3738–3745
American Oil Chemists’ Society (AOCS) (2009) Official methods and recommended practices of the American Oil Chemists’ Society. Method Cd 8b–90. AOCS Press, Champaign, IL
Volden J, Bjelanovic M, Vogt G, Slinde E, Skaugen M, Nordvi B, Egelandsdal B (2011) Oxidation progress in an emulsion made from metmyoglobin and different triacylglycerols. Food Chem (in press). doi:10.1016/j.foodchem.2011.03.106
Olsen E, Vogt G, Saarem K, Greibrokk T, Nilsson A (2005) Autoxidation of cod liver oil with tocopherol and ascorbyl palmitate. J Am Oil Chem Soc 82:97–103
Jacobsen C (2010) Challenges when developing omega-3 enriched foods. Ol., Corps Gras, Lipides 17:251–258
Marlett JA, McBurney MI, Slavin JL (2002) Position of the American Dietetic Association: health implications of dietary fiber. J Am Diet Assoc 102:993–1000
Jumaa M, El Saleh F, Hassan I, Müller BW, Kleinebudde P (2000) Influence of cellulose type on the properties of extruded pellets. Part I. Physicochemical characterisation of the cellulose types after homogenisation. Colloid Polym Sci 278:597–607
Eidhin DN, Burke J, O’Beirne D (2003) Oxidative stability of omega3-rich camelina oil and camelina oil-based spread compared with plant and fish oils and sunflower spread. J Food Sci 68:345–353
Giogios I, Grigorakis K, Nengas I, Papasolomontos S, Papaioannou N, Alexis MN (2009) Fatty acid composition and volatile compounds of selected marine oils and meals. J Sci Food Agric 89:88–100
Let MB, Jacobsen C, Frankel EN, Meyer AS (2003) Oxidative flavour deterioration of fish oil enriched milk. Eur J Lipid Sci Technol 105:518–528
Abramovič H, Butinar B, Nikolič V (2007) Changes occurring in phenolic content, tocopherol composition and oxidative stability of Camelina sativa oil during storage. Food Chem 104:903–909
Frankel EN (1993) Formation of headspace volatiles by thermal decomposition of oxidized fish oils vs. oxidized vegetable oils. J Am Oil Chem Soc 70:767–772
Council for Responsible Nutrition (CRN) (2002) CRN proposed monograph on omega-3 EPA and DHA: explanatory notes from Sam Zeller, Chairman of the Technical Committee. March 2002. CRN, Washington, DC
Belitz H-D, Grosch W, Schieberle P, Burghagen MM (2004) Food chemistry, 3rd edn. Springer, Berlin, Heidelberg, New York, pp 191–232
Lee H, Kizito SA, Weese SJ, Craig-Schmidt MC, Lee Y, Wei C-I, An H (2003) Analysis of headspace volatile and oxidized volatile compounds in DHA-enriched fish oil on accelerated oxidative storage. J Food Sci 68:2169–2177
Acknowledgments
Hanne Devle, Dag Ekeberg, and Kari Olsen at the Department of Chemistry, Biotechnology and Food Science at the Norwegian University of Life Sciences (UMB) are thanked for their invaluable assistance. The technical assistance of Elin Ørmen at the Department of Plant and Environmental Sciences at the UMB is also greatly appreciated. Aina Lundon, Berit Nordvi, and Gjermund Vogt at Bioforsk Øst Apelsvoll, TINE SA, and Nofima Mat AS are thanked for their cooperation. Financial support was given by the Research Council of Norway (Project no. NFR184846/I10).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Saga, L.C., Rukke, EO., Liland, K.H. et al. Oxidative Stability of Polyunsaturated Edible Oils Mixed With Microcrystalline Cellulose. J Am Oil Chem Soc 88, 1883–1895 (2011). https://doi.org/10.1007/s11746-011-1865-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11746-011-1865-1