
Abstract
To develop an easy and robust method for creating genetically stable and easily detectable Arabidopsis mutants, we adopted the polycistronic tRNA–gRNA CRISPR/Cas9 (PTG/Cas9) system, a multiplex gene-editing tool in rice, with PTOX as the reporter gene. The PTG/Cas9 system has a great potential in generating large deletions detectable by PCR, which greatly simplifies the laborious work of mutant screening. We constructed a PTOX–PTG/Cas9 system with five gRNAs and introduced it into Arabidopsis. At T1 generation, 24.4% of transgenic plants were chimeric with PCR-detectable deletions in PTOX locus, but no homozygous mutant was found, indicating that gene editing occurred predominantly in somatic cells. After a self-cross propagation, 60% of T1 chimeric plants were able to produce homozygous, heterozygous, or bi-allelic ptox offsprings. Inheritable homozygous ptox mutants without Cas9 gene can be obtained earliest at T2 generation. We further targeted five other genes using the same procedure and achieved homozygous Cas9-free mutants with large deletions for all genes within three generations. We established a standard and reliable protocol to generate stable inherited deletion mutants in 2–3 generations along with simple PCR screening methods. We conclude that the rice PTG/Cas9 system is an efficient, easy, and rapid tool to edit genes in Arabidopsis. We propose that it could be applied to other genes in Arabidopsis, and it might have the potential to edit genes in other plant species as well.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Collections of T-DNA insertional mutants have made vital contributions to determining functions of Arabidopsis genes. However, there are still 12% of Arabidopsis genes lacking T-DNA insertion and 8% genes with a single insertion in publically available T-DNA insertional mutant libraries (O’Malley and Ecker 2010; Kleinboelting et al. 2015, 2017). Moreover, T-DNA favors inserting into the regions of transcriptional initiation and polyadenylation, which usually results in a partial suppression of gene’s functions (Li et al. 2006). Therefore, there still is a large need to create gene knockout mutants in Arabidopsis; and gene-editing technologies provide powerful tools to fulfil this imperative requirement (Liu et al. 2013). Today, the RNA-guided nuclease complex, the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated nuclease 9 (Cas9), is the most powerful and widely used tool to edit genes in diverse organisms due to its high efficiency and great specificity (Jinek et al. 2012; Doudna and Charpentier 2014).
The CRISPR/Cas9 system is derived from the type II CRISPR adaptive immune system in Streptococcus pyogenes, which detects and degrades invasive DNAs from bacteriophages and plasmids (Jinek et al. 2012; Fineran and Charpentier 2012; Gasiunas et al. 2012). It consists of two components, an endonuclease Cas9 and a synthetic guide RNA (gRNA) harboring a 20-nt spacer and a 76-nt Cas9-binding scaffold. After being expressed in plants, Cas9 is introduced to the genomic site that is base-paired with the gRNA spacer, binds to its downstream protospacer adjacent motif (PAM) with a consensus sequence of NGG, and creates a double-strand break (DSB) at a site ~ 3 bp upstream of PAM (Jinek et al. 2012; Doudna and Charpentier 2014). Then, DSB is repaired through either an error-free homologous recombination mechanism or an error-prone non-homologous end-joining pathway. The latter process often leads to insertions, deletions, or substitutions at the target sites (Mali et al. 2013; Cong et al. 2013).
A number of CRISPR/Cas9 systems have been developed and successfully applied to edit genes in Arabidopsis (Ma et al. 2016; Ding et al. 2016; Liu et al. 2017). The early versions usually encode a single gRNA driven by a Pol III promoter and a Cas9 gene driven by the 35S promoter (Shan et al. 2013; Li et al. 2013; Nekrasov et al. 2013; Feng et al. 2013). However, the CRISPR/Cas9 system-directed gene-editing efficiency is much lower in Arabidopsis compared to other plant species, probably because the Arabidopsis gene transformation method does not require a tissue regeneration process, which is usually applied to most plant species. In addition, the generated mutations are often somatic and rarely transmitted to the next generation. Single gRNA CRISPR-Cas9 systems normally generate SNP or short indel mutations at target sites, which can be detected by time-consuming and laborious methods, like DNA sequencing or PCR–RFLP if the mutation destroys a restriction enzyme site. A number of advanced methods were developed to detect SNP or small indel events, including Indel Detection by Amplicon Analysis (IDAA, Yang et al. 2015), qPCR/dPCR (Mock et al. 2016; Falabella et al. 2017), amplicon next-generation sequencing (NGS, Sentmanat et al. 2018), inference of CRISPR edits (ICE, Hsiau et al. 2018), and tracking indels by decomposition (TIDE, Brinkman et al. 2018); however, they are quite expensive and require special equipment or sequencing.
In recent years, many efforts were made to increase the editing capacity and efficiency in Arabidopsis. One way is to promote the expression of Cas9 in stem cells using egg cell-specific promoters like the Yao or EC1.2 promoter instead of the constitutively expressing promoters like the 35S or ubiquitin promoter, leading to an increase of the inheritable mutation rate (Yan et al. 2015; Wang et al. 2015). The other way is to raise the number of genomic target sites by expressing multiple gRNAs simultaneously in plants. Multiplex gene-editing systems could be constructed by assembling several independent gRNA expression cassettes in one vector (Ma et al. 2015; Zhang et al. 2016), or by compacting multiple gRNAs into a single expression cassette and recruiting endogenous post-transcriptional RNA processing systems to generate multiple gRNAs (Gao and Zhao 2014; Nissim et al. 2014; Xie et al. 2015). Multiplex gene-editing systems are not only able to increase the editing efficiency, but also can simplify the mutation identification, because they are capable of generating large PCR-detectable deletions between two target sites. Therefore, using multiplex gene-editing systems followed by a PCR examination on large deletion events, we are able to develop an efficient method to robustly generate target mutants and boost the application of CRISPR/Cas9 systems in Arabidopsis.
The polycistronic tRNA–gRNA (PTG)/Cas9 system originally developed for rice is a powerful multiplex gene-editing system with a frequency of up to ~ 100% (Xie et al. 2015). It clusters multiple tRNA–gRNA architectures into one expression cassette, designated as PTG gene, each gRNA carrying a different spacer sequence. Through plant endogenous tRNA-processing system, tRNA fragments are excised out from the primary transcripts of the PTG gene, releasing all the gRNAs to guide gene editing (Xie et al. 2015). This system has been successfully used in maize (Qi et al. 2016) and kiwifruit (Wang et al. 2018) after some modifications like switching the rice tRNA or promoters to corresponding elements from the host plant species. Compared to other multiplex gene-editing systems, the PTG/Cas9 system is relatively easier in vector construction and transformation, especially when the number of gRNAs increases (Ma et al. 2015; Zhang et al. 2016).
The rice PTG/Cas9 system could also work in Arabidopsis, since all components essential for this system are conserved in plants (Xie et al. 2015). In this work, we applied this rice PTG/Cas9 in Arabidopsis for several genes, and it was indeed able to create large PCR-detectable deletions at desired loci. Based on this property, we were able to identify and isolate stably heritable Arabidopsis Cas9-free mutants in two or three generations through simple PCR screening. In addition, we summarized a reliable and standard protocol to generate and identify Arabidopsis deletion mutants using the rice PTG/Cas9 editing system and the PCR screening method.
Materials and methods
Primers
All the primers used in this experiment are listed in Table S1.
Plant materials and growth conditions
Arabidopsis thaliana wild-type Columbia-0 (Col-0), mutant im (At4g22260), and PTG/Cas9 transgenic plants in Col-0 background were planted in soil or half-strength MS (1/2MS) medium supplemented with 1% (w/v) sucrose. Seedlings were germinated and grown under low light condition (23 °C, 30 μmol m−2 s−1, 16-h light/8-h dark daylight cycle) for 10 days, and then transferred to the normal light condition (23 °C, 100 μmol m−2 s−1, 16-h light/8-h dark daylight cycle).
Constructing gene-specific PTG/Cas9 system
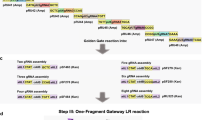
The PTOX-PTG gene contains 5 gRNAs that are interspaced by tRNA fragment and named as gRNA1-5, respectively. It was assembled in order from six fragments amplified by PCR from the pGTR vector individually with specific primers and ligated into the BsaI site of pRGEB32 to generate pRGEB32-PTOX using the Golden Gate Assembly strategy (Fig. 1b).

Schematic diagram of pRGEB32-PTOX. a Diagram of the PTOX gene with five gRNA target sites. The black and white blocks represent protein-coding region and untranslated regions, respectively. Five gRNA targeting sites and two primers (PTOX-F and PTOX-R; black arrows) for examining the PTOX structure are marked at corresponding positions. The number beside the corresponding gRNA target is the relative position of the gRNA first nucleotide to the translation start site. b Diagram of the pRGEB32-PTOX construction procedure. The PTOX-PTG consisting of five gRNAs is first amplified in six fragments by PCR with pGTR as a template, which are digested by BsaI and ligated in order. The pre-assembled PTOX-PTG fragment is re-amplified by PCR and cut with FokI before ligation into BsaI-digested binary vector pRGEB32 to generate pRGEB32-PTOX (see details in "Materials and methods"). The fragment number (Frag. #) and primer pairs are labeled on the sides of the corresponding fragment
Fragment 1 with composition of (5′) FokI adaptor::tRNA::5′ half of gRNA1 spacer::BsaI (3′) was amplified using primers 5′L-F and gRNA1-R. The intermediate fragments were amplified with primers gRNA(n − 1)-F and gRNAn-R, generating a structure of (5′) BsaI::3′ half of gRNA(n − 1) spacer::gRNA scaffold::tRNA::5′ half of gRNAn spacer::BsaI (3′). The last fragment, fragment 6, was amplified with primers gRNA5-F and 3′L-R, resulting in the composition of (5′) BsaI::3′ half of gRNA5 spacer::gRNA scaffold::FokI adaptor (3′). The PCR reaction in this step was performed in a 50-μl mix containing 0.1-ng pGTR plasmid, 1 × Primer STAR Max DNA polymerase (TAKARA, Japan), and 2 μM of each forward and reverse primer. The PCR program was as follows: 98 °C for 2.5 min, 35 cycles of 98 °C for 10 s, 50 °C for 15 s, and 72 °C for 30 s, and a final extension of 72 °C for 10 min. The PCR products were separated by electrophoresis on 1.2% agarose gel in TAE buffer (40-mM Tris–Acetate, 1-mM EDTA, pH 8.0) and purified from gel with Universal DNA Purification Kit (TIANGEN, China).
Primary assembly of these six fragments was conducted by simultaneous digestion and ligation. The reaction was performed in a 20-µl mixture including 50 ng of each fragment, 1 × T7 ligase buffer, 0.1-mg/ml BSA, 1500-U T7 ligase (NEB, USA), and 5-U BsaI (NEB, USA), with program: 40 cycles of 37 °C for 5 min and 20 °C for 10 min, followed by a final incubation at 20 °C for 60 min. The ligation mixture after a 105- fold dilution was subjected to nested-PCR with primers 5′S-F and 3′S-R, to specifically amplify the assembled PTOX-PTG fragment. 1-μl diluted ligation mix was used as template, and the PCR reaction was performed with the condition described above, except that the annealing temperature was increased to 60 °C.
Finally, the full-length PTOX-PTG fragment was purified from agarose gel, digested by FokI (NEB, USA), and then inserted into the BsaI site of pRGEB32 with T4 ligase (TAKARA, Japan) following the standard manufactory protocols. The generated pRGEB32-PTOX construct was confirmed through Sanger sequencing with primers Crispr_F and Crispr_R.
For genes At1g18170, At1g70270, At1g73655, At1g23965, and At1g74070, the binary vectors carrying desired PTG/Cas9 systems were constructed similarly, while the number and primers of gRNAs were different for different genes.
Plant transformation
Empty vector pRGEB32 and pRGEB32 carrying desired PTG/Cas9 were transformed to Agrobacterium tumefaciens strain GV3101, respectively, through the freeze–thaw method. Then, each of them was transformed into Col-0 through Agrobacterium-mediated transformation with the floral dipping method (Clough and Bent 1998). T1 generation seedlings were screened on 1/2 MS medium supplemented with 1% (w/v) sucrose and 20 µg/ml hygromycin. Positive transgenic plants were transferred to soil after 2 weeks.
Mutant screening via PCR analysis
Genomic DNA samples were extracted from Arabidopsis leaves according to the Edward’s Method (Edwards et al. 1991). PCR analysis was applied for examining the targeted gene structure with gene-specific primers. The presence of T-DNA was examined by PCR with the primer pair of Hyg_F/Hyg_R specific for Hygr or Cas9_F/Cas9_R specific for Cas9. The PCR condition was: 30-ng genomic DNA, 1x exTaq Buffer, 1U exTaq (TAKARA, Japan), 2.5 mM of each dNTP, and 0.5 μM of each primer in 15-μl mixture. The PCR reaction followed the program: 94 °C 2.5 min (94 °C 15 s, 58 °C 15 s, and 72 °C 2.5 min) × 35 cycles, 72 °C 10 min. The PCR products were separated on 1% agarose gel in TAE buffer and stained with ethidium bromide. Selected bands were eluted from the gel, and then sequenced directly or cloned into pMD18T vector (TAKARA, Japan) for sequencing. The sequencing chromatograms were viewed with 4Peaks program (v1.7.2).
To quantify the somatic deletion rate of target genes, the stained gels were imaged using a Gel Documentation and Image Analysis System ChampGel 6000 (Beijing Sagecreation, China). Band intensity was quantified in Adobe Photoshop by multiplying the band area with the mean intensity after background subtraction. The somatic deletion rate of each sample was calculated as the intensity percentage of entire shorter bands in all lanes.
Immunoblot analysis
Total protein was extracted from rosette leaves and boiled for 10 min in sampling buffer containing 125-mM Tris–HCl (pH 6.8), 4% (w/v) SDS, 2% (v/v) 2-mercaptoethanol, 0.001% (w/v) bromphenol blue, and 20% (v/v) glycerol. The protein samples were separated on a 12% SDS-PAGE gel. Immunoblot analysis against PTOX was done as described previously using a rabbit polyclonal antibody (Fu et al. 2012). Chemiluminescent signals were generated using the ECL Plus reagent (Tanon, China) and visualized using the Tanon-5200 Chemiluminescent Imaging System (Tanon, China).
Bioinformatics analysis
Six U3 snoRNA promoter sequences were analyzed by multiple sequence alignment software ClustalW (Thompson et al. 1994). Five sequences from A. thaliana (At), Oryza sativa (Os), Triticum aestivum (Ta), and Zea mays (Zm) were obtained from the NCBI website (www.ncbi.nlm.nih.gov), including AtU3b_p (accession No. X52629), AtU3d_p (accession No. X58068), OsU3_p (accession No. X79685), TaU3_p (accession No. X63065), and ZmU3_p (accession No. Z29641). The U3 snoRNA promoter sequence of Gossypium barbadense L. (GbU3_p) was reported by Lei et al. in 2016 (Lei et al. 2016).
Results
Constructing a PTOX-PTG/Cas9 system to edit the PTOX gene in Arabidopsis
The Arabidopsis phytoene desaturase encoding gene, PDS, is often used as a target to examine gene-editing efficiency because of the albino phenotype caused by pds mutation (Ma et al. 2015; Xie et al. 2015). However, pds mutants are lethal, making further analysis impossible. In this work, we chose Arabidopsis PTOX as the report gene to test gene-editing efficiency. Its product, PTOX, is a plastoquinol oxygen oxidoreductase residing on the chloroplast thylakoid membrane, and plays an important role in balancing the redox state of thylakoid membrane (Fu et al. 2009; Wang and Fu 2016). The Arabidopsis PTOX-deficient mutant, immutans (im), shows a striking green and white variegation phenotype on its leaves (Fig. 2a), making mutant screening process convenient. Moreover, this mutant is viable and able to produce large amounts of seeds, allowing us to analyze gene transmission from generation to generation.

Gene-editing efficiency of PTOX-PTG/Cas9 system in T1 transgenic plants. a Plants of Col-0, immutans (im), and transgenic T1 carrying PTOX-PTG/Cas9 system. Plants were photographed 4 weeks after germination. Most of T1 plants were green like Col-0 (wild-type like T1); a few of T1 plants showed mild variegation which is pointed with white arrow; bar = 1 cm. b The band pattern of PTOX in T1 plants. With primers PTOX_F and PTOX_R, 93 T1 plants were amplified out a single wild-type sized band like plant #1 and #2; 30 T1 plants were amplified multiple bands like #3, #4, #34, and #46. The deletion rate is calculated according to the intensity of the short bands relative to the total intensity of the entire amplicons. Three short ptox bands were cut from gel for sequence analysis. c Schematic structures of three ptox variants from b. All three variants result from a large fragment deletion between two gRNA target sites (highlighted in grey). The breakpoints are numbered according to the corresponding PAM site (red letters). The deleted regions are represented as dotted line
In the rice PTG/Cas9 system, expressions of the PTG gene and Cas9 are under the control of a rice U3 snoRNA promoter (OsU3p) and an ubiquitin promoter (OsUBIp), respectively (Xie et al. 2015). We analyzed the conservation of U3 snoRNA promoters (U3p) from several dicotyl and monocotyl land plants using the ClustalW program. It revealed that two key elements, the upstream sequence element (USE, RTCCCACATCG) at position − 60 and the TATA box at position − 25, are highly conserved (Fig. S1), suggesting that OsU3p would function well in Arabidopsis. In addition, the ubiquitin promoter (UBIp) is also conserved in plants (Monia et al. 1990; Ozkaynak et al. 1987). The conservation of U3p and UBIp hinted that the PTG/Cas9 system could be applied directly for gene editing in Arabidopsis. Therefore, we tested whether the original version of the rice PTG/Cas9 system in Arabidopsis works without replacing OsU3p and OsUBIp with their Arabidopsis counterparts, AtU3p and AtUBIp.
In this study, we constructed a PTOX-PTG structure containing five gRNAs evenly targeting the PTOX gene. In detail, gRNA1 and gRNA4 were designed according to the DNA negative strand, and the other three (gRNA2, gRNA3, and gRNA5) were based on the positive strand (Fig. 1a). The PTOX-PTG architecture was amplified in six fragments by PCR with vector pGTR as a template and pre-assembled following the Golden Gate (GG) assembly method described by Xie et al. 2015 (Fig. 1b). Then, the resulting architecture was cloned into the binary vector pRGEB32 at a site downstream of OsU3p, generating vector pRGEB32-PTOX for transforming Arabidopsis (Fig. 1b).
Gene editing of Arabidopsis PTOX in T1 generation
The pRGEB32-PTOX was introduced into Arabidopsis Col-0 through the Agrobacterium-mediated flower-dipping transformation method. We obtained 123 T1 transgenic plants by screening for hygromycin resistance and further PCR confirmation. The majority of transgenic plants looked like wild-type plants, but notably, six plants produced variegated spots in restricted areas (Fig. 2a; Fig. S2a), implying that gene editing of PTOX gene had occurred.
To evaluate the editing efficiency of PTOX–PTG/Cas9 system, we performed a primary analysis on PTOX gene sequence in T1 plant leaf tissue by a simple PCR method with PTOX specific primers (Fig. 1a). Out of these plants, 93 only had a single wild-type sized band (~ 2.5-kb), and 30 (24.4%) produced a few additional shorter bands (Fig. 2b; Fig. S2b). We randomly sequenced three shorter bands from different T1 lines, revealing that they were PTOX variants with different deletions caused by the activity of PTOX-PTG/Cas9 system (Fig. 2b, c). It is noticeable that deletions residing in three examined bands all started from the target site of gRNA1, but ended at the target sites of different gRNAs, i.e., band 1 in gRNA3, band 2 in gRNA4, and band 3 in gRNA5, generating 1069-bp, 1593-bp, and 2093-bp deletions, respectively (Fig. 2c). The distances from the deletion breakpoints to the corresponding PAM sites were within 10 bp except that the 3′-end breakpoint of deletion in band 1 was 50-bp upstream of its corresponding PAM site (Fig. 2c). It suggests that different gRNAs could guide Cas9 to cut their target sites simultaneously, creating two or more DSBs in one DNA molecule and, consequently, resulting in fragmental deletions.
Different PTOX deletion patterns were observed across T1 plants and each ptox variant had a lower intensity compared to the wild-type sized band, indicating that they resulted from somatic gene-editing events and were only presented in a small portion of cells (Fig. 2b; Fig. S2b). We calculated the somatic deletion rate according to the intensity of the short bands, and found that it ranged from 0 to 87.9% (Fig. 2b; Fig. S2b; Table S2). However, the actual somatic editing rate should be higher, because there should be some SNP or small indel mutations, which could not be resolved by gel electrophoresis, as observed in this and previous studies (Xie et al. 2015).
Taken together, the results showed that the rice PTG/Cas9 system was able to edit the PTOX gene in Arabidopsis as we expected, probably due to the conservation of U3p and UBIp promoters in higher plants. In our case, 24.4% of T1 transgenic plants harbored PCR-detectable deletions in the PTOX gene. However, these were all chimeric plants resulting from somatic gene-editing events, and no homozygous ptox mutant was obtained in this generation. Our results agree with previous reports that gene editing by CRISPR/Cas9 mainly occurred at the somatic level in Arabidopsis (Ma et al. 2015; Feng et al. 2014).
Screening homozygous PTOX null mutants in T2 generation
The ultimate goal to apply the PTG/Cas9 system in Arabidopsis is to create homozygous null mutants for further functional research. Although we did not get any homozygous ptox mutant at the T1 generation, 30 T1 plants harboring somatic editing events raised the possibility of achieving the inheritable mutants in their T2 offspring, especially the ones derived from lines with a high ratio of somatic editing events.
We performed an initial screening on the T2 offspring from 118 surviving T1 transgenic lines (~ 100 seeds per line) including 25 lines with somatic deletions and 93 lines without obvious somatic deletions. Here, all seedlings grew on soil without antibiotic selection. Of 25 T1 lines with detectable deletions, we observed variegated im-like T2 plants in 15 lines (at a ratio of 60%) (Fig. 3a; Table S2). By contrast, in the offspring of 93 T1 plants carrying single wild-type sized PTOX, we only discovered one im-like plant (line #1) (Fig. 3a; Table S2). The ratio of im-like plants in each line ranged mostly from 0 to 7.55%, except that line #46 produced about 45% variegated offspring (Table S2). None of the segregation fit the Mendelian 3:1 ratio, confirming that these T1 plants are genetically heterogeneous (Table S2).

Characterization of putative homozygous ptox mutants in T2 generation. a Phenotypes of five putative homozygous ptox T2 plants. These five plants developed an im-like variegated phenotype. Photos were taken from 3-week-old plants; bar = 1 cm. b Immunoblot analysis of the PTOX protein in five putative homozygous mutants. Wild-type Col-0 and the PTOX null mutant im were used as controls. The asterisk symbol marks unspecific bands detected by the PTOX antiserum. The filter stained by Ponceau S is a loading control. c The PTOX patterns in five putative homozygous mutants and Col-0. d Schematic diagrams of ptox alleles in five mutant lines. Red circles mark nucleotides deleted in the mutants; red letters represent inserted nucleotides; red dashes underline the large deleted regions
We further performed PCR analysis on the PTOX gene of 69 im-like T2 plants from 11 families, including 46 plants from line #46, and 23 plants from 10 other lines (Fig. S3a, b). As a result, 55 plants produced a single ~ 2.5-kb band; seven plants generated a smaller sized single band (Fig. S3a, b). It indicated that these 62 plants were putative homozygotes. In addition, a couple of plants (88-1 and 29-4) produced double bands (Fig. S3a, b), suggesting that they carried two different ptox alleles and were bi-allelic heterozygous mutants. Notably, a small portion of T2 im-like plants (5 plants) still displayed multiple-band patterns, suggesting that somatic editing of the PTOX gene by the PTOX-PTG/Cas9 system could occur in this generation, as well (Fig. S3a, b).
We selected five putative homozygous mutants for further analyses, including three carrying a wild-type sized band (1-1, 29-3, and 78-1), and two with shorter PTOX form (46-21 and 46-22) (Fig. 3a, c). Immunoblot analysis revealed no PTOX protein in these plants, illustrating that they were PTOX null mutants (Fig. 3b). The PCR product amplified by PTOX primers from each putative mutant was sequenced, and results showed that all five plants harbored mutated ptox genes (Fig. 3d), explaining their im-like variegated phenotypes. In these mutants, the sequencing chromatograms of ptox amplicons from plants 1-1, 46-21, 46-24, and 78-1 displayed a high quality and clean signals without any double peak in the whole-gene region, demonstrating that these plants are homozygous mutants. The mutated sites in all four ptox alleles occurred adjacent to the target sites of the designed gRNAs (Fig. 3d). The ptox allele in plant 1-1 (ptox1-1) carried an additional C at the target site of gRNA3. Allele ptox78-1 carried a 1-bp and a 4-bp deletion at the target sites of gRNA3 and gRNA5, respectively. Alleles ptox46-21 and ptox46-24 both harbored a ~ 1-kb deletion between target sites of gRNA3 and gRNA5, and a single-nucleotide insertion at the target site of gRNA2 or gRNA1.
On the contrary, the chromatogram of plant 29-3 had overlapping signals in the region between the target sites of gRNA4 and gRNA5, suggesting that the sample is heterogeneous (Fig. S4a). Therefore, we subcloned the amplicon into the vector pMD-18T, and randomly sequenced ten colonies. The result showed that the PCR product from plant 29-3 consisted of two PTOX isoforms with sequences different around the target sites of gRNA4 and gRNA5 (Fig. 3d, Fig. S4), revealing that plant 29-3 was a bi-allelic mutant exhibiting an im-like phenotype. These two alleles, ptox29-3a and ptox29-3b, both contained a single-nucleotide deletion 4 bp upstream of the gRNA2 PAM site, and ptox29-3b carried two additional indels at the target sites of gRNA4 and gRNA5, which matched the region displaying overlapping sequencing signals (Fig. 3d, Fig. S4).
Altogether, we successfully obtained PTOX homozygous mutants in T2 generation. The five gRNAs seemed to work randomly and unbiasedly in guiding Cas9 to edit the targets. The mutations in T2 homozygous mutants likely originated from somatic gene-editing events occurring in T1 generation. Therefore, T1 plants with a higher somatic editing rate have a higher chance to produce homozygous mutants in their offspring, like in the case of plant #46.
Identifying PTG/Cas9-free homozygous PTOX null mutants
The presence of CRISPR/Cas9 in gene-edited plant will increase the off-target risk, making further complementation test difficult and functional analysis complicated (Gao et al. 2016). It is necessary to remove the PTG/Cas9 system as soon as the mutations are produced. Due to the random integration, the T-DNA carrying PTG/Cas9 is most often not tightly linked with the target gene; therefore, we can easily separate the PTG/Cas9 system from the desired mutant lines simply through natural chromosomal recombination and sequential gene segregation during self-cross.
To obtain homozygous Cas9-free ptox mutants, we examined nine variegated T2 plants from line #46 by PCR. Two plants, 46-2 and 46-9, were found to be T-DNA and Cas9-free, because the Cas9 and Hygr genes were not detected in their genomes (Fig. 4a). The ptox gene in both plants appeared in wild-type size (Fig. 4a), but sequencing revealed that these two mutants identically harbor a 15-bp insertion and a 4-bp deletion at the target sites of gRNA3 and gRNA4, respectively (Fig. 4c), suggesting that they originally descended from the same cell. Since the Hygr and Cas9 genes are co-presented in the T-DNA, we can easily screen the Cas9-free mutant by checking their resistance against hygromycin. When sown on 1/2 MS plates supplemented with hygromycin, T3 offspring of 46-2 and 46-9 was highly sensitive to hygromycin, confirming that they were stable homozygous T-DNA and Cas9-free lines (Fig. 4b; Table S3). In addition, all T3 offspring plants from these two lines developed a variegated im-like phenotype, proving that the ptox allele was stably transmitted from the T2 to the T3 generation (Fig. 4b; Table S3).

Screening Cas9-free homozygous ptox mutants in the T2 progeny of line #46. a Examination of PTOX, Hygr, and Cas9 genes in im-like T2 plants by PCR. Plant 46-2 and 46-9 are putative homozygotes free of T-DNA insertion. b Inheritance of im-like variegation and hygromycin sensitivity of plant 46-2 in T3 generation. The seeds from plant 46-2 were planted on 1/2 MS with or without hygromycin (Hyg) selection. c Diagram of ptox allele in plant 46-2 and 46-9. Mutant alleles in two plants are the same, carrying a 15 bp insertion (red letters) at the gRNA3 target site and a 4 bp deletion (red dashes) in gRNA4. PAM site is in magenta
As to the lines failing to obtain Cas9-free plants in T2 generation, we continued to screen the presence of T-DNA in T3 generation. For instance, we further characterized two putative homozygous mutants carrying obviously shorter ptox alleles (46-4 and 46-5; Fig. 4a) on 1/2 MS medium with hygromycin in T3 generation. Both plants produced 100% of variegated offspring, indicating that they were homozygous. Moreover, plant 46-4 produced ~ 25% hygromycin sensitive and Cas9-free offsprings (Table S3). Since hygromycin only suppresses plant growth but does not kill plants, these sensitive Cas9-free homozygous mutants can be transplanted onto 1/2 MS plate without hygromycin for recovery as they continue to grow in soil.
Gene editing of other Arabidopsis genes with the PTG/Cas9 system
We successfully created Cas9-free PTOX null mutants in Arabidopsis with the rice PTG/Cas9 system, and we intended to expand its application to other genes in Arabidopsis. We focused on five genes that we are highly interested in, while no T-DNA insertional knockout mutants are publically available. By coincidence, all of them are located in chromosome one. Target genes At1g18170, At1g73655, and At1g74070 encode immunophilins; At1g23965 and At1g70270 encode bHLH-like transcription factors. For each gene, 4–6 gRNAs were designed for editing (Fig. 5a, b).

Cas9-free homozygous mutants of 5 Arabidopsis genes created with the PTG/Cas9 system. a The gRNA design and the mutant alleles of three immunophilin genes. b The gRNA design and the mutant alleles of two genes encoding bHLH-like transcription factors. For each gene, the wild-type and mutant gene structures are presented on the left side; the PCR results of the target genes and Hygr in mutants are presented on the right. For PCR analysis, Col carrying empty pRGEB32 plasmid (EP) was used as control. Schemes of gene structures are labeled as in Fig. 1a. The red dotted lines indicate deleted region in mutant alleles
Following the same procedure applied to the PTOX gene, we made a specific PTG/Cas9 construct for each gene and generated corresponding transgenic plants. We started screening T1 transgenic plants for large DNA fragment deletions by PCR. For each gene, we found 15–20 individuals showing multiple bands out of 100 T1 plants. T2 offspring from 2 to 3 T1 plants with somatic deletion rate greater than 20% were analyzed further for the target gene structure and the presence of T-DNA insertion by PCR. We examined about 100 T2 plants in total for each gene. For At1g23965, we found two different Cas9-free homozygous mutant lines. For At1g74070 and At1g70270, we obtained one homozygous and one heterozygous Cas9-free mutant lines. For At1g73655 and At1g18170, we only observed two types of heterozygous mutants. Therefore, we continued the examination on the T3 progeny from these heterozygous T2 mutants to follow the inheritance of the mutation and the presence of T-DNA. Finally, we obtained two different homozygous T-DNA-free mutants for At1g73655 and At1g18170 in T3 generation. All the mutant alleles were sequenced, revealing that the deletions happened between two targets (one of the mutant alleles for each gene is displayed in Fig. 5). Taken together, the rice PTG/Cas9 system was demonstrated to be an efficient gene-editing tool for Arabidopsis.
Discussion
The PTG/Cas9 system contains a repetitive tRNA-gRNA structure, mimicking the original bacterial CRISPR structure. After being expressed in plant cells, the primary transcript of polycistronic tRNA–gRNA will be processed by endogenous tRNA-processing RNases, producing multiple different gRNAs, which guide Cas9 to edit multiple target sites independently and greatly increase the editing efficiency (Xie et al. 2015). When two or more gRNA-Cas9 complexes cut different sites of a targeted gene simultaneously, it will likely generate DNA fragmental deletions that are suitable for regular PCR examination based on amplicon length polymorphisms of the target genes. Taking advantage of this property, we intentionally developed this PCR-based method to identify desired null mutants created by the PTG/Cas9 system.
Efficiency of the rice PTG/Cas9 system in creating Arabidopsis mutants with large deletion
In this study, we evaluated the rice PTG/Cas9 editing efficiency in creating large deletions in Arabidopsis by a conventional PCR method. In Arabidopsis, we observed that PTOX-PTG/Cas9 resulted in about 25% of T1 transgenic plants generating different extents of PCR-detectable deletions in target regions (Fig. 2b; Fig. S2b; Table S2). In T2 generation, about 20% of the mutants (14 out of 69) carried PCR-detectable deletions (Fig. S3). In rice, only 0–11.76% of T1 transgenic plants showed chromosomal fragment deletion between two targets within a gene (Xie et al. 2015). A comparable ratio was observed in T1 transgenic rice carrying another multiplex editing system (Ma et al. 2015). A probable reason for the high deletion rate in this work is that we designed five gRNAs to target PTOX, whereas the other experiments only applied 2–3 gRNAs for one gene. We reasoned that different gRNAs would guide Cas9 to cut their targets independently, and the deletion between two sites would be generated only when two gRNA-Cas9 complexes land on and cut the same gene at the same time. Increasing the gRNA number for one gene could raise this chance and consequently promote the deletion rate.
The PCR-based examination of large deletions is an easy, quick, and cheap method for screening mutants. However, a downside of this approach is that it would miss all the SNPs or small indels if these mutations do not provoke obvious abnormal phenotypes. According to our observation in T2 generation, most of the mutants (55 out of 69) only carried PCR-undetectable small-scale mutations (Fig. 3; Fig. 4; Fig. S3), just like in the case of rice (Xie et al. 2015). Therefore, this method cannot evaluate the actual overall editing efficiency and editing events. For the purpose, we can conduct the direct Sanger sequencing or PCR–RFLP analysis. Nevertheless, several advanced approaches have proven to be more sensitive, for example, the IDAA method (Yang et al. 2015), the combined qPCR/dPCR analysis of the target genes (Mock et al. 2016; Falabella et al. 2017), and NGS analysis of the target gene amplicons (Sentmanat et al. 2018). In addition, a few algorithms have been developed to robustly verify and quantify the CRISPR/Cas9 edits using Sanger sequencing data, such as ICE (Hsiau et al. 2018) and TIDE (Brinkman et al. 2018). For those who wish to evaluate overall gene-editing events or to identify as many mutants as possible in a limited number of transformants, these methods could be better choices.
High somatic editing rate in T1 plants is crucial for achieving homozygous Arabidopsis mutants
Similar to previous reports (Ma et al. 2015; Feng et al. 2014), most editing events in T1 plants with PTOX-PTG/Cas9 happened in somatic cells. We did not observe homozygous or heterozygous ptox mutant in T1 generation based on PTOX band patterns and their phenotypic segregation patterns in T2 progenies (Fig. 2b; Fig. S2b; Table S2). Therefore, the probability of generating heterozygous or homozygous mutant in T2 generation highly relies on whether T1 somatic editing events occurred in stem cells that would further develop to form gametes. In our work, we used PCR-based examination to roughly estimate and compare the editing efficiency among different transformants carrying the PTG/Cas9 system, because the large deletion rate should be positively correlated with the overall editing efficiency. Our results show that when the somatic deletion ratio is higher than 20%, the plants consistently produce variegated im-like offspring (Table S2). Most of the im-like plants are homozygous plants, and about 20% of them carry alleles with large deletion (Fig. 4; Fig. S3). In contrast, among the T1 lines with a lower somatic deletion ratio (0% < ratio < 20%), only 5 out of 15 lines produced putative homozygous mutants (Table S2). Moreover, out of 93 T1 lines without obvious somatic deletions, only one line produced im-like mutants. Therefore, a high somatic editing rate in T1 plants is crucial for achieving homozygous Arabidopsis mutants successfully and rapidly, i.e., in the first generation or second generation.
A protocol to apply PTG/Cas9 in Arabidopsis
With this PTG/Cas9 system, we successfully created deletion mutants in PTOX and other five genes, demonstrating that the PTG/Cas9 system is a reliable gene-editing tool for Arabidopsis. According to our experiences, we established a simple protocol to create and obtain the aimed homozygous Cas9-free deletion mutants in Arabidopsis (Fig. 6). The first step is to design and construct a binary vector carrying the gene-specific PTG/Cas9 system, with gRNA number varying from 3 to 6. Second, the vector is transformed into wild-type plants via Agrobacterium infection and positive transgenic T1 plants are selected after screening against hygromycin. The third step is to evaluate the somatic deletions of target genes in T1 transgenic plants by PCR with gene-specific primers. Normally after screening about 100 T1 transgenic plants, we will obtain more than three lines with deletion rate greater than 20%, and we only need to focus on the offspring of 2–3 lines during the next screening step. T2 plants arising from T1s with a high somatic deletion rate are examined for homozygous or heterozygous mutants by PCR. Plants carrying one short sharp band or two sharp bands corresponding to the target genes are considered as homozygous or heterozygous mutants, respectively. The short bands will be sequenced either directly or after being subcloned into T vector for confirming the mutation. These putative T2 mutants would be examined for T-DNA insertion by PCR with Hygr or Cas9-specific primers. Following this simple protocol, we can obtain homozygous Cas9-free mutants and verify their genetic inheritances in T3 based on the segregation of phenotypes and genotypes. The operations in this protocol, including vector construction, gene transformation, seed propagation, PCR screening, and segregation of transgenes, can be performed routinely in a basic Arabidopsis molecular laboratory. The key point in this protocol is to search for T1 transgenic plants with a high somatic editing rate. The germline cells in those lines have a higher probability to contain mutations and pass them onto the next generation. If a line has a very low somatic editing rate, it is not likely to produce inheritable mutant offspring.

A flowchart for creating Cas9-free homozygous mutant in Arabidopsis
Possible optimizing strategies for applying the PTG/Cas9 system in Arabidopsis
In our work, we directly applied the rice PTG/Cas9 system to edit genes in Arabidopsis without any modification on the major scaffold. It turned out that the rice PTG/Cas9 system could edit genes well in Arabidopsis, probably due to the functional conservation of U3p, tRNA and UBIp between rice and Arabidopsis. However, the processes involved in PTG/Cas9 directed gene editing always have subtle differences amongst plant species, especially the expression and post-transcriptional processing of Cas9 and tRNA–gRNA, which is a prerequisite for the successful application of the PTG/Cas9 system. Rice promoters and tRNAs may not be able to drive the PTG/Cas9 system work with optimal efficiency in Arabidopsis. Therefore, substitution of the rice promoters and tRNAs with those from Arabidopsis would be a good strategy to further optimize this system. Successful application of the PTG/Cas9 system in maize (Qi et al. 2016) and kiwifruit (Wang et al. 2018) are two examples. In these two works, the rice-specific elements in the PTG/Cas9 system, including OsU3p, OstRNA, and OsUBIp, were replaced by corresponding elements from maize and kiwifruit, respectively. The resulting modified systems in both species exhibited a higher editing efficiency compared to the original method in rice.
In addition, increasing the editing efficiency in stem cells would improve the stable transmission of the DNA change, and, therefore, increase the possibility of getting heterozygous or homozygous mutants in fewer generations. It has been shown that the editing efficiency in T1 generation is increased when Cas9 is driven by a promoter of egg cell-specific or meristem-specific genes like EC1.2 (Wang et al. 2015) and Yao (Yan et al. 2015; Li et al. 2018). Heterozygous and homozygous mutants were identified as early as in T1 generation with Cas9 driven by the Yao promoter (Li et al. 2018).
Conclusion
The PTG/Cas9 system was first designed for rice genome editing, and has been used for editing genes in maize and kiwifruit after modification. In this study, we demonstrated that this system also works in Arabidopsis, further indicating that this system has a great potential to edit genes in other plant species. Moreover, by taking advantage of the multiplex PTG/Cas9 system for creating large deletions, we developed a rapid and reliable strategy to identify the aimed Arabidopsis null mutants in three generations with a simple PCR method. Compared to the methods involving mainly screening short indels or SNPs, it reduces DNA sequencing steps, overcomes the limitation of the PCR–RFLP method in selecting suitable gRNA target sites, and minimizes the effort of detecting mutants. This method could also facilitate the mutant screening and identification in other species with any multiplex gene-editing system.
Author contribution statement
MX, AF, YW, and LH designed research; LH, MZ, JH, YH (Yingying Hu), YH (Yanshuang Huo), HH, YH (Yaqi Hao), and WZ performed research; LH, MX, YW, and AF analyzed data; LH, MX, and AF wrote the paper with contributions and approval from all authors.
Abbreviations
- CRISPR:
-
Clustered regularly interspaced short palindromic repeat
- Cas9:
-
CRISPR-associated nuclease 9
- gRNA:
-
Guide RNA
- PAM:
-
Protospacer adjacent motif
- DSB:
-
Double-strand break
- PTG/Cas9:
-
Polycistronic tRNA–gRNA/Cas9
- PTOX:
-
Plastid terminal oxidase
- U3p :
-
U3 snoRNA promoters
- UBIp :
-
Ubiquitin promoter
References
Brinkman EK, Kousholt AN, Harmsen T, Leemans C, Chen T, Jonkers J et al (2018) Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res 46:e58
Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–743
Cong L, Ran FA, Cox DM, Lin S, Barretto RP, Habib N et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Ding Y, Li H, Chen L, Xie K (2016) Recent advances in genome editing using CRISPR/Cas9. Front Plant Sci 7:703
Doudna JA, Charpentier E (2014) The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096
Edwards K, Johnstone C, Thompson C (1991) A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res 19:1349
Falabella M, Sun L, Barr J, Pena AZ, Kershaw EE, Gingras S et al (2017) Single-step qPCR and dPCR detection of diverse CRISPR-Cas9 gene editing events in vivo. G3 (Bethesda) 7:3533–3542
Feng Z, Zhang B, Ding W, Liu X, Yang D, Wei P et al (2013) Efficient genome editing in plants using a CRISPR/Cas system. Cell Res 23:1229–1232
Feng Z, Mao Y, Xu N, Zhang B, Wei P, Yang D et al (2014) Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in Arabidopsis. Proc Natl Acad Sci USA 111:4632–4637
Fineran PC, Charpentier E (2012) Memory of viral infections by CRISPR-Cas adaptive immune systems: acquisition of new information. Virology 434:202–209
Fu A, Aluru M, Rodermel SR (2009) Conserved active site sequences in Arabidopsis plastid terminal oxidase (PTOX): in vitro and in planta mutagenesis studies. J Biol Chem 284:22625–22632
Fu A, Liu H, Yu F, Kambakam S, Luan S, Rodermel S (2012) Alternative oxidases (AOX1a and AOX2) can functionally substitute for plastid terminal oxidase in Arabidopsis chloroplasts. Plant Cell 24:1579–1595
Gao Y, Zhao Y (2014) Self-processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing. J Integr Plant Biol 56:343–349
Gao X, Chen J, Dai X, Zhang D, Zhao Y (2016) An effective strategy for reliably isolating heritable and Cas9-free Arabidopsis mutants generated by CRISPR/Cas9-mediated genome editing. Plant Physiol 171:1794–1800
Gasiunas G, Barrangou R, Horvath P, Siksnys V (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA 109:15539–15540
Hsiau T, Maures T, Waite K, Yang J, Kelso R, Holden K et al (2018) Inference of CRISPR edits from Sanger trace data. bioRxiv. https://doi.org/10.1101/251082
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Kleinboelting N, Huep G, Appelhagen I, Viehoever P, Li Y, Weisshaar B (2015) The structural features of thousands of T-DNA insertion sites are consistent with a double-strand break repair-based insertion mechanism. Mol Plant 8:1651–1664
Kleinboelting N, Huep G, Weisshaar B (2017) Enhancing the GABI- Kat Arabidopsis thaliana T-DNA insertion mutant database by incorporating Araport11 annotation. Plant Cell Physiol 58:e7
Lei J, Xu X, Dai P, Li J, Zhang J, Liu X (2016) Functional analysis of different truncated U3 promoters in cotton. Cotton Sci 28:307–314 (in Chinese)
Li Y, Rosso MG, Ulker B, Weisshaar B (2006) Analysis of T-DNA insertion site distribution patterns in Arabidopsis thaliana reveals special features of genes without insertions. Genomics 87:645–652
Li J, Norville JE, Aach J, Mccormack M, Zhang D, Bush J et al (2013) Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotechnol 31:688–691
Li C, Chen C, Chen H, Wang S, Chen X, Cui Y (2018) Verification of DNA motifs in Arabidopsis using CRISPR/Cas9-mediated mutagenesis. Plant Biotechnol J. https://doi.org/10.1111/pbi.12886
Liu W, Yuan JS, Stewart CN (2013) Advanced genetic tools for plant biotechnology. Nat Rev Genet 14:781–793
Liu X, Wu S, Xu J, Sui C, Wei J (2017) Application of CRISPR/Cas9 in plant biology. Acta Pharmaceutica Sinica B 7:292–302
Ma X, Zhang Q, Zhu Q, Liu W, Chen Y, Qiu R et al (2015) A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Molecular Plant 8:1274–1284
Ma X, Zhu Q, Chen Y, Liu Y (2016) CRISPR/Cas9 platforms for genome editing in plants: developments and applications. Mol Plant 9:961–974
Mali P, Yang L, Esvelt KM, Aach J, Guell M, Dicarlo JE et al (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826
Mock U, Hauber I, Fehse B (2016) Digital PCR to assess gene-editing frequencies (GEF-dPCR) mediated by designer nucleases. Nat Protoc 11:598–615
Monia BP, Ecker DJ, Crooke ST (1990) New perspectives on the structure and function of ubiquitin. Nat Biotechnol 8:209–215
Nekrasov V, Staskawicz BJ, Weigel D, Jones JD, Kamoun S (2013) Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat Biotechnol 31:691–693
Nissim L, Perli SD, Fridkin A, Perezpinera P, Lu TK (2014) Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol Cell 54:698–710
O’Malley RC, Ecker JR (2010) Linking genotype to phenotype using the Arabidopsis unimutant collection. Plant J 61:928–940
Ozkaynak E, Finley D, Solomon MJ, Varshavsky A (1987) The yeast ubiquitin genes: a family of natural gene fusions. EMBO J 6:1429–1439
Qi W, Zhu T, Tian Z, Li C, Zhang W, Song R (2016) High-efficiency CRISPR/Cas9 multiplex gene editing using the glycine tRNA-processing system-based strategy in maize. BMC Biotechnol 16:58
Sentmanat MF, Peters ST, Florian CP, Connelly JP, Pruett-Miller SM (2018) A survey of validation strategies for CRISPR-Cas9 editing. Scientific Rep 8:888
Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z et al (2013) Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol 31:686–688
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Wang D, Fu A (2016) The plastid terminal oxidase is a key factor balancing the redox state of thylakoid membrane. Enzymes 40:143–171
Wang Z, Xing H, Dong L, Zhang H, Han C, Wang X et al (2015) Egg cell-specific promoter-controlled CRISPR/Cas9 efficiently generates homozygous mutants for multiple target genes in Arabidopsis in a single generation. Genome Biol 16:144
Wang Z, Wang S, Li D, Zhang Q, Li L, Zhong C et al (2018) Optimized paired-sgRNA/Cas9 cloning and expression cassette triggers high-efficiency multiplex genome editing in kiwifruit. Plant Biotechnol J 16:1424–1433
Xie K, Minkenberg B, Yang Y (2015) Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc Natl Acad Sci USA 112:3570–3575
Yan L, Wei S, Wu Y, Hu R, Li H, Yang W et al (2015) High-efficiency genome editing in Arabidopsis using YAO promoter-driven CRISPR/Cas9 system. Mol Plant 8:1820–1823
Yang Z, Steentoft C, Hauge C, Hansen LK, Thomsen AL, Niola F et al (2015) Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res 43:e59
Zhang Z, Mao Y, Ha S, Liu W, Botella JR, Zhu J (2016) A multiplex CRISPR/Cas9 platform for fast and efficient editing of multiple genes in Arabidopsis. Plant Cell Rep 35:1519–1533
Acknowledgements
We thank Dr. Kabin Xie (Huazhong Agriculture University) for providing pGTR and pRGEB32 vectors. We also like to thank Professor Jean-David Rochaix (University of Geneva) for revising the manuscript. This project was supported by National Natural Science Foundation of China (NSFC), No. 31471261 and No. 31270284 to AF and No. 31371226 to MX, Shaanxi Province Education Department Key Laboratory Research Project (No. 13JS108) to MX, and Shaanxi Province Education Department Special Scientific Research Project (No. 16JK1792) to WZ.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by J. Van Huylenbroeck.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hui, L., Zhao, M., He, J. et al. A simple and reliable method for creating PCR-detectable mutants in Arabidopsis with the polycistronic tRNA–gRNA CRISPR/Cas9 system. Acta Physiol Plant 41, 170 (2019). https://doi.org/10.1007/s11738-019-2961-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-019-2961-3