
Abstract
Sepsis is among the most important causes of mortality, particularly within the elderly population. Sepsis prevalence is on the rise due to different factors, including increasing average population age and the concomitant rise in the prevalence of frailty and chronic morbidities. Recent investigations have unveiled a "trimodal" trajectory for sepsis-related mortality, with the ultimate zenith occurring from 60 to 90 days until several years after the original insult. This prolonged temporal course ostensibly emanates from the sustained perturbation of immune responses, persevering beyond the phase of clinical convalescence. This phenomenon is particularly associated with the aging immune system, characterized by a broad dysregulation commonly known as "inflammaging." Inflammaging associates with a chronic low-grade activation of the innate immune system preventing an appropriate response to infective agents. Notably, during the initial phases of sepsis, neutrophils—essential in combating pathogens—may exhibit compromised activity. Paradoxically, an overly zealous neutrophilic reaction has been observed to underlie multi-organ dysfunction during the later stages of sepsis. Given this scenario, discovering treatments that can enhance neutrophil activity during the early phases of sepsis while curbing their overactivity in the later phases could prove beneficial in fighting pathogens and reducing the detrimental effects caused by an overactive immune system. This narrative review delves into the potential key role of neutrophils in the pathological process of sepsis, focusing on how the aging process impacts their functions, and highlighting possible targets for developing immune-modulatory therapies. Additionally, the review includes tables that outline the principal potential targets for immunomodulating agents.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sepsis is currently defined as a life-threatening organ dysfunction caused by a dysregulated host response to infection, with organ dysfunction defined by Sequential Organ Failure Assessment (SOFA) score ≥ 2 [1]. This new definition marks a deep line from the traditional association of infection and inflammation, dismissing the old terms "systemic inflammatory response syndrome" and "severe sepsis". Septic shock represents a specific subset of sepsis, clinically characterized by the requirement of vasopressor therapy and serum lactate concentration exceeding 2 mmol/L. This condition is associated with high mortality rates, reaching up to 40% [1].
Previously, sepsis-related mortality was described as having a biphasic pattern, with an early peak attributed to inadequate fluid resuscitation and a late peak caused by persistent organ injury or failure. However, recent evidence has presented a shift toward a triphasic pattern for sepsis-related mortality [2]. Notably, advances in intensive care protocols, coupled with the increasing average lifespan, have likely reduced the impact of the early peak and brought attention to the emergence of a third peak. This third peak occurs from 60 to 90 days and can persist for several years after the onset of sepsis, as illustrated in Fig. 1.

Sepsis mortality rates. Sepsis-related mortality was reported to have a biphasic pattern, characterized by an early first peak due to inadequate antibiotic therapy and fluid challenge and a second peak (from day 1 to first weeks) due to multi-organ failure. In the last years, thanks to the improvement of standard of care, especially in the intensive care unit department, the mortality rate showed a triphasic pattern. The third peak, starting from 60 to 90 days to several years, is more typical in a population of elderly subjects with the burden of multiple comorbidity
The worldwide population of individuals aged 60 years or older is consistently increasing. In the late 1950s, this age group accounted for approximately 5% of the total population, but it is projected to rise significantly, reaching up to 21% by the year 2050 [3]. Elderly patients represent a significant portion of the deaths occurring during the third peak as they face a high risk of persistent, recurrent, secondary, or nosocomial infections. Among different pathophysiological changes occurring with age, a large number of predisposing factors (such as polytherapy and malnutrition), together with the surge of chronic low-grade systemic inflammatory response—the so-called “inflamm-aging”—deeply impact on the physiological response to microbial infections with the final effect of reducing survival rates [4,5,6].
The aging of the immune system, known as immuno-senescence, is considered a critical factor contributing to inflamm-aging. This process is believed to be responsible for the increased susceptibility of the elderly to infections and potentially to autoimmune diseases and cancer [7]. Although the precise pathophysiological mechanisms are not yet fully clarified, immunosenescence ultimately leads to dysregulation of immune functionality, favoring a chronic, low-grade, and sterile activation of innate immune cells. Consequently, the adaptive immune response becomes compromised, and the immune system struggles to respond effectively to appropriate immunogenic stimuli [8].
Given the critical role of innate immune cells—in particular, neutrophils—as the first line of defense against pathogens and their connection to the development of inflamm-aging, this review article aims to provide a comprehensive overview of the role of these cells in the pathophysiology of sepsis in the elderly population but also sheds light on potential targets for the development of immune-modulatory therapies.
A shared common pathway between aging, chronic diseases, and sepsis
While the classic definition of the elderly population used to be set at 65 years old, more recent studies have shifted this age threshold to 75 years. The clinical management of elderly individuals during sepsis is more complex due to limited evidence (as most trials include younger subjects to avoid confounders), blurred symptoms, poly-therapy, and presence of comorbidities.
Aging, chronic diseases, and acute infections all share a common underlying mechanism: an abnormal and sustained inflammatory response that impairs the normal immune function. The elderly often experience a chronic low-grade inflammatory state, impacting their immune response and leading to constant hyper-stimulation. This persistent over-stimulation induces changes in immune cell behavior and differentiation, potentially explaining the progression and development of numerous chronic diseases and heightened susceptibility to infectious complications, particularly from opportunistic bacteria and fungi [9].
Also, this immune dysregulation contributes to decreased quality of life, increased hospital readmission rates, higher recurrence of infections, and poor survival outcomes [10]. On the other hand, sepsis can be regarded as an accelerator of immuno-senescence, the process of immune system aging, which promotes the development of chronic inflammatory diseases.
Apart from immuno-senescence, several other mechanisms have been identified as potential contributors to the onset of inflammaging. These mechanisms include increased production and reduced disposal of misfolded and misplaced proteins, epigenetic modifications, metabolic alterations, dysbiosis (imbalance in the gut microbiota), mitochondrial dysfunction, oxidative stress, and genetic predisposition. By highlighting these factors and their interconnectedness, we gain a deeper understanding of the complex dynamics between aging, inflammation, and sepsis in the elderly population. This knowledge can pave the way for better-targeted interventions and improved management strategies for sepsis in older adults.
A distinct time course of the immune system response is observed between young and elderly individuals [6]. While this phenomenon is often considered a para-physiological process, it can have detrimental effects in the elderly. During the initial pro-inflammatory response against an infection, if the pathogen is promptly eliminated, the immune system's homeostasis is quickly restored. However, in the case of sepsis or septic shock, especially in elderly patients, the dysfunctional immune system mounts a response that extends beyond pathogen eradication and can persist for months or even years, altering the immune system homeostasis. Therefore, the balance between immunosuppression and sustained inflammation may differentiate the course of infection, both at short and long term and is crucial to prevent the immune response from becoming counterproductive [5].
The similarities between immunosenescence in elderly patients and the immunosuppression induced by sepsis suggest common pathways that act synergistically. Nevertheless, a comprehensive theory that unifies these mechanisms is still lacking, and this gap contributes to the absence of specific targeted therapies for sepsis in older adults. In summary, understanding the differential immune responses in young and elderly individuals during sepsis is crucial for developing effective and tailored therapeutic strategies.
How does immune function change in the elderly?
In younger adults, the innate immune system mounts a well-coordinated and controlled response to infections, effectively clearing the invading pathogens while minimizing host tissue damage. However, sepsis disrupts this immune response by altering the functionality and quality of effector cells [11].
As we age, the dysregulation of the innate immune system becomes more pronounced, leading to persistent inflammation and a range of impairments in immune cell function. This includes reduced cytokine production in response to appropriate antigenic stimuli, contracted antigen presentation, decreased phagocytosis and chemotaxis, altered reactive oxygen species (ROS) production, chronic catabolism, and defective cellular apoptosis. Among the many affected cell populations are polymorphonuclear neutrophils (PMN) (as summarized in Table 1). Despite the preservation of immune cell counts in the elderly even at extreme ages, these cells undergo age-dependent functional impairments [12].
Usually, in course of sepsis, the host's response to infection is initiated by the systemic activation of the innate immune system mainly via the interaction between pathogen-associated molecular patterns (PAMPs) and pattern recognition receptors (PRRs). Additionally, the release of endogenous damage-associated molecular patterns (DAMPs), traditionally associated with sterile inflammation, appears to have an emerging role in sepsis by perpetuating inflammatory signals and delaying damage resolution. Of interest, in the context of sepsis, DAMPs can be released by death cells but also actively by activated cells. PAMPs and DAMPs are then recognized by (pattern recognition receptors) PRRs resulting in numerous downstream effects designed to promote the host's immune response to the newly identified pathogen and resulting in a severe and persistent inflammatory response through the release of IL-1, TNF, IL-6, and IL-17 [14]. Although the cytokine storm is essential during the early phase of the host response against pathogens, these cytokines also hold the potential to cause extensive tissue damage when not properly regulated. Consequently, the cytokine storm is associated with the first peak of early mortality in sepsis [2].
In the elderly, the first phase of sepsis is typically altered both qualitatively and quantitatively. The release of pro-inflammatory cytokines lasts longer and is increased, favoring a higher degree of tissue damage [11, 14].
Subsequently, or even simultaneously, an anti-inflammatory compensatory state begins, involving immunomodulatory cytokines, such as IL-10, IL-4, and transforming growth factor beta-β [15]. In younger patients, this response results in reduced inflammatory and cellular activation, facilitating the resolution of the inflammatory response. However, in the elderly, this immunomodulatory response is defective and skewed toward chronic low levels of inflammation. Recent research suggests that the two phases likely overlap, and the strict distinction between pro- and anti-inflammatory cytokines may be too simplistic, as many cytokines exert opposite functions depending on the microenvironment [11].
As individuals age, innate immune cells show defective antigen-presenting functions, and macrophages exhibit reduced expression of Human Leukocyte Antigen—DR isotype (HLA-DR), leading to a blunted stimulation of major histocompatibility complex class II [16]. Further, the expression of toll-like receptors on various immune cells is decreased in the aged population, rendering them more susceptible to microbial pathogens. Furthermore, innate immune cells move their metabolism from oxidative phosphorylation toward an increase in aerobic glycolysis and consequent lactate production [17]. As a result, detecting high serum levels of lactic acid becomes a strong indicator of worse outcomes.
One major consequence of the hyper-inflammatory state is the generation of free radicals, produced by the activation of neutrophils, macrophages, and endothelial cells in response to pathogen contact or indirectly via cytokine-mediated activation. However, in the elderly, the production of ROS is impaired, resulting in an ineffective oxidative burst and reduced bactericidal capacity of PMN cells [18]. Concomitantly, this alteration disrupts the homeostatic balance and leads to lipid peroxidation and damage to DNA, proteins, and mitochondria [19].
Another impaired mechanism associated with aging is apoptosis, a crucial process in maintaining the balance between survival and death of damaged cells. Increasing evidence suggests that apoptosis is widely involved in immunosenescence. Accordingly, TNF-α and the typical sepsis-related cellular damage can enhance the apoptotic process, leading to alterations in T cell populations, increased risk of autoimmunity, and accumulation of effector/memory cells. Recent evidence indicates that the long-life, constant stimulation of the immune system and the oxidative burst can modify the behavior of apoptotic lymphocytes and antigen-presenting cells [20].
Lastly, in recent years, the role of inflammasomes has emerged as a critical signaling pathway in the innate immune system and the progression of inflammation. Inflammasomes are assembled upon PRR activation. Specifically, five protein receptors, including members of the nucleotide-binding oligomerization domain (NOD), leucine-rich repeat (LRR)-containing protein (NLR) family—particularly the pyrin domain-containing 1–3 and CARD domain-containing 4 members (NLRP1, NLRP3, and NLRC4)—as well as the proteins absent in melanoma 2 (AIM2) and pyrin, have been identified with the capability to assemble inflammasomes. PRR activation generates a complex that converts pro-inflammatory caspases into their mature form, inducing a significant release of pro-inflammatory cytokines. Notably, inflammasome activation of the active protease Caspase-1 triggers a downstream response, resulting in the release of IL-1β and IL-18. Additionally, activated Caspase-1 cleaves Gasdermin-D inducing pore formation on the membrane. Such mechanism facilitates cytokines release and can eventually lead to pyroptosis, a form of programmed cell death [21].
A study conducted in vitro and in vivo demonstrated that neutrophils, conventionally viewed as the target of IL-1β, can activate the NLRC4 inflammasome during acute Salmonella infection [22]. Here, neutrophils emerge as major producers of IL-1β in vivo, with distinctive resistance to undergo pyroptosis. This enables neutrophils to support IL-1β production at the infection site without compromising their essential antimicrobial function, which would be lost if they rapidly underwent cellular lysis after Caspase-1 activation. Accordingly, after infection, resident macrophages react to infection by releasing a swift burst of IL-1β within the initial hour, whereas neutrophils might maintain IL-1β levels in the subsequent hours [22].
A growing body of evidence indicate that in the elderly, the NLRP3 inflammasome is overactivated. Studies on elderly patients infected with SARS-CoV-2 report increased basal levels of pro-inflammatory cytokines in pulmonary-resident aged macrophages compared to younger patients, leading to an accumulation of oxidative damage and mitochondrial dysfunction [23]. Yet, specific data on neutrophil inflammasome function in the elderly are missing. Experimentally, NLRP3 inhibition enhances overall health, mitigating multiple age-related changes associated with inflammaging. Mice with defective NLRP3 function exhibit better glycemic control, improved cognitive function, [24] decreased autophagy in neutrophils, increased phagocytosis, clearance of bacteria, and improved survival [25]. These findings have been replicated in animal models during sepsis with associated lung damage, [26] and acute hepatic damage, [27] demonstrating reduced cytokine levels, decreased neutrophilic infiltrate, and attenuated macrophage pyroptosis.
The aged neutrophils
Neutrophils, which were traditionally considered a homogeneous population, actually consist of different phenotypes with distinct functions [28]. These various sub-populations of neutrophils likely originate from a common progenitor, but it's not yet clear whether they differentiate centrally in the bone marrow or whether they do this when in peripheral tissues. Each phenotype appears to have peculiar functions and behaves differently at different stages of the inflammatory process.
For example, one subtype of neutrophils leads to a "pro-angiogenic" response, promoting the formation of new blood vessels and aiding in the clearance of infection by generating high concentrations of matrix metalloproteinase 9, which helps remodel the extracellular matrix to facilitate tissue recovery [29]. Besides, other neutrophil subtypes appear to modulate T cells, [30] other are involved in tumor surveillance, [28] or have the ability to transmigrate from inflammatory tissues back into the bloodstream and return to the marginated pool. In homeostasis conditions, two subpopulations of neutrophils have been disclosed, although their precise functions are still unknown: cluster of differentiation (CD) 177 + neutrophils appear to be involved in anti-neutrophil cytoplasmic antibody-associated vasculitis, [31] while neutrophils expressing Olfactomedin-4, a candidate neutrophil subset marker, are associated with an increased risk of death in patients with septic shock, [32] although no substantial differences have been observed between neutrophils expressing Olfactomedin-4 and those that do not.
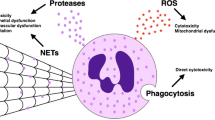
The aging of both cells and hosts is closely linked, and host aging leads to specific changes in neutrophil expression and functions. While PMNs do not divide, their senescence may lead to a process of phenotypic change with altered cytokine secretion, degranulation, cell migration, inflammation, apoptosis, and phagocytosis, making them more prone to inflammation and detrimental effects. This has been linked to organ dysfunction, diseases, and poor host outcomes. A comparison between young and aged neutrophils is illustrated in Fig. 2.

Comparison between the young and aged neutrophil. Neutrophils play a crucial role in the immune system's response to infections. In young neutrophil, there is an efficient antimicrobial activity against pathogens. In contrast, in the aged neutrophil, the immunological response might be impaired favoring several detrimental effects. The impact of aging on neutrophils can be significant and could be influenced by various factors, including overall health, genetics, and lifestyle. Understanding the changes in neutrophil function with aging is crucial for developing strategies to support immune function and mitigate age-related immuno-senescence. BCL-xl, B cell lymphoma extra large; CXCR-4, C-X-C chemokine receptor type 4; DNA, Deoxyribonucleic acid, Mcl-1, myeloid cell leukemia 1; ICAM-1, intercellular adhesion molecule 1; NET, neutrophil extracellular trap; PI3K, phosphoinositide 3-kinase ROS, reactive oxygen species
The aging of the host causes functional deficits in neutrophils that are similar to those induced by sepsis. With age, there are changes in the expression of surface markers on neutrophils, such as impaired expression of C-X-C Motif Chemokine Receptor (CXCR)-2 and CXCR4. Usually, during sepsis, neutrophils express low levels of CXCR4 to promote their redistribution toward infection sites [33]. However, in the elderly, the expression of CXCR4 increases on circulating neutrophils, directing them toward stromal-derived-factor-1-rich environments such as the bone marrow, a peculiar mechanism known as "reverse" transmigration [34].
Moreover, in aged mice, hematopoietic stem cells show reduced regenerative potential, and the proliferative responsiveness of immature neutrophils to granulocyte-colony-stimulating factor is altered with aging [35]. Despite this defective regenerative ability, the number of circulating neutrophils remains similar throughout life, indicating that the impairments in PMN function are mainly qualitative rather than quantitative (Fig. 3).

Take-home message: A shared common pathway between sepsis, aging, and inflammation. In the elderly, the chronic low-level systemic inflammation referred to as "inflammaging" is potentially linked to the development of pathological conditions such as sepsis or age-related diseases. These stimulations can disrupt the delicate equilibrium of inflammatory burden, giving rise to an elevated susceptibility to persistent, recurring, secondary, and nosocomial infections. Consequently, this heightened vulnerability contributes to amplified rates of hospital re-admissions and mortality, while concurrently diminishing the overall quality of life
Indeed, accuracy of neutrophil adhesion and diapedesis mechanisms appear affected by aging. Adhesion molecules, like selectins (e.g., L-selectin) and integrins (e.g., CD11b/CD18), play a crucial role in neutrophil trafficking and recruitment to sites of inflammation. With aging, there can be variations in the expression levels and functionality of these adhesion molecules on neutrophils, which can influence their adhesion capacity and migration, potentially impacting their ability to reach infection sites.
Reduced neutrophil chemotaxis, or their ability to migrate toward inflammatory sites, has been linked to decreased expression of intercellular adhesion molecule 1 (ICAM-1). During inflammation, endothelial cells upregulate ICAM-1 expression in response to pro-inflammatory cytokines and chemokines. ICAM-1 interacts with β2 integrins (such as lymphocyte function antigen-1 and macrophage-1 antigen) expressed on neutrophils surface, facilitating their adhesion to the endothelium and subsequent transmigration into the inflamed tissue. However, in some animal studies, aging has been associated with a decline in ICAM-1 expression on endothelial cells, particularly in tissues prone to age-related pathologies. This reduction in ICAM-1 expression can impair neutrophil adhesion and subsequent transmigration across the endothelium, leading to decreased neutrophil infiltration at the site of inflammation [36].
After extravasation, following the chemo-attractive gradient, neutrophils release ROS and proteinase in order to create a path through the extracellular matrix. However, the accuracy of migration through the inflammation site is reduced in older individuals, likely due to excessive activity of phosphoinositide 3-kinases (PI3K). In response to chemotactic signals, such as chemokines or other chemoattractants, PI3K is activated and generates phosphatidylinositol (3,4,5)-trisphosphate (PIP3) from phosphatidylinositol (4,5)-bisphosphate, molecules predisposed to recruits and activates downstream effectors. An excessive PI3K activity can impact the neutrophil’s migration accuracy. Actually, a neutrophil’s overactivity is the result of elevated PIP3 levels, altering the distribution and timing of signaling events within the migrating neutrophils, leading to misdirected or prolonged migration paths [37]. Furthermore, PI3K signaling influences actin polymerization, cell polarization, and cytoskeletal rearrangements necessary for cell migration. Excessive PI3K activity can disturb these processes, resulting in impaired cell motility and reduced migration accuracy. PI3K can also interact with other signaling pathways involved in neutrophil migration, such as G protein-coupled receptors, and dysregulated PI3K activity can disrupt the balance of crosstalk between different signaling pathways, affecting the migratory response to specific chemo-attractants. The dysregulated PI3K activity and subsequent impairment of neutrophil migration accuracy observed in aging may contribute to decreased immune surveillance, delayed wound healing, and impaired resolution of inflammation [37].
Indeed, during the initial response of the immune system to pathogens, neutrophils may show reduced migration accuracy, which can impact their ability to effectively reach the site of infection. However, even when they arrive at the infection site, the resolution of inflammation can become prolonged due to several factors. One contributing factor is the defective discharge of neutrophils from the inflamed tissue, which can disrupt proper neutrophil trafficking. Neutrophils that have migrated into the site of inflammation need to be cleared or exit the tissue to prevent excessive accumulation and tissue damage. Delayed apoptosis, altered chemokine gradients, and changes in the extracellular matrix composition can all impact neutrophil egress. Indeed, altered expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases can affect the degradation and turnover of extracellular matrix components, thereby impairing neutrophil egress. However, it's worth noting that the mechanisms governing these processes may not be uniform, as studies examining lung endothelia have reported contradictory findings, with ICAM-1 expression sometimes being more pronounced in the elderly, which can favor the lack of resolution of the inflammatory response [38]. During different phases of inflammation, same markers may be overexpressed while others reduced. This highlights the complexity of the aging immune system and the need to consider timing and context when studying immune modulation.
Another well-documented phenomenon in the elderly is the decreased phagocytosis of opsonized bacteria, which can be influenced by several factors altered with aging, including impaired receptor expression, such as Fc receptors and complement receptors [39]. Furthermore, dysregulated signaling pathways involving protein kinases can also impair the coordination and execution of phagocytic processes [40]. Also, increased oxidative stress and ROS production in aged neutrophils can further disrupt phagocytic machinery, leading to impaired phagocytosis and decreased intracellular killing capacity [18].
Interestingly, an increase in granule mobilization, as evidenced by the over-regulation of CD63, a primary granule marker, has been reported [37]. This suggests that certain aspects of neutrophil function may be enhanced in the elderly, while others are compromised, resulting in a complex and multifaceted effect on the immune response.
A relatively recent discovery concerns the ability of neutrophils to extrude into the extracellular environment a network of chromatin fibers and histones coated by granular proteins, such as lactoferrin, elastases, proteinase-3, and myeloperoxidase, forming structures called (NETs). The alteration of NETosis induced by aging has been poorly studied, and the results are controversial. While some studies have identified reduced NET formation by neutrophils from older donors, the current understanding suggests an increase in NET production in elderly subjects [41]. However, although the overall production of NETs is higher in elderly individuals, functionality and effectiveness of these NETs may be compromised due to alterations in their composition and impaired clearance mechanisms.
In aging, neutrophils' production of ROS becomes dysregulated, even in the absence of infection or stimuli. This increased ROS production can trigger spontaneous NET release, contributing to the elevated basal levels of NET formation in aged individuals [42]. Such a phenomenon is probably related to the chronic low-grade inflammation and altered neutrophil activation observed in older individuals. Consequently, the increased release of NETs in the absence of infection or appropriate stimuli can lead to prolonged inflammation and tissue damage, forming a vicious cycle. Furthermore, NETs released by aged neutrophils generally have an unbalanced composition of antimicrobial proteins, including altered levels of DNA and histones [43]. For example, increased citrullination of histones, mediated by peptidyl-arginine deiminase enzymes, has been reported in aged neutrophils. These modifications can affect the ability of histones to bind DNA and promote chromatin de-condensation, potentially influencing NET structure and function.
While NETs are designed to trap and eliminate pathogens, dysregulated NET formation in aging may compromise their antimicrobial function. Altered composition, reduced antimicrobial capacity, and impaired clearance can undermine the ability of NETs to effectively control infections, leading to increased susceptibility to microbial pathogens. Moreover, in elderly patients exposed to infection, NETs may exhibit an enhanced pro-inflammatory phenotype. This can be attributed to alterations in the composition of NETs, including increased exposure of pro-inflammatory molecules such as high-mobility group box 1 protein [13, 44]. These modified NETs can activate immune cells, including macrophages and dendritic cells, leading to sustained inflammation and tissue damage. As such, the increased basal levels of NET formation in aging individuals can have both beneficial and detrimental effects. On one hand, NETs can help in the clearance of pathogens and contribute to immune defense. On the other hand, the elevated levels of NETs in the absence of specific stimuli can lead to prolonged inflammation, tissue damage, and potentially contribute to age-related pathologies.
How sepsis impacts on the innate immune system: a focus on neutrophil function
During the course of sepsis, several alterations in the immune system have been reported, affecting both innate and adaptive responses [8]. Sepsis-related immune suppression is marked by significant impairments that share similarities with immuno-senescence, as shown in Table 1. As mentioned earlier, sepsis exerts its impact on the immune system by disrupting the functionality and quality of effector cells responsible for maintaining homeostasis [11].
In sepsis, an initial over-expression of pro-inflammatory cytokines by various components of the innate immune system was reported. This exaggerated and uncontrolled activation of the inflammatory response can promote the activation of the coagulation system and the formation of micro-thrombi, potentially causing dangerous conditions such as disseminated intravascular coagulation [45]. The reciprocal interaction between immune system cells and platelets, endothelial cells, and clotting factors is referred to as "immunothrombosis" with roles in preventing pathogen spread. During sepsis, immune cells interact with endothelial cells, inducing a pro-adhesive and pro-thrombotic phenotype rich of tissue factor (TF), E-selectin, ICAM-1, vascular cell adhesion molecule 1 (VCAM-1), and von Willebrand factor (vWF). In conditions featuring endothelial dysfunction, such as cardiovascular disease and sepsis, the induction of NETosis further fuels immuno-thrombosis mechanisms. Specifically NETs, harbor IL-1α and other DAMPs on their chromatin web, causing upregulation of vWF and TF expression by the endothelium, increasing thrombin production from platelets, and blunting the production of endogenous anti-coagulant factors [46, 47].
Examining various neutrophilic phenotypes has revealed that the presence of aged neutrophils in the bloodstream is harmful to tissues and promotes vascular inflammation. In an ischemia–reperfusion model study, the depletion of aged neutrophils prevented micro-clot formation and improved survival after myocardial infarction [48]. While NETs may contribute to thrombosis, this study did not identify them as the primary instigator, as two different NET inhibitors failed to prevent clot formation in re-perfused microcirculation. Furthermore, no evidence was found to suggest that the proliferation and apoptosis of endothelial cells are directly altered by neutrophils, indicating that aged neutrophils do not compromise basal vascular integrity before a direct insult [48].
However, the severity of organ dysfunction and the increased mortality cannot be solely explained by the pro-inflammatory status alone. Simultaneously, sepsis induces an increased release of anti-inflammatory cytokines that alter the ability of innate immune system cells to respond to infections, leading to a reduction in several essential functions. For example, during sepsis, there is a decreased expression of the HLA-DR isotype in the bone marrow, which impairs the antigen-presenting capabilities of monocytes, further exacerbating immunosuppression and reducing the ability to combat pathogens [49].
Other mechanisms of immune system dysregulation during sepsis include the reduction of antimicrobial activities of macrophages and neutrophils. These alterations result in impaired recruitment and migration to the site of infection, as well as impaired ability to phagocytose foreign materials. In addition, there is also an increased loss of effector immune cells, wherein the process of apoptosis is affected, increased in macrophages and dendritic cells, and delayed in PMNs. Generally, apoptosis aims to eliminate damaged cells in order to maintain homeostasis but, in sepsis, the death receptor ligands increase the expression of Fas/Fas-ligand on the surface of the peripheral blood mononuclear cells leading to downstream activation of Caspase-8 and ultimately Caspase-3, also called as the extrinsic apoptosis pathway [50]. As a result, the immune system faces greater challenges in fighting pathogens, leaving the patient more vulnerable to secondary infections. In elderly individuals, these mechanisms of immunosuppression, resulting from both sepsis and inflammaging, overlap and act synergistically, likely remaining among the main causes of poor outcomes in septic patients.
After the migration toward the site of infection, neutrophils employ various mechanisms to protect the body. Some of these mechanisms, like phagocytosis, oxidative burst, cytokine release, and degranulation, have long been known, while others, such as neutrophil extracellular trap formation and microvesicle release, have been more recently characterized. While in the early stages of infection, especially in non-severe disorders, the antimicrobial response of neutrophils is adequate and effective, in the more severe forms such as septic shock, under a condition of prolonged stimulation and immune suppression, the same mechanism can become impaired. In fact, during severe infection, neutrophils show defective recruitment and migration to the infection site, which can be attributed to a higher proportion of immature cells in the bloodstream [51]. This condition is influenced by the activation of pro-inflammatory cytokines and toll-like receptors, leading to the desensitization of chemokine C-X-C motif ligand 12, a key regulator of neutrophil mobilization, and promoting their release into the bloodstream. Furthermore, the receptor CXCR2 appears to be involved as an antagonist of CXCR4, further promoting neutrophil mobilization [33].
Moreover, in the course of sepsis, C–C chemokine receptor type (CCR) 2 signaling is implicated in recruiting neutrophils to infected tissues. The release of CCR2 ligands, particularly monocyte chemoattractant protein-1, from infected tissues promotes the activation and migration of neutrophils through a CCR2-dependent mechanism. While this is crucial for host defense against pathogens, dysregulated CCR2 signaling can lead to excessive and inappropriate neutrophil infiltration, causing tissue damage [52].
Furthermore, the chemokine gradient is vital for neutrophil diapedesis through endothelial tight junctions. However, during severe sepsis, neutrophils exhibit reduced affinity for chemoattractants, possibly related to pro-inflammatory products that, together with chemokines released from the luminal part of the endothelium, induce conformational changes on the surface of the neutrophils [53].
While some populations of the innate immune system, such as natural killer cells, dendritic cells, and macrophages, increase their apoptosis rate during sepsis, neutrophils demonstrate delayed apoptosis, attributed to the increased amount of anti-apoptotic proteins, particularly myeloid cell leukemia 1 (Mcl-1) [54]. Pro-inflammatory mediators like lipopolysaccharide activate anti-apoptotic pathways, resulting in higher expression of anti-apoptotic proteins, such as B cell lymphoma-extra-large and Mcl-1 proteins [55]. This might explain the increased neutrophils’ lifespan during sepsis [56]. Moreover, the antimicrobial activities of the innate system are severely impaired during sepsis. This is characterized by reduced production of pro-inflammatory cytokines and a tendency for the reduction of anti-inflammatory mediators. In certain cases, microorganism-specific stimuli (e.g., during Pseudomonas aeruginosa sepsis) may activate the complement system, increasing component 5a while decreasing ROS and nitric oxide production, as well as phagocytosis [56].
Relatively recent studies have indicated dysregulated NET formation following severe infection [57]. While NETs play a crucial role in defense mechanisms, their dysregulation during sepsis can have both positive and negative consequences. Although they prevent bacterial dissemination in the early stage of infection, promoting the physical containment of microbes, their presence during sepsis is associated with more severe disease and organ damage [58]. Additionally, NETs may contribute to coagulation disorders in sepsis, particularly in the liver sinusoids and pulmonary capillaries, as their components interact with activated platelets, promoting adhesion, activation, and aggregation, leading to thrombosis, cellular ischemia, and organ injury [59].
Lastly, degranulation is qualitatively impaired during sepsis, resulting from an alteration in granule contents and balance in neutrophils. This leads to heterogeneity in granules and subsequent impairment of related neutrophil functions, such as migration and diapedesis [60].
Time is the key element for immunomodulation of neutrophils during sepsis
Sepsis and septic shock are serious public health concerns. To address this issue, implementing a diagnostic process for sepsis is necessary to identify patients at higher risk of death, optimizing therapeutic strategies to reduce mortality and associated costs. Nevertheless, a large number of randomized trials were organized in the hypothesis of modulating the septic response to infection, but none of these demonstrated to increase long-term survival.
Nowadays, a significant portion of sepsis-related mortality occurs several days or months after a patient's discharge, probably due to the immunosuppression state typical of septic patients, coupled with their comorbidities. This phenomenon is particularly pronounced in the elderly population, possibly due to immuno-senescence. As a result, ongoing trials are focusing on immune-modulatory therapies. Understanding how sepsis affects the functions of the innate immune system could be the key to identifying appropriate therapeutic strategies that can prevent long-term mortality. Neutrophils play a crucial role in the fight against pathogens, exhibiting different activities depending on the phase of the septic process [61]. Targeting these cells may help restore a balanced immune response to the infection.
Immuno-modulating agents are commonly used in clinical practice and have significantly improved the prognosis of several diseases, such as inflammatory bowel diseases, rheumatoid arthritis, psoriasis, chronic obstructive pulmonary disease, and anti-neutrophil cytoplasmic antibody-associated vasculitis. Several trials are currently focusing on targeting neutrophils as a therapeutic strategy for different diseases, but little emphasis has been given to their modulation in sepsis. Some attempts have been made to use immunosuppressant treatments for neutrophils or their environment in septic patients, but uncertainty surrounds their effectiveness in recent years [62]. Many trials failed to show the benefits of using such treatments in clinical practice. For example, trials concerning the granulocyte–macrophage colony-stimulating factor, which augments circulating neutrophil levels, did not prove useful despite promising preclinical results [63]. Similarly, activated protein C, which can reduce integrin-mediated migration and NETs formation in neutrophils in preclinical settings, in recent trials did not show the clinical benefits of immunomodulation in septic patients [64].
Similarly, anti-TNF-α drugs, which are effective in treating chronic diseases like rheumatoid arthritis, have been considered as potential targets in animal models of sepsis [65]. However, skepticism arose due to their seemingly irrelevant or even harmful effects in the clinical setting. Though, immuno-modulating agents might require several days before determining substantial effects, especially if a large quantity of cytokines is produced abruptly as it happens in sepsis [62].
In fact, one of the main differences between experiments in animal models and clinical trials is the time of anti-TNF-α administration. In most of the pre-clinical studies, anti-TNF-α drugs are administered around the onset of sepsis [62]. Conversely, the belated administration of these drugs is ineffective even in animal models [66]. Thus, an earlier administration of anti-TNF-α might be investigated in future clinical studies even though finding the right time of administration could be challenging. A relatively recent meta-analysis showed that the use of anti-TNF-α antibodies reduces 28-day mortality compared with controls in septic patients [67]. Thus, the use of anti-TNF-α might require further evaluation, especially concerning the right time of administration with adequate sample size trials. Stratification of patients based on early markers of sustained inflammatory response might further help in findings people who would benefit the most from immunomodulatory approaches.
Actually, the early hours of sepsis are crucial for initiating an appropriate treatment even though only modest symptoms might appear in the initial phases. Especially in the elderly, only an obnubilated mental status can be the very first signal of sepsis [68]. Typical symptoms tend to manifest later in the course of the disease in these frail patients, with the risk of milder response to the administered treatments. In the elderly, the rationale might be that of increasing neutrophils’ activity during the very early phases for favoring the immune response toward pathogens, and that of reducing neutrophils over-activity during the late phases to prevent detrimental effects.
Restoring neutrophils activity
Table 2 presents recent preclinical evidence regarding new potential neutrophil-related targets or treatments to restore their activity. One example is IL-33, a cytokine belonging to the IL-1 family, mainly known for its role in TH2 polarization. Sepsis is associated with a reduction of the IL-33 receptor during the early phases, leading to a decreased number of neutrophils at the site of infection [69]. The administration of IL-33 or other molecules that counterbalance its activity promotes higher neutrophil infiltration at the infection site and results in improved prognosis in animal models of sepsis, as reported in Table 2.
Conversely, immunosuppression contributes to increased mortality in the later stages of sepsis. Detecting the up and down regulation of surface molecules on PMNs’ can aid in the stratification of patients’ risk of mortality. Among these molecules, CD24 and CD279 appear to be optimal biomarkers for predicting subsequent sepsis and worse outcomes. CD24 is a small, heavily glycosylated cellular surface protein predominantly expressed most significantly in neutrophils, connected to the membrane. CD279, also known as programmed death 1 (PD-1), is a protein found on the surface of B and T lymphocytes that regulate T cell responses, acting as a suppressor of neutrophil stimulation. In sepsis, the cross-linking of CD24 on the neutrophil surface is downregulated, resulting in delayed apoptosis [70]. Conversely, higher expressions of CD24 and CD279 on neutrophils predict clinical worsening of sepsis, whereas lower expressions are associated with earlier discharge from the hospital [71]. Similar to the promising results seen in cancer treatment with immune checkpoint targeting, targeting PD-1 in sepsis seems promising based on recent evidence [72, 73]. Also in progressive multifocal leukoencephalopathy, PD-1 blockade with pembrolizumab could reduce viral load and increase lymphocyte activities [74]. An observational study demonstrated that anti-PD-1 and programmed death-ligand 1 (PD-L1) antibodies enhance neutrophil functionality [75].
Lastly, in older adults, the accuracy of neutrophil migration through the inflammatory site may be reduced, possibly due to excessive PI3K activity. Blocking PI3K, particularly the inhibition of PI3Kγ or PI3Kδ, has been linked to the restoration of cell migration ability [37].
Reducing neutrophils detrimental effects
As the fight against pathogens reiterates, the risk of tissue and organ damage increases. In general, neutrophils’ activity can determine several multi-organ failure in different ways: releasement of proteolytic enzymes, NETs, and blood flow occlusion because of excess neutrophils’ binding to the endothelium. As shown in Table 3, recent evidence demonstrates that animal models of sepsis with reduced production of NETs have a better prognosis, fewer organ damages, and improved survival.
The increase of neutrophil apoptosis is another potential therapeutic approach. Normally, neutrophils initiate the apoptotic process after eliminating pathogens, leading to the resolution of inflammation [76]. However, apoptosis of neutrophils tends to be decreased in septic patients [77]. Possible explanations include hyper-activation of nuclear factor-kappa B and downregulation of caspases-9 and -3 in neutrophils of septic patients, as well as the accumulation of cytoplasmic myeloid nuclear differentiation antigen, which slows the apoptotic process in these neutrophils [78]. Treatment with molecules that prevent apoptosis, such as caspase inhibitors or anti-apoptotic cytokines, has been shown to improve survival in sepsis, especially in monocytes [78].
Another potential target process is the neutrophil recruitment and infiltration into the site of infection. CCR2 signaling on neutrophils triggers a cascade of intracellular events that facilitate their migration. Upon ligand binding, CCR2 activates downstream signaling pathways, such as the mitogen-activated protein kinase pathway PI3K pathway, leading to actin polymerization and cytoskeletal rearrangement. These events promote neutrophil chemotaxis and adhesion to endothelial cells, facilitating their transmigration into the infected tissues. Preclinical studies showed that blockade of CCR2, either genetically or pharmacologically, has the potential to reduce the inappropriate infiltration of neutrophils during sepsis, leading to organ damage and mortality [52].
Recent studies show that propofol, one of the most commonly used anesthetic drugs, can prevent neutrophil overactivity by inhibiting formyl peptide receptor 1 [79]. In fact, the formyl peptide receptor 1 is related to neutrophils chemotaxis, superoxide production, elastase, and leukotriene B4 release [79]. However, not all studies have found propofol to be an anti-neutrophil drug, as low-dose treatments have been shown to have a pro-survival effect on neutrophils. [80].
Another therapeutic strategy involves removing leukocytes or pro-inflammatory molecules from the blood of septic patients. A prospective, multicenter study demonstrated that patients treated with hemodiafiltration had longer survival rates compared to their predicted survival based on the APACHE II (Acute Physiology and Chronic Health Evaluation II) score [81]. On the contrary, leukofiltration showed no difference in terms of mortality but improved renal function in septic patients [82]. However, generalizations are not possible at the present moment due to limited data availability, as reported in a recent meta-analysis [83]. These therapeutic strategies hold promise in addressing the complexities of sepsis and its impact on the immune system, but further research is needed to establish their effectiveness and safety in clinical settings.
In conclusion, a novel therapeutic approach could target inflammasomes, acting upstream of the production of specific cytokines. Specifically, inhibiting the activation of inflammasome NLRP3 in response to the accumulation of DAMPs holds promise for controlling systemic chronic inflammation in the elderly and addressing age-related pathologies, extending beyond the neutrophilic level.
Pre-clinical studies provide evidence that the administration of Hemin, [26] an inhibitor of heme oxygenase-1 (HO-1), effectively hinders the activation of inflammasome NLRP3. This intervention results in reduced levels of IL-1β and IL-18, accompanied by the inhibition of neutrophil recruitment. Notably, this limits the inflammatory response and mitigates acute lung damage induced by sepsis. Additionally, exploring the potential of gene silencing of NLRP3 [27] reveals promising outcomes, as it appears to diminish neutrophilic infiltrate and associated liver damage induced by sepsis. These findings suggest the potential of targeting inflammasome NLRP3 as a strategy to address age-related pathologies and sepsis-induced complications.
Concluding remarks and future perspectives
Functional neutrophils are of pivotal importance for host defense during infective conditions. Yet, sepsis itself may cause neutrophil dysfunction reducing the appropriate release of pro-inflammatory mediators in the early phase in favor of a more prolonged action with deleterious side-effects. Such an effect is particularly seen in the elderly as it couples with the general aging of the inflammatory system, which has similar effects on PMN function.
A more profound understanding of how the innate immune system becomes impaired during sepsis and aging is essential in order to improve patients’ health. In the near future, advancements in understanding neutrophil functions are eagerly awaited to develop potential immunomodulatory therapies that can enhance clinical outcomes. Even though this concept may be counterintuitive, we here reported how sustained inflammation and neutrophil activation especially in the late phase of an infection can be deleterious. However, the short life of these cells together with the presence of different phenotypes (including regulatory ones) does not encourage approaches targeting those cells at broad. Most probably, a personalized therapy taking into account the age of the patients as well as a tight timing control targeting specific mediators/mechanisms (i.e., NETosis or inflammasomes) may be the avenue to follow.
Data availability
This is a review article. It does not include any original data.
References
Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M et al (2016) The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315(8):801–810
Delano MJ, Ward PA (2016) The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol Rev 274(1):330–353
Camici GG, Liberale L (2017) Aging: the next cardiovascular disease? Eur Heart J 38(21):1621–1623
Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F et al (2007) Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev 128(1):92–105
Liberale L, Montecucco F, Tardif JC, Libby P, Camici GG (2020) Inflamm-ageing: the role of inflammation in age-dependent cardiovascular disease. Eur Heart J 41(31):2974–2982
Liberale L, Badimon L, Montecucco F, Luscher TF, Libby P, Camici GG (2022) Inflammation, aging, and cardiovascular disease: JACC review topic of the week. J Am Coll Cardiol 79(8):837–847
Pawelec G (2018) Age and immunity: What is “immunosenescence”? Exp Gerontol 105:4–9
Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH et al (2011) Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 306(23):2594–2605
Otto GP, Sossdorf M, Claus RA, Rodel J, Menge K, Reinhart K et al (2011) The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care 15(4):R183
Jones TK, Fuchs BD, Small DS, Halpern SD, Hanish A, Umscheid CA et al (2015) Post-acute care use and hospital readmission after sepsis. Ann Am Thorac Soc 12(6):904–913
Hotchkiss RS, Monneret G, Payen D (2013) Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 13(12):862–874
Shaw AC, Goldstein DR, Montgomery RR (2013) Age-dependent dysregulation of innate immunity. Nat Rev Immunol 13(12):875–887
Denning NL, Aziz M, Gurien SD, Wang P (2019) DAMPs and NETs in Sepsis. Front Immunol 10:2536
Chousterman BG, Swirski FK, Weber GF (2017) Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol 39(5):517–528
Liu D, Huang SY, Sun JH, Zhang HC, Cai QL, Gao C et al (2022) Sepsis-induced immunosuppression: mechanisms, diagnosis and current treatment options. Mil Med Res 9(1):56
Seidler S, Zimmermann HW, Bartneck M, Trautwein C, Tacke F (2010) Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol 11:30
Palsson-McDermott EM, O’Neill LA (2013) The Warburg effect then and now: from cancer to inflammatory diseases. BioEssays 35(11):965–973
Drew W, Wilson DV, Sapey E (2018) Inflammation and neutrophil immunosenescence in health and disease: targeted treatments to improve clinical outcomes in the elderly. Exp Gerontol 105:70–77
Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D et al (2019) Reactive Oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev 2019:5080843
Cao C, Yu M, Chai Y (2019) Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death Dis 10(10):782
Broz P, Dixit VM (2016) Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16(7):407–420
Chen KW, Gross CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ et al (2014) The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell Rep 8(2):570–582
Lara PC, Macias-Verde D, Burgos-Burgos J (2020) Age-induced NLRP3 Inflammasome Over-activation Increases Lethality of SARS-CoV-2 Pneumonia in Elderly Patients. Aging Dis 11(4):756–762
Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A et al (2013) Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab 18(4):519–532
Jin L, Batra S, Jeyaseelan S (2017) Deletion of Nlrp3 augments survival during polymicrobial sepsis by decreasing autophagy and enhancing phagocytosis. J Immunol 198(3):1253–1262
Luo YP, Jiang L, Kang K, Fei DS, Meng XL, Nan CC et al (2014) Hemin inhibits NLRP3 inflammasome activation in sepsis-induced acute lung injury, involving heme oxygenase-1. Int Immunopharmacol 20(1):24–32
Wu Y, Ren J, Zhou B, Ding C, Chen J, Wang G et al (2015) Gene silencing of non-obese diabetic receptor family (NLRP3) protects against the sepsis-induced hyper-bile acidaemia in a rat model. Clin Exp Immunol 179(2):277–293
Hellebrekers P, Vrisekoop N, Koenderman L. Neutrophil phenotypes in health and disease. Eur J Clin Invest. 2018;48 Suppl 2(Suppl Suppl 2):e12943.
Christoffersson G, Vagesjo E, Vandooren J, Liden M, Massena S, Reinert RB et al (2012) VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood 120(23):4653–4662
Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers JW et al (2012) A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest 122(1):327–336
Jerke U, Rolle S, Dittmar G, Bayat B, Santoso S, Sporbert A et al (2011) Complement receptor Mac-1 is an adaptor for NB1 (CD177)-mediated PR3-ANCA neutrophil activation. J Biol Chem 286(9):7070–7081
Alder MN, Opoka AM, Lahni P, Hildeman DA, Wong HR (2017) Olfactomedin-4 Is a Candidate Marker for a Pathogenic Neutrophil Subset in Septic Shock. Crit Care Med 45(4):e426–e432
Eash KJ, Greenbaum AM, Gopalan PK, Link DC (2010) CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest 120(7):2423–2431
Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM (2003) Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 19(4):583–593
Chatta GS, Andrews RG, Rodger E, Schrag M, Hammond WP, Dale DC (1993) Hematopoietic progenitors and aging: alterations in granulocytic precursors and responsiveness to recombinant human G-CSF, GM-CSF, and IL-3. J Gerontol 48(5):M207–M212
Brubaker AL, Rendon JL, Ramirez L, Choudhry MA, Kovacs EJ (2013) Reduced neutrophil chemotaxis and infiltration contributes to delayed resolution of cutaneous wound infection with advanced age. J Immunol 190(4):1746–1757
Sapey E, Greenwood H, Walton G, Mann E, Love A, Aaronson N et al (2014) Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood 123(2):239–248
Nomellini V, Brubaker AL, Mahbub S, Palmer JL, Gomez CR, Kovacs EJ (2012) Dysregulation of neutrophil CXCR2 and pulmonary endothelial icam-1 promotes age-related pulmonary inflammation. Aging Dis 3(3):234–247
Gasparoto TH, Dalboni TM, Amor NG, Abe AE, Perri G, Lara VS et al (2021) Fcgamma receptors on aging neutrophils. J Appl Oral Sci 29:e20200770
Mansfield PJ, Shayman JA, Boxer LA (2000) Regulation of polymorphonuclear leukocyte phagocytosis by myosin light chain kinase after activation of mitogen-activated protein kinase. Blood 95(7):2407–2412
Sabbatini M, Bona E, Novello G, Migliario M, Reno F (2022) Aging hampers neutrophil extracellular traps (NETs) efficacy. Aging Clin Exp Res 34(10):2345–2353
Wang Y, Wang W, Wang N, Tall AR, Tabas I (2017) Mitochondrial oxidative stress promotes atherosclerosis and neutrophil extracellular traps in aged mice. Arterioscler Thromb Vasc Biol 37(8):e99–e107
Tsourouktsoglou TD, Warnatsch A, Ioannou M, Hoving D, Wang Q, Papayannopoulos V (2020) Histones, DNA, and citrullination promote neutrophil extracellular trap inflammation by regulating the localization and activation of TLR4. Cell Rep 31(5):107602
Feldman N, Rotter-Maskowitz A, Okun E (2015) DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res Rev 24(Pt A):29–39
Tang D, Wang H, Billiar TR, Kroemer G, Kang R (2021) Emerging mechanisms of immunocoagulation in sepsis and septic shock. Trends Immunol 42(6):508–522
Maneta E, Aivalioti E, Tual-Chalot S, Emini Veseli B, Gatsiou A, Stamatelopoulos K et al (2023) Endothelial dysfunction and immunothrombosis in sepsis. Front Immunol 14:1144229
Zhu S, Yu Y, Qu M, Qiu Z, Zhang H, Miao C et al (2023) Neutrophil extracellular traps contribute to immunothrombosis formation via the STING pathway in sepsis-associated lung injury. Cell Death Discov 9(1):315
Adrover JM, Del Fresno C, Crainiciuc G, Cuartero MI, Casanova-Acebes M, Weiss LA, et al. A Neutrophil Timer Coordinates Immune Defense and Vascular Protection. Immunity. 2019;50(2):390–402 e10.
Leijte GP, Rimmele T, Kox M, Bruse N, Monard C, Gossez M et al (2020) Monocytic HLA-DR expression kinetics in septic shock patients with different pathogens, sites of infection and adverse outcomes. Crit Care 24(1):110
Luan YY, Yao YM, Xiao XZ, Sheng ZY (2015) Insights into the apoptotic death of immune cells in sepsis. J Interferon Cytokine Res 35(1):17–22
Kovach MA, Standiford TJ (2012) The function of neutrophils in sepsis. Curr Opin Infect Dis 25(3):321–327
Souto FO, Alves-Filho JC, Turato WM, Auxiliadora-Martins M, Basile-Filho A, Cunha FQ (2011) Essential role of CCR2 in neutrophil tissue infiltration and multiple organ dysfunction in sepsis. Am J Respir Crit Care Med 183(2):234–242
Colbert JF, Schmidt EP (2016) Endothelial and Microcirculatory Function and Dysfunction in Sepsis. Clin Chest Med 37(2):263–275
Lucas CD, Dorward DA, Tait MA, Fox S, Marwick JA, Allen KC et al (2014) Downregulation of Mcl-1 has anti-inflammatory pro-resolution effects and enhances bacterial clearance from the lung. Mucosal Immunol 7(4):857–868
Zhu CL, Wang Y, Liu Q, Li HR, Yu CM, Li P et al (2022) Dysregulation of neutrophil death in sepsis. Front Immunol 13:963955
Shen XF, Cao K, Jiang JP, Guan WX, Du JF (2017) Neutrophil dysregulation during sepsis: an overview and update. J Cell Mol Med 21(9):1687–1697
Mulet M, Osuna-Gomez R, Zamora C, Artesero I, Arus M, Vera-Artazcoz P, et al. (2023) Dysregulated neutrophil extracellular traps formation in sepsis. Immunology
Czaikoski PG, Mota JM, Nascimento DC, Sonego F, Castanheira FV, Melo PH et al (2016) Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PLoS ONE 11(2):e0148142
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr et al (2010) Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 107(36):15880–15885
Martin-Fernandez M, Vaquero-Roncero LM, Almansa R, Gomez-Sanchez E, Martin S, Tamayo E et al (2020) Endothelial dysfunction is an early indicator of sepsis and neutrophil degranulation of septic shock in surgical patients. BJS Open 4(3):524–534
Sonego F, Castanheira FV, Ferreira RG, Kanashiro A, Leite CA, Nascimento DC et al (2016) Paradoxical roles of the neutrophil in sepsis: protective and deleterious. Front Immunol 7:155
Brown KA, Brown GA, Lewis SM, Beale R, Treacher DF (2016) Targeting cytokines as a treatment for patients with sepsis: A lost cause or a strategy still worthy of pursuit? Int Immunopharmacol 36:291–299
Frydrych LM, Bian G, Fattahi F, Morris SB, O’Rourke RW, Lumeng CN et al (2019) GM-CSF administration improves defects in innate immunity and sepsis survival in obese diabetic mice. J Immunol 202(3):931–942
Annane D, Timsit JF, Megarbane B, Martin C, Misset B, Mourvillier B et al (2013) Recombinant human activated protein C for adults with septic shock: a randomized controlled trial. Am J Respir Crit Care Med 187(10):1091–1097
Xu C, Chang A, Hack BK, Eadon MT, Alper SL, Cunningham PN (2014) TNF-mediated damage to glomerular endothelium is an important determinant of acute kidney injury in sepsis. Kidney Int 85(1):72–81
Marquez-Velasco R, Bojalil R, Buelna A, Flores-Guzman F, Estevez-Ramirez J, Laguna J et al (2006) Anti-tumor necrosis factor alpha F(ab’)2 antibody fragments protect in murine polymicrobial sepsis: concentration and early intervention are fundamental to the outcome. Inflamm Res 55(9):378–384
Lv S, Han M, Yi R, Kwon S, Dai C, Wang R (2014) Anti-TNF-alpha therapy for patients with sepsis: a systematic meta-analysis. Int J Clin Pract 68(4):520–528
Liang SY (2016) Sepsis and other infectious disease emergencies in the elderly. Emerg Med Clin North Am 34(3):501–522
Babic ZM, Zunic FZ, Pantic JM, Radosavljevic GD, Jovanovic IP, Arsenijevic NN et al (2018) IL-33 receptor (ST2) deficiency downregulates myeloid precursors, inflammatory NK and dendritic cells in early phase of sepsis. J Biomed Sci 25(1):56
Parlato M, Souza-Fonseca-Guimaraes F, Philippart F, Misset B, Captain Study G, Adib-Conquy M, et al. CD24-triggered caspase-dependent apoptosis via mitochondrial membrane depolarization and reactive oxygen species production of human neutrophils is impaired in sepsis. J Immunol. 2014;192(5):2449–59
Shankar-Hari M, Datta D, Wilson J, Assi V, Stephen J, Weir CJ et al (2018) Early PREdiction of sepsis using leukocyte surface biomarkers: the ExPRES-sepsis cohort study. Intensive Care Med 44(11):1836–1848
Nakamori Y, Park EJ, Shimaoka M (2020) Immune deregulation in sepsis and septic shock: reversing immune paralysis by targeting PD-1/PD-L1 pathway. Front Immunol 11:624279
Monneret G, Gossez M, Venet F (2016) Sepsis in PD-1 light. Crit Care 20(1):186
Cortese I, Muranski P, Enose-Akahata Y, Ha SK, Smith B, Monaco M et al (2019) Pembrolizumab treatment for progressive multifocal leukoencephalopathy. N Engl J Med 380(17):1597–1605
Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, Hotchkiss RS (2016) Frontline Science: Defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J Leukoc Biol 100(6):1239–1254
Savill J (1997) Apoptosis in resolution of inflammation. J Leukoc Biol 61(4):375–380
Tamayo E, Gomez E, Bustamante J, Gomez-Herreras JI, Fonteriz R, Bobillo F, et al. Evolution of neutrophil apoptosis in septic shock survivors and nonsurvivors. J Crit Care. 2012;27(4):415 e1–11
Fotouhi-Ardakani N, Kebir DE, Pierre-Charles N, Wang L, Ahern SP, Filep JG et al (2010) Role for myeloid nuclear differentiation antigen in the regulation of neutrophil apoptosis during sepsis. Am J Respir Crit Care Med 182(3):341–350
Okuno T, Koutsogiannaki S, Ohba M, Chamberlain M, Bu W, Lin FY et al (2017) Intravenous anesthetic propofol binds to 5-lipoxygenase and attenuates leukotriene B(4) production. FASEB J 31(4):1584–1594
Hsing CH, Chen CL, Lin WC, Lin CF (2015) Propofol treatment inhibits constitutive apoptosis in human primary neutrophils and granulocyte-differentiated human HL60 cells. PLoS ONE 10(6):e0129693
Shiga H, Hirasawa H, Nishida O, Oda S, Nakamura M, Mashiko K et al (2014) Continuous hemodiafiltration with a cytokine-adsorbing hemofilter in patients with septic shock: a preliminary report. Blood Purif 38(3–4):211–218
Rubino AS, Serraino GF, Mariscalco G, Marsico R, Sala A, Renzulli A (2011) Leukocyte depletion during extracorporeal circulation allows better organ protection but does not change hospital outcomes. Ann Thorac Surg 91(2):534–540
Snow TAC, Littlewood S, Corredor C, Singer M, Arulkumaran N (2021) Effect of extracorporeal blood purification on mortality in sepsis: a meta-analysis and trial sequential analysis. Blood Purif 50(4–5):462–472
Mishra HK, Johnson TJ, Seelig DM, Walcheck B (2016) Targeting ADAM17 in leukocytes increases neutrophil recruitment and reduces bacterial spread during polymicrobial sepsis. J Leukoc Biol 100(5):999–1004
Kobayashi Y, Iwata A, Suzuki K, Suto A, Kawashima S, Saito Y et al (2013) B and T lymphocyte attenuator inhibits LPS-induced endotoxic shock by suppressing Toll-like receptor 4 signaling in innate immune cells. Proc Natl Acad Sci USA 110(13):5121–5126
Craciun FL, Schuller ER, Remick DG (2010) Early enhanced local neutrophil recruitment in peritonitis-induced sepsis improves bacterial clearance and survival. J Immunol 185(11):6930–6938
Hasan Z, Palani K, Rahman M, Thorlacius H (2011) Targeting CD44 expressed on neutrophils inhibits lung damage in abdominal sepsis. Shock 35(6):567–572
Xu H, Xu J, Xu L, Jin S, Turnquist HR, Hoffman R et al (2018) Interleukin-33 contributes to ILC2 activation and early inflammation-associated lung injury during abdominal sepsis. Immunol Cell Biol 96(9):935–947
Kasten KR, Prakash PS, Unsinger J, Goetzman HS, England LG, Cave CM et al (2010) Interleukin-7 (IL-7) treatment accelerates neutrophil recruitment through gamma delta T-cell IL-17 production in a murine model of sepsis. Infect Immun 78(11):4714–4722
Pelletier M, Ratthe C, Girard D (2002) Mechanisms involved in interleukin-15-induced suppression of human neutrophil apoptosis: role of the anti-apoptotic Mcl-1 protein and several kinases including Janus kinase-2, p38 mitogen-activated protein kinase and extracellular signal-regulated kinases-1/2. FEBS Lett 532(1–2):164–170
Alves-Filho JC, Sonego F, Souto FO, Freitas A, Verri WA Jr, Auxiliadora-Martins M et al (2010) Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med 16(6):708–712
Lan F, Yuan B, Liu T, Luo X, Huang P, Liu Y, et al. Interleukin-33 facilitates neutrophil recruitment and bacterial clearance in S. aureus-caused peritonitis. Mol Immunol. 2016;72:74–80.
Lin X, Luo H, Yan X, Song Z, Gao X, Xia Y et al (2018) Interleukin-34 ameliorates survival and bacterial clearance in Polymicrobial sepsis. Crit Care Med 46(6):e584–e590
Hall SR, Tsoyi K, Ith B, Padera RF Jr, Lederer JA, Wang Z et al (2013) Mesenchymal stromal cells improve survival during sepsis in the absence of heme oxygenase-1: the importance of neutrophils. Stem Cells 31(2):397–407
Chen J, Wang B, Lai J, Braunstein Z, He M, Ruan G et al (2018) Trimetazidine attenuates cardiac dysfunction in endotoxemia and sepsis by promoting neutrophil migration. Front Immunol 9:2015
Vago JP, Nogueira CR, Tavares LP, Soriani FM, Lopes F, Russo RC et al (2012) Annexin A1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. J Leukoc Biol 92(2):249–258
Rahman M, Zhang S, Chew M, Syk I, Jeppsson B, Thorlacius H (2013) Platelet shedding of CD40L is regulated by matrix metalloproteinase-9 in abdominal sepsis. J Thromb Haemost 11(7):1385–1398
Duffin R, Leitch AE, Sheldrake TA, Hallett JM, Meyer C, Fox S et al (2009) The CDK inhibitor, R-roscovitine, promotes eosinophil apoptosis by down-regulation of Mcl-1. FEBS Lett 583(15):2540–2546
Wang K, Hampson P, Hazeldine J, Krystof V, Strnad M, Pechan P et al (2012) Cyclin-dependent kinase 9 activity regulates neutrophil spontaneous apoptosis. PLoS ONE 7(1):e30128
Ode Y, Aziz M, Wang P (2018) CIRP increases ICAM-1(+) phenotype of neutrophils exhibiting elevated iNOS and NETs in sepsis. J Leukoc Biol 103(4):693–707
Zhang CY, Dong X, Gao J, Lin W, Liu Z, Wang Z. Nanoparticle-induced neutrophil apoptosis increases survival in sepsis and alleviates neurological damage in stroke. Sci Adv. 2019;5(11):eaax7964.
Bakele M, Joos M, Burdi S, Allgaier N, Poschel S, Fehrenbacher B et al (2014) Localization and functionality of the inflammasome in neutrophils. J Biol Chem 289(8):5320–5329
Weber GF, Chousterman BG, He S, Fenn AM, Nairz M, Anzai A et al (2015) Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science 347(6227):1260–1265
Asaduzzaman M, Zhang S, Lavasani S, Wang Y, Thorlacius H (2008) LFA-1 and MAC-1 mediate pulmonary recruitment of neutrophils and tissue damage in abdominal sepsis. Shock 30(3):254–259
Herter JM, Rossaint J, Spieker T, Zarbock A (2014) Adhesion molecules involved in neutrophil recruitment during sepsis-induced acute kidney injury. J Innate Immun 6(5):597–606
Wu Z, Sawamura T, Kurdowska AK, Ji HL, Idell S, Fu J (2011) LOX-1 deletion improves neutrophil responses, enhances bacterial clearance, and reduces lung injury in a murine polymicrobial sepsis model. Infect Immun 79(7):2865–2870
Hirano Y, Ode Y, Ochani M, Wang P, Aziz M (2018) Targeting junctional adhesion molecule-C ameliorates sepsis-induced acute lung injury by decreasing CXCR4(+) aged neutrophils. J Leukoc Biol 104(6):1159–1171
Mai SH, Khan M, Dwivedi DJ, Ross CA, Zhou J, Gould TJ et al (2015) Delayed but not early treatment with dnase reduces organ damage and improves outcome in a murine model of sepsis. Shock 44(2):166–172
Martinod K, Fuchs TA, Zitomersky NL, Wong SL, Demers M, Gallant M et al (2015) PAD4-deficiency does not affect bacteremia in polymicrobial sepsis and ameliorates endotoxemic shock. Blood 125(12):1948–1956
Biron BM, Chung CS, Chen Y, Wilson Z, Fallon EA, Reichner JS et al (2018) PAD4 deficiency leads to decreased organ dysfunction and improved survival in a dual insult model of hemorrhagic shock and sepsis. J Immunol 200(5):1817–1828
Li Y, Liu Z, Liu B, Zhao T, Chong W, Wang Y et al (2014) Citrullinated histone H3: a novel target for the treatment of sepsis. Surgery 156(2):229–234
El Kebir D, Gjorstrup P, Filep JG (2012) Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci U S A 109(37):14983–14988
Lin R, Zhang Y, Pradhan K, Li L (2020) TICAM2-related pathway mediates neutrophil exhaustion. Sci Rep 10(1):14397
Rahman M, Gustafsson D, Wang Y, Thorlacius H, Braun OO (2014) Ticagrelor reduces neutrophil recruitment and lung damage in abdominal sepsis. Platelets 25(4):257–263
Futosi K, Nemeth T, Pick R, Vantus T, Walzog B, Mocsai A (2012) Dasatinib inhibits proinflammatory functions of mature human neutrophils. Blood 119(21):4981–4991
Goncalves-de-Albuquerque CF, Rohwedder I, Silva AR, Ferreira AS, Kurz ARM, Cougoule C et al (2018) The Yin and Yang of tyrosine kinase inhibition during experimental Polymicrobial sepsis. Front Immunol 9:901
Acknowledgements
None.
Funding
Open access funding provided by Università degli Studi di Genova within the CRUI-CARE Agreement. None.
Author information
Authors and Affiliations
Contributions
All authors actively contributed to the conception, drafting and revision of text, tables, and figures included in the submission.
Corresponding author
Ethics declarations
Conflict of interest
LL is co-inventor on the International Patent WO/2020/226993 filed in April 2020. The patent relates to the use of antibodies which specifically bind IL-1α to reduce various sequelae of ischemia–reperfusion injury to the central nervous system. LL reports speaker fees outside of this work from Daiichi-Sankyo. FM received a round table fee from Astra Zeneca. The other authors have no direct financial or personal interests to declare.
Ethical Statements and Human and animal rights statement
This is a review article, as it does not include animal or human data this is not necessary.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ramoni, D., Tirandi, A., Montecucco, F. et al. Sepsis in elderly patients: the role of neutrophils in pathophysiology and therapy. Intern Emerg Med 19, 901–917 (2024). https://doi.org/10.1007/s11739-023-03515-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11739-023-03515-1