
Abstract
Kunitz trypsin inhibitor (KTI) is one of the widely studied protease inhibitors (PIs) and has been reported to take part in plant defense mechanism during pathogenesis. Numerous plant-origin recombinant KTIs have been described to exhibit anti-pathogenic properties and were used to fight pathogens in the field of pharmacology and agriculture. In this study, a novel Kunitz trypsin inhibitor gene, ClKTI, was isolated from a medicinal herb plant, turmeric, Curcuma longa. The full-length ClKTI gene is 754 bp long (Accession No. KF889322.1 in NCBI database) and it was obtained using 5′/3′ rapid amplification of cDNA ends (RACE) technique. ClKTI has an open reading frame of 639 bp length which encodes for 213 amino acids and contains the Kunitz-family motif, (V-X-D-X2-G-X2-L-X5-Y-X-I) and an altered reactive site motif, (G/E-I-S). Sequence similarity search using BLASTX showed that ClKTI shared the highest similarity to KTI from Theobroma cacao with 58% max identity while conserved domains search resulted in ClKTI having specific hits with Kunitz-family soybean trypsin inhibitor (STI). Phylogenetic studies suggested that ClKTI is related to T. cacao while protein homology modeling analysis indicated that it has 12 β-sheets with three disulfide bridges. Using real-time quantitative PCR, the ClKTI gene expression pattern in five different tissues (flower, basal stem, stem, rhizome and root) treated with methyl-jasmonate (MeJA) was studied where MeJA was suggested to regulate expression of PI genes in plants. The results indicated that the expression of ClKTI generally increased in the MeJA-treated tissues with the root tissues possessing the highest expression and stem tissues showed the highest expression fold-change.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Protease inhibitors (PI) are proteins that are capable to inhibit proteases by regulating proteolytic activities inside cells. They form stable complex with target proteases resulting in either blockage or alteration of active structures of the proteases (Habib and Fazili 2007; Oddepally et al. 2013). PIs are commonly found in a diverse group of microorganisms, animals and also plants (Lawrence and Koundal 2005; Haq et al. 2004; Habib and Fazili 2007). In fact, a large amount of plant-origin PIs has been identified and characterized (Bijina et al. 2011; de Oliveira et al. 2012) and most of them were isolated from storage organs such as seeds and tubers. In plant system, PIs involve in numerous physiological roles ranging from plant development to stress-related responses. One of the interesting roles of PIs is their involvement in plant defense mechanism. Their concentration were significantly increased at the sites of pathogen attack, followed by subsequent accumulation at all other parts of the plant (Lawrence and Koundal 2005; Haq et al. 2004; Fan and Wu 2005; Habib and Fazili 2007). In another case, PIs that were fed to larvae of flour beetle were shown to be toxic and retarded the larvae growth (Habib and Fazili 2007; Koiwa et al. 1997). Therefore, it was concluded that proteases and other proteolytic enzymes secreted by the pathogens during pathogenesis had been deactivated by the PIs. The PI characteristic of being able to deactivate pathogenic proteases has since attracted many researches to develop recombinant PIs (Lawrence and Koundal 2005). PI genes could be incorporated into crop plants and expressed as a defensive mechanism tool against insects and pathogens. Recombinant PIs could also be synthesized as medicines to treat viral and bacterial infections (Haq et al. 2004; Oddepally et al. 2013).
PIs are classified based on the DNA sequence homology of their inhibitory domains. Common PI families that gain the interest of researchers include Bowman–Birk, serine protease inhibitors (Serpin), cysteine protease inhibitors (cystatins) and Kunitz type. These PIs families have been reported to display anti-pathogenic, anti-fungal, anti-viral and anti-feedant properties (Habib and Fazili 2007; Haq et al. 2004). PIs in Kunitz family are small molecules with size ranging between 18 and 24 kDa and contain serine and trypsin inhibitor domains. Kunitz-type trypsin inhibitor (KTI) is a PI from the Kunitz family that has low cysteine content. It comprises one or two polypeptide chains with two disulfide bridges formed from four cysteine residues. KTI’s single reactive site is found in one of its protein loop that contains an arginine residue (Habib and Fazili 2007; Huang et al. 2010; Oliva et al. 2010; de Oliveira et al. 2012; Oddepally et al. 2013). However, there are KTIs that have more than one inhibitory domain, in which a single protein has both activities against serine and cysteine proteases (Migliolo et al. 2010). Enzymes that are inhibited by KTIs include chymotrypsin, α-amylase, human plasmin and HIV reverse transcriptase (Birk 2010; Fang et al. 2010; Oddepally et al. 2013). KTIs were also reported to show bioinsecticidal activity as it was able to inhibit trypsin-like activities in the larval midgut of several pest insects and leads to retardation of larvae growth (Cruz et al. 2013; Garcia et al. 2004; Oliva et al. 2010).
Turmeric, Curcuma longa, is a medicinal plant that belongs to the Zingiberaceae family. It is abundantly found in Asian countries including India, China, Thailand and Malaysia. It has been used as folk medicine for common illness due to its anti-oxidant, anti-fungal and anti-inflammatory medicinal properties (Apisariyakul et al. 1995; Araújo and Leon 2001; Lantz et al. 2005; Selvam et al. 1995). Generally, compounds found in medicinal plants are usually utilized for drug discovery, especially towards inhibiting viral infection (Mukhtar et al. 2008; Baskaran et al. 2015). Sookkongwaree et al. (2006) reported that phenolic extracts of plants from Zingiberaceae family, including C. longa, exhibited inhibition activities toward viral proteases from human immunodeficiency virus-1 (HIV-1), hepatitis C virus (HCV) and human cytomegalovirus (HCMV). The report, however, does not specify the exact compound that contributed to the inhibition which has led us to consider on the presence and roles of protease inhibitors as one of the potential inhibitory compound. However, at this time point this is merely a speculation.
In the recent years, genes coding for anti-pathogenic compound (such as Bt toxin) have been incorporated into crop plants to provide protection against pathogens and pest insects. However, the effectiveness of the currently used anti-pathogenic compound is slowly decreasing as pathogens start to adapt and gain resistance towards them. Discovery of PIs from novel sources, such as in turmeric could be helpful in reducing the resistance built up by pathogens as they have not been exposed to the pathogens and pest insects before (Lawrence and Koundal 2005; Haq et al. 2004; Habib and Fazili 2007). Curcuma longa is a suitable novel source candidate to discover novel PIs; owing to its medicinal properties and has been proven to exhibit inhibition on viral proteases in its crude extracts.
However, to the best of our knowledge, there are limited reports on the characterization of PIs from C. longa. Previously, we have reported the characterization of cysteine protease inhibitor genes found in turmeric (Chan et al. 2014). In this current study, we report the isolation of a full-length complementary-DNA (cDNA) of Kunitz-type trypsin inhibitor from C. longa (designated ClKTI) and its sequence analysis. A phylogenetic tree was generated to study the relationships between ClKTI and KTIs of other plant species and homology modeling was conducted to predict the theoretical tri-dimensional structure of the putative KTI. Finally, gene expression profiling of ClKTI in five different tissues of C. longa plant treated with methyl-jasmonate (MeJA) was conducted using real-time quantitative polymerase chain reaction (RT-qPCR) as MeJA was reported in several studies to induce the gene expression of KTI (Haruta et al. 2001; Major and Constabel 2008; Oliva et al. 2010).
Results
Molecular cloning and characterization of novel KTI cDNA from Curcuma longa
A partial cDNA fragment of ClKTI with the length of 378 bp was amplified from total RNA of C. longa leaf tissues. Based on the sequence of this partial fragment, a new set of PCR primers was designed to amplify a full-length ClKTI gene. Using 5′ and 3′ rapid amplification of cDNA ends (RACE) method, a full-length ClKTI gene with the total length of 754 bp nucleotides was successfully isolated with a predicted open reading frame (ORF) of 639 bp length. A start codon, ATG, is located at nucleotide position 3 while a stop codon, TAG, is located at position 642–645. The ORF encodes for a putative ClKTI protein; consisting of 213 amino acids with a calculated molecular mass of 22.8 kDa and a theoretical pI of 5.06. A signal peptide is found on the putative ClKTI protein and the cutoff point is located between the 24th and 25th amino acids (Fig. 1).

A full-length sequence of ClKTI with deduced amino acid sequence. A start codon, ATG, is located at nucleotide position 3 while a stop codon, TAG, is located at position 642–644. The motif of Kunitz trypsin inhibitor (KTI) ([L,I,V,M]-X-D-X2-G-X2-[L,I,V,M]-X5-Y-X-[L,I,V,M]) and the reactive site motif (G/E-I-S) were detected as boxed. Signal peptide was detected (SignalP 4.1, http://www.cbs.dtu.dk/services/SignalP/) on the sequence, spanned by a length of 24 amino acids and the calculated cutoff point is between the 24th and 25th amino acids (AEA-TS), as indicated by the black arrow
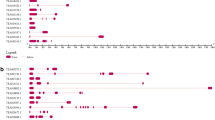
From the sequence analysis, the isolated ClKTI cDNA was confirmed to be a Kunitz-type protease inhibitor as it has the Kunitz-family motif, ([L,I,V,M]-X-D-X2-G-X2-[L,I,V,M]-X5-Y-X-[L,I,V,M]), and an altered reactive site motif, (G/E-I-S), (Fig. 1) which are commonly present in this family. The conserved domain (CD) search has confirmed that ClKTI belongs to the Kunitz family as it has specific conserved domains that is normally been found in Kunitz-family soybean trypsin inhibitor (STI). Moreover, multiple sequence alignment of ClKTI with KTI of other plant species and BLAST analysis has also shown that the ClKTI shares consensus motifs found in other KTI members (Fig. 2). ClKTI has the highest similarity to KTI protein isolated from Theobroma cacao (Accession No. XP_007029360) with a similarity of 58% and the E value of 3E−49. Therefore, our ClKTI cDNA sequence has been deposited into National Center for Biotechnology Information (NCBI) GenBank database (http://www.ncbi.nlm.nih.gov) with the Accession No. KF88932.1.

Multiple sequence alignments of deduced amino acids sequence of ClKTI with sequences of KTI from different plant species. The reactive site of KTI is boxed. The entries are Nicotiana tabacum (ACL12055.1), Arabidopsis thaliana (NP565061.1), ClKTI (AHJ25677.1), Pisum sativum (O82711.1) and Theobroma cacao (XP007029360.1). The identical amino acids (conserved) between the sequences, including the KTI motif, were highlighted in yellow. The similar block amino acids were highlighted in green while conservative amino acids were highlighted in blue
Evolution relationships and homology modeling
An unrooted phylogenetic tree was constructed using neighbor-joining method using deduced amino acid sequence of ClKTI (Accession No.: AHJ25677.1) and sequences of KTI genes from other plants. The resulted phylogenetic dendrogram (Fig. 3) shows that there are three main clades formed from the sequences and ClKTI is grouped together with Theobroma cocao, Arabidopsis thaliana and Nicotiana tabacum under the same clade. On the other hand, there are two other clades that contain plants from Salicaceae (Populus tremula, Populus alba, Populus balsamifera, Populus nigra, Salix triandra and Salix myrsinifolia) and Fabaceae families (Glycine max and Bauhinia rufa).

Phylogenetic tree was constructed using neighbor-joining method with 100 bootstrapping using deduced amino acids sequence of ClKTI (Accession No.: AHJ25677.1, Curcuma longa) with other KTI sequences from different plant species. ClKTI shows the closest evolutionary relationship with KTI belonging to Theobroma cacao
Tri-dimensional protein homology modeling is conducted to predict a protein structure of a given amino acid sequence using a homologue protein with resolved experimentally tri-dimensional structure as template (Biasini et al. 2014). The theoretical tri-dimensional protein structure of ClKTI was modeled (Fig. 4) using a trypsin inhibitor isolated from Murraya koenigii as template (PDB ID: 3zc9, UniProt Accession No.: D2YW43). Our ClKTI 3D protein was modeled with 80% sequence coverage and has a 40% similarity with the template. The 3D model also contains 12 beta-sheets and three disulfide bridges which were formed from six cysteine residues.

Ribbon representation of the predicted tri-dimensional protein model of ClKTI based on homology modeling. Cysteine residues were represented in green ball-and-stick and disulfide bridges were indicated in lines and labeled. The theoretical ClKTI tri-dimensional protein structure was modeled using a trypsin inhibitor from Murraya koenigii as model template (SWISS-MODEL software, http://swissmodel.expasy.org/)
RT-qPCR analysis
Identification and validation of reference gene candidates for qPCR in Curcuma longa
To date there has been no reports on RT-qPCR experiments or reference gene validation studies being conducted on C. longa. Reference genes play an important role in normalizing the expression level of the target gene that is being studied. In our experiment, we chose three most commonly used reference gene candidates for the gene expression studies of other plant species. Actin, alpha-tubulin and ubiquitin (ubox) genes were selected for the expression stability validation in five different tissues of C. longa. PCR primers for each reference gene candidate and the gene of interest are shown in Table 1. It is important to ensure the specificity and efficiency of the primers coding for the reference genes are suitable to be used prior to validation.
A single band of PCR products was generated when tested with cDNAs from five different tissues (flower, stem, basal stem, rhizome and root) as template, confirming the specificity of the primers. Melting curve analysis also showed that a single peak was formed for the reference gene and the gene of interest. For the efficiency test, standard curves were plotted from the RT-qPCR amplification data and the results showed that the efficiencies of all the primers were in the range of 98–109% with r 2 value of 0.98–0.99 (Table 1). These results indicated that all of the primers had fulfilled the minimum requirement of primer specificity and efficiency for RT-qPCR expression analysis.
Then, the expression level of three reference gene candidates was determined in five different C. longa tissues with RT-qPCR. Reference genes’ expression stability test was performed using the quantification cycle value, Cq, of the reference genes in the different tissues and the results were analyzed using RefFinder program (http://fulxie.0fees.us/). The results indicated that Actin was the most stable reference gene among the other candidates and was selected to be used in the expression profiling of ClKTI (Fig. 5).

Expression stability of the reference gene candidates and their stability ranking generated by RefFinder. The quantification cycle value, Cq, of the three reference genes expressed in the five different tissues, namely flower, stem, basal stem, rhizome and root was used in the analysis. a RefFinder incorporates different software and algorithms to calculate the expression stability and ranked them. b A comprehensive result generated by RefFinder and actin was determined as the most stable reference gene
ClKTI expression profiling
The expression profile of ClKTI gene in control plant (non-treated) and MeJA-treated C. longa tissues was conducted with Actin used as the reference gene. The target tissues used in the study were flower, stem, basal stem, rhizomes and root tissues. As shown in Fig. 6, generally the normalized fold expression of ClKTI in the MeJA-treated tissues is higher than the expression in control tissues. Root tissues have the highest value of normalized fold expression while basal stem tissues are the lowest in both treatments. Then, using ∆∆CT method, the increase in expression fold of ClKTI in comparison to the control and MeJA tissues was calculated. The highest increase of ClKTI is in the MeJA-treated stem tissues with the expression value of 9.78-fold (Table 2).

Normalized fold expression of ClKTI in methyl-jasmonate (MJ)-treated and -non-treated (NT) tissues by real-time qPCR using Actin as reference gene for normalization. Generally, the expression of ClKTI was higher in methyl-jasmonate-treated tissues as compared to control tissues
Discussion
The discovery of new protease inhibitors from novel sources would have a tremendous effect in decelerating resistance evolution in pathogens. Protease inhibitors, especially from Kunitz family, are constantly under research in the field of pharmacology and agriculture (Habib and Fazili 2007; Haq et al. 2004; Huang et al. 2010; Major and Constabel 2008) due to their potential inhibitory properties against pathogens. In this study, a novel full-length cDNA encoding a Kunitz-type trypsin inhibitor from C. longa was isolated and characterized.
The 754-bp-long ClKTI gene sequence which was obtained from leaf tissues of C. longa through 5′/3′ RACE method contains two start codons at the 5′ end and a poly A tail at 3′ end with three stop codons. This has suggested that a full-length cDNA of the gene was successfully isolated with complete open reading frames (ORFs). The putative protein deduced from the longest ORF has a calculated mass of 22 kDA, which is a common protein size found within Kunitz-type inhibitors (Habib and Fazili 2007; Oliva et al. 2010). Homology sequence search of ClKTI through protein databases in NCBI reveals that it possesses 58% sequence similarity with Kunitz-type inhibitors isolated from T. cacao. The similarity percentage, however, is relatively low as protein sequences of protease inhibitors in Kunitz family were reported to be highly diverse and can go as low as 25% in similarity (Major and Constabel 2008; Oliva et al. 2010).
Sequence analysis has shown that our ClKTi gene contains several motifs and characteristics that are commonly found in Kunitz-type PIs family. They are ([L,I,V,M]-X-D-X2-G-X2-[L,I,V,M]-X5-Y-X-[L,I,V,M]) and the reactive site, glycine/glutamate-isoleucine-serine (G/E-I-S) motif. The reactive site motif found in ClKTI is the glycine/serine residues, G/S, which is a distinct alteration from the common reactive site motif consisting Arg63-Ile64 (STI numbering) (Habib and Fazili 2007; Koide et al. 1973; Major and Constabel 2008; Oliva et al. 2010; Srinivasan et al. 2005). One interesting characteristic that was found in our ClKTI is that it possesses six cysteine residues which could potentially form three disulfide bridges (Fig. 4). Unlike other members in Kunitz-type PIs family that usually have four cysteine residues (which could form only two disulfide bridges), ClKTI has six cysteine residues that would form three disulfide bridges. It is, therefore, being speculated that ClKTI has a very stable tertiary protein structure due to the presence of these three disulfide bridges. Nevertheless, these variations do not have any major or obvious negative effects on the reactivity of the proteins. In fact, KTIs that are devoid of disulfide bonds such as BrTI (B. rufa trypsin inhibitor) and KTI with single disulfide bond, like SWTI (Swartzia pickelli trypsin inhibitor), were reported to show positive inhibitory activities to trypsin activities (Do Socorro et al. 2002; Oliva et al. 2010). As for the alterations in reactive site motifs, KTIs isolated from chickpea G/E-I-S motif were still shown to be functional and maintained their inhibitory activity (Do Socorro et al. 2002; Oliva et al. 2010; Srinivasan et al. 2005).
The differences in number of cysteine residues, disulfide bonds and sequence alteration in the reactive sites of KTI could be due to adaptations of the plants in defending themselves against pathogens. Sequence and structure modifications of proteins in plants is the result of evolution from their initial ancestral precursors (Lingaraju and Gowda 2008; Oliva et al. 2010) and they are crucial for plants to constantly adapt and become resistant towards insect and pathogen attacks. Since the similarity of KTI sequences, apart from the conserved region, is low within KTIs family, the characteristics that determine the evolution divergence are based on the presence or absence of single or multiple disulfide bonds (Lingaraju and Gowda 2008; Oliva et al. 2010). This may also explain the behavior of the phylogenetic tree that was generated with our ClKTI and KTIs from other plant species (Fig. 3). The tree does not have a distinctive clade grouping of the KTIs except for those of the same species (Populus sp. and Salix sp.) or with similar or close characteristics. From the phylogenetic tree analysis, KTIs from G. max and B. rufa were grouped together as they are both single chains and have one disulfide bond (Oliva et al. 2010).
In the gene expression profiling, RT-qPCR analysis is one of the most common and extensively used techniques. In this analysis, it is important to use validated reference genes for expression normalization. Reference genes play a major role in normalizing the expression level of the gene of interest in the expression profiling. Suitability of DNA primers for RT-qPCR is also equally important. Efficiencies and specificity of the primers must be in accordance with the MIQE guidelines (Bustin et al. 2009; Taylor et al. 2010). However, for C. longa, there are not many studies reported with regard to RT-qPCR analysis. Therefore, we selected a few genes that were commonly used in RT-qPCR analysis performed onto other Zingiberaceae family such as actin, alpha-tubulin and ubiquitin (ubox) as the reference gene candidates. The primers were designed (Table 1) based on the nucleotide sequences from Arabidopsis and subjected to specificity and PCR efficiency tests. Results obtained from melting curve analysis, standard curve analysis and r 2 value (Table 1), concluded that the specificity, PCR efficiency and r 2 value of the primers were in the acceptable range (90–110%). Efficiencies lower than the range suggest the presence of contaminants of PCR or inactive Taq polymerase while efficiencies higher than the range might be due to the formation of primer dimers (Taylor et al. 2010). These tests eliminated the possibility of false-positive result and to ensure that the primers were capable of producing a reaction with a twofold increase of amplicons per cycle (100%).
As the reference genes play a major role in normalizing the expression level of the gene of interest in the gene expression profiling, the most expression stable reference gene need to be validated. Once the efficiencies and specificity of the primers were confirmed, validation of the most expression stable reference genes among the selected candidates were calculated using quantification cycle value, Cq. Commonly used programs to calculate the stability of reference genes are Bestkeeper (Pfaffl et al. 2004), NormFinder (Andersen et al. 2004), GeNorm (Vandesompele et al. 2002) and the comparative delta-ct method (Silver et al. 2006). Whilst all these programs are equally competent to calculate the stability of reference genes, each of them utilizes different computational method and possibly produced a varied outcome (Xie et al. 2012). Therefore, we used RefFinder, a web-based program (URL: http://fulxie.0fees.us/) to evaluate and determine the most stable reference gene for our study. RefFinder integrates the results from Bestkeeper, NormFinder, GeNorm and the comparative delta-ct method and provides a comprehensive final overall by allocating appropriate weight to each reference genes based on the rankings obtained from each program (Xie et al. 2012). From the results, Actin was ranked as the most expression stable reference gene among the candidate genes (Fig. 5). Zhang et al. (2015) has reported that RefFinder was successfully being employed in their experiment to identify and validate the stability ranking of the reference gene candidates in the samples of Helicoverpa armigera.
Expression profiling of ClKTI in the five different tissues was performed with Actin as the reference gene for normalization. As shown in Fig. 6, the samples treated with MeJA, which mimic the effect of pathogens and pest insects attack, showed a higher ClKTI expression level when compared to the non-treated samples. A similar expression pattern was observed in the studies of KTI genes from different parts of poplar trees (flower, apical shoot and young leaves) in which their gene expression was induced in the treatment by MeJA when compared to untreated samples (Haruta et al. 2001; Major and Constabel 2008). In our study, the root tissues showed the highest normalized expression in both MeJA-treated and untreated (control) samples, while stem tissues had the highest increment in fold-change ratio of treated to untreated. These observations are consistent with the study reported by Koiwa et al. 1997 where jasmonate was shown to play a part in the PI synthesis pathway (also known as octadecanoid pathway) and the pathway was triggered by wounding and oral secretion of insects. As PIs have been postulated to be involved in plant defense mechanisms, a higher concentration of ClKTI in the roots as compared to other tissues of the plants was expected. Roots are exposed to a higher chance of pathogen attacks and they are among the first line of defense for plant during pathogen attack. Meanwhile, tremendous increment of fold-change was shown in the stem tissues. This could be due to the systemic transportation of PIs (including ClKTI) through the stem tissues to all parts of the plants after the increased of PIs production which was triggered earlier by the MeJA treatment. It has been suggested that after the attacks by pathogens and pest insects, local signals would induce the production of PIs as part of the plant defense response and distal defense responses are induced through systemic signals mobilized through vascular system (Koiwa et al. 1997).
In conclusion, our study has shown that with the presence of the relevant motifs and characteristics, the isolated full-length novel cDNA has been confirmed as a Kunitz-type trypsin inhibitor. The predicted ORF was expected to be expressed as a functional and inhibitory reactive recombinant protein as it shared sequence homology with other KTIs especially on the reactive site and the occurrence of disulfide bonds. Reference gene candidates for RT-qPCR in C. longa had been validated and the expression profiling of the isolated ClKTI was conducted. The increased abundance of ClKTI in MeJA-treated tissues in comparison to non-treated tissues could suggest the involvement of PIs in plant defense mechanism. More studies could be carried out on ClKTI to further deepen our understanding on plant PIs and possibly lead to the production of a new multi-spectrum anti-pathogenic compound that can be used or modified to combat pests and pathogens.
Materials and methods
Plant materials and treatment
Turmeric (C. longa) plants were grown and maintained in a botanical garden at Universiti Putra Malaysia (UPM), Malaysia. The plants were planted individually in pots. Six-month-old plants were chosen for methyl-jasmonate (MeJA) treatment prior to sampling. The plants were placed inside a sealed growth chamber and treated with cotton ball soaked with 5 mL of 10% MeJA in ethanol for 24 h (Hildmann et al. 1992; Farmer et al. 1992; Lomate and Hivrale 2012; Moura and Ryan 2001). After 24 h of incubation, tissues of the plant were excised at the stalk with a sterilized blade and collected, washed with distilled water and ground immediately in liquid nitrogen using pre-chilled mortar and pestle. About 200 mg of the samples were aliquoted into 2-mL microcentrifuge tubes and was used for total RNA extraction.
Data mining and primer design
Nucleic acid sequences of different plants’ KTIs, including raw NGS transcriptomic data of C. longa were mined at NCBI website (http://www.ncbi.nlm.nih.gov). Multiple sequence alignment was performed using AlignX, Vector NTI Advance 11 (Invitrogen, USA) and the conserved regions with high similarity were screened. A pair of primer (KTI_F, 5′-GGGCCACGAGTACTACATCC-3′ and KTI_R, 5′-CCAGCTTGTAGTCCCTCTCC-3′) was then designed based on the conserved regions and the expected size of the amplified fragment was around 380 bp.
Total RNA extraction
Total RNA from the leaf tissues was extracted using RNazol RT (Molecular Research Center Inc., USA) according to the manufacturer’s protocol while the tissues from rhizomes and roots were extracted using modified CTAB method (Chan et al. 2014; Jamalnasir et al. 2013). Integrity of the extracted RNA was determined by gel electrophoresis with 1.0% agarose gel stained with ethidium bromide and visualized under ultraviolet (UV) light. Quality and quantity of the total RNA were determined using NanoDrop ND 1000 (Thermo Scientific, USA) where only total RNAs with A 260/280 ratio of 1.80–2.00 were used for further applications. Presence of DNA contamination was removed using DNase I, Amplification Grade (Thermo Scientific, USA) according to the manufacturer’s protocol and high-purity total RNA was kept in −80 °C for further use.
cDNA synthesis and polymerase chain reaction (PCR)
About 1 ug of high-integrity and -purity total RNA was used for cDNA synthesis using iScript™ Reverse Transcription Supermix for RT-qPCR (Bio-Rad, CA, USA) by following the manufacturer’s protocol. First-round PCR was performed in a thermocycler in a 20-μL reaction mixture using the synthesized cDNA as template (GoTaq®Flexi DNA polymerase kit, Promega, USA). The PCR was carried out with an initial denaturing step at 94 °C for 3 min, 30 rounds of 94 °C for 45 s, 65 °C for 45 s, 72 °C for 45 s and a final elongation step at 72 °C for 7 min. The PCR product was purified from agarose gel using QIAquick PCR product purification kit (Qiagen, USA). The purified PCR product was ligated into a cloning vector, pGEM-T easy (Promega, USA) and cloned into TOP10 E. coli cells according to the manufacturer’s protocol. Blue/white screening was conducted and colony containing the insert was cultured for plasmid extraction. The plasmid extraction was carried out with GeneJETplasmid mini-prep kit (Thermo Scientific, USA) and the plasmid harboring the ligated product was sequenced. The sequence of 378 bp length was subjected to Basic Local Alignment Search Tool (BLAST) at NCBI website and the identity of the gene was determined.
Molecular cloning of ClKTI cDNA and sequence analysis
Initially, nucleotide sequences of plant KTI of different species (obtained from the NCBI database) were aligned to identify conserved domains of KTIs. A pair of polymerase chain reaction (PCR) primers was generated, KTI_F (5′-GGGCCACGAGTACTACATCC-3′) and KTI_R (5′-CCAGCTTGTAGTCCCTCTCC-3′), based on the conserved domains and a cDNA fragment was obtained. 5′/3′ RACE (Rapid Amplification of cDNA Ends) method was used to obtain the full-length cDNA of ClKTI. Specific primers towards 5′ end, KTI5_R (5′-AGCCACTGCGAGCGGGCATGCGAA-3′), and 3′ end, KTI3_F (5′-TTGCGTGCAGTCGACGGTGTGG-3′), were designed based on the 378-bp-long partial cDNA sequence obtained from the first-round PCR. All 5′/3′ RACE were carried out using SMARTer RACE kit (Clontech, CA, USA) and it was according to the manufacturer’s protocol. The 5′ and 3′ RACE products were analyzed, purified and sent for sequencing as described above. Using the sequences obtained from 5′/3′ RACE, a pair of primer KTIF_F (5′-ACATGGGGAACATCGCTGACA-3′) and KTIF_R (5′-TGACAAGGAAAATCCACATCT-3′) was designed to isolate the full-length ClKTI from 5′ end’s extremity to poly A tail. The PCR started with an initial heating at 94 °C for 3 min, 30 cycles of 94 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s and final elongation at 72 °C for 7 min. The PCR product was analyzed in 1% agarose gel and VC 100-bp marker (Vivantis, Malaysia) was used to determine the size of the band and was sequenced. Signal peptide analysis was carried out with SignalP 4.1 program (http://www.cbs.dtu.dk/services/SignalP/) (Petersen et al. 2011). Conserved domain (CD) search was conducted at http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi.
Phylogenetic tree construction
Amino acid sequences of KTIs from other plant species were selected from NCBI databases and aligned. From the alignment, the sequences were analyzed and trimmed before constructing the phylogenetic tree using neighbor-joining method with 100 bootstrapping. All the works were conducted using MEGA 5.2 software (Tamura et al. 2011).
Homology modeling
Homology modeling was performed using amino acid sequence of the longest ORF in ClKTI by submitting to SWISS-MODEL software (http://swissmodel.expasy.org/) (Arnold et al. 2006; Bordoli et al. 2009; Guex et al. 2009; Biasini et al. 2014). The model was viewed using RasMol software (Sayle and Milner-Whit 1995).
Real-time quantitative polymerase chain reaction (RT-qPCR) analysis
Selection of reference gene candidates and primer design
Nucleotide sequences of reference gene candidates were screened and obtained from GenBank Database in NCBI website and the primers were designed based on the recommendations in MIQE guidelines (Bustin et al. 2009; Taylor et al. 2010). The designed primer sequences are as shown in Table 1.
Specificity and efficiency test
The designed primers were initially used for PCRs to determine its specificity by examining the band formed on agarose gel (1%) by electrophoresis of respective PCR products. The PCR protocol started with initial heating at 94 °C for 3 min, 40 cycles of 94 °C for 15 s, 60 °C for 60 s and final elongation at 72 °C for 10 min. Next, florescence-based RT-qPCR was conducted using SensiFAST SYBR Hi-ROX kit (Bioline, USA) for standard curve and melting curve plotting in accordance with the manufacturer’s protocol with Applied Biosystems StepOnePlus qRT-PCR machine (ThermoFisher Scientific, USA). The standard curve and melting curve results were analyzed with Applied Biosystems StepOnePlus qRT-PCR software.
Ranking of expression stable reference gene candidates
The expression of reference gene candidates were studied on the cDNA of five C. longa tissues (flower, basal stem, stem, rhizome and root) with RT-qPCR using SensiFast SYBR Hi-ROX kit (Bioline, USA) according to the manufacturer’s protocol using Bio-RAD CFX96 real-time PCR system (Bio-Rad, USA). The reaction condition is as described above. Raw quantification cycle, Cq, values for each reference gene candidates in each tissue cDNA were obtained and the expression stability ranking was evaluated using RefFinder. RefFinder incorporates and calculates the geometric mean of the weights of the results derived from Bestkeeper, GeNorm, Normfinder and the comparative ∆CT method and determines the overall ranking by assigning appropriate weights to each gene (Xie et al. 2012).
Expression profiling
Expression profiling of ClKTI in the five different tissues of C. longa was performed with RT-qPCR and Actin was used as the reference gene for normalization of the expression fold. The protocol for RT-qPCR is as described above. It was conducted with SensiFast SYBR Hi-ROX kit (Bioline, USA) in accordance with the manufacturer’s protocol and using Bio-RAD CFX96 real-time PCR system (Bio-Rad, USA). The results were analyzed using the Bio-RAD CFX96 Manager software (Bio-Rad, USA) and the expression profiling graph of ClKTI was plotted.
Author contribution statement
Idea and design of experiments: CSN, NAB, NAS. Conducting the experiments: CSN, NMD. Data analysis and interpretations: CSN, NAB, MM, HCL, NAS. Preparation of manuscript: CSN, NAS.
References
Andersen CL, Jensen JL, Orntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64:5245–5250
Apisariyakul A, Vanittanakom N, Buddhasukh D (1995) Antifungal activity of turmeric oil extracted from Curcuma longa (Zingiberaceae). J Ethnopharmacol 49(3):163–169
Araújo CC, Leon LL (2001) Biological activities of Curcuma longa L. Mem I Oswaldo Cruz 96(5):723–728
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22(2):195–201
Baskaran G, Salvamani S, Ahmad SA, Shaharuddin NA, Pattiram PD, Shukor MY (2015) HMG-CoA reductase inhibitory activity and phytocomponent investigation of Basella alba leaf extract as a treatment of hypercholesterolemia. Drug Des Devel Ther 9:509–517
Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T et al (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42(Web Server issue):W252–W258. doi:10.1093/nar/gku340
Bijina B, Chellappan S, Krishna JG, Basheer SM, Elyas KK, Bahkali AH, Chandrasekaran M (2011) Protease inhibitor from Moringa oleifera with potential for use as therapeutic drug and as seafood preservative. Saudi J Biol Sc 3:273–281
Birk Y (2010) Plant protease inhibitors: significance in nutrition, plant protection, cancer prevention and genetic engineering. Springer, Berlin, p 170
Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T (2009) Protein structure homology modelling using SWISS-MODEL workspace. Nat Protoc 4:1–13
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55(4):611–622
Chan SN, Abu Bakar N, Mahmood M, Ho CL, Shaharuddin NA (2014) Molecular cloning and characterization of novel phytocystatin gene from turmeric, Curcuma longa. BioMed Res Int. doi:10.1155/2014/973790
Cruz ACB, Massena FS, Migliolo L, Macedo LLP, Monteiro NKV, Oliveira AS, Macedo FP, Uchoa AF, Grossi de Sa MF, Vasconcelos IM, Murad AM, Franco OL, Santos EA (2013) Bioinsecticidal activity of a novel Kunitz trypsin inhibitor from Catanduva (Piptadenia moniliformis) seeds. Plant Physiol Biochem 70:61–68
De Oliveira CFR, Vasconcelos IM, Aparicio R, Freire MDGM, Baldasso PA, Marangoni S, Macedo MLR (2012) Purification and biochemical properties of a Kunitz-type trypsin inhibitor from Entada acaciifolia (Benth.) seeds. Process Biochem 47(6):929–935
Do Socorro M, Cavalcanti M, Oliva MLV, Fritz H, Jochum M, Mentele R, Sampaio M et al (2002) Characterization of a Kunitz trypsin inhibitor with one disulfide bridge purified from Swartzia pickellii. Biochem Biophys Res Commun 291(3):635–639
Fan SG, Wu GJ (2005) Characteristics of plant proteinase inhibitors and their applications in combating phytophagous insects. Bot Bull Acad Sin 46:273–279
Fang EF, Wong JH, Ng TB (2010) Thermostable Kunitz trypsin inhibitor with cytokine inducing, antitumor and HIV-1 reverse transcriptase inhibitory activities from Korean large black soybeans. J Biosci Bioeng 109(3):211–217
Farmer EE, Johnson RR, Ryan CA (1992) Regulation of expression of proteinase inhibitor genes by methyl jasmonate and jasmonate acid. Plant Physiol 98:995–1002
Garcia VA, MdasGM Freire, Novello JC, Marangoni S, Macedo MLR (2004) Trypsin inhibitor from Poecilanthe parviflora Seeds: purification, characterization, and activity against pest proteases. Protein J 23(5):343–350
Guex N, Peitsch MC, Schwede T (2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 30(1):162–173
Habib H, Fazili KM (2007) Plant protease inhibitors: a defense strategy in plants. Biotechnol Mol Biol Rev 2(3):68–85
Haq SK, Atif SM, Khan RH (2004) Protein proteinase inhibitor genes in combat against insects, pests, and pathogens: natural and engineered phytoprotection. Arch Biochem Biophys 431(1):145–159
Haruta M, Major IT, Christopher ME, Patton JJ, Constabel CP (2001) A Kunitz trypsin inhibitor gene family from trembling aspen (Populus tremuloides Michx.): cloning, functional expression, and induction by wounding and herbivory. Plant Mol Biol 46(3):347–359
Hildmann T, Ebneth M, Peña-Cortés H, Sánchez-Serrano JJ, Willmitzer L, Prat S (1992) General roles of abscisic and jasmonic acids in gene activation as a result of mechanical wounding. Plant Cell 4(9):1157–1170
Huang H, Qi SD, Qi F, Wu CA, Yang GD, Zheng CC (2010) NtKTI1, a Kunitz trypsin inhibitor with antifungal activity from Nicotiana tabacum, plays an important role in tobacco’s defense response. FEBS J 277(19):4076–4088
Jamalnasir H, Wagiran A, Shaharuddin NA, Samad AA (2013) Isolation of high quality RNA from plant rich in flavonoids, Melastoma decemfidum Roxb ex. Jack. AJCS 7(7):911–916
Koide T, Tsunasawa S, Ikenaka T (1973) Studies on soybean trypsin inhibitors, 2, amino-acid sequence around the reactive site of soybean trypsin inhibitor (Kunitz). Eur J Biochem 416:408–416
Koiwa H, Bressan RA, Hasegawa PM (1997) Regulation of protease inhibitors. Trends Plant Sci 2(10):379–384
Lantz RC, Chen GJ, Solyom AM, Jolad SD, Timmermann BN (2005) The effect of turmeric extracts on inflammatory mediator production. Phytomedicine 12(6–7):445–452
Lawrence PK, Koundal KR (2005) Plant protease inhibitors in control of phytophagous insects. Electron J Bioctechnol 5(1):93–109
Lingaraju MH, Gowda LR (2008) A Kunitz trypsin inhibitor of Entada scandens seeds: another member with single disulfide bridge. Biochim Biophys Acta 1784(5):850–855
Lomate PR, Hivrale VK (2012) Wound and methyl jasmonate induced pigeon pea defensive proteinase inhibitor has potency to inhibit insect digestive proteinases. Plant Physiol Bioch 57:193–199
Major IT, Constabel CP (2008) Functional analysis of the Kunitz trypsin inhibitor family in poplar reveals biochemical diversity and multiplicity in defense against herbivores. Plant Physiol 146:888–903
Migliolo L, de Oliveira AS, Ea Santos, Franco OL, de Sales MP (2010) Structural and mechanistic insights into a novel non-competitive Kunitz trypsin inhibitor from Adenanthera pavonina L. seeds with double activity toward serine- and cysteine-proteinases. J Mol Graph Model 29(2):148–156
Moura DS, Ryan CA (2001) Wound-inducible proteinase inhibitors in pepper. Differential regulation upon wounding, systemin, and methyl jasmonate. Plant Physiol 126(1):289–298
Mukhtar M, Arshad M, Ahmad M, Pomerantz RJ, Wigdahl B, Parveen Z (2008) Antiviral potentials of medicinal plants. Virus Res 2:111–120
Oddepally R, Sriram G, Guruprasad L (2013) Phytochemistry purification and characterization of a stable Kunitz trypsin inhibitor from Trigonella foenum-graecum (fenugreek) seeds. Phytochemisty 96:26–36
Oliva MLV, Silva MCC, Sallai RC, Brito MV, Sampaio MU (2010) A novel subclassification for Kunitz proteinase inhibitors from leguminous seeds. Biochimie 92(11):1667–1673
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8(10):785–786. doi:10.1038/nmeth.1701
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol Lett 26:509–515
Sayle R, Milner-Whit EJ (1995) RasMol: biomolecular graphics for all. Trends Biochem Sci 20(9):374
Selvam R, Subramanian L, Gayathri R, Angayarkanni N (1995) The anti-oxidant activity of turmeric (Curcuma longa). J Ethnopharmacol 2:59–67
Silver N, Best S, Jiang J, Thein SL (2006) Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol Biol 7:33
Sookkongwaree K, Geitmann M, Roengsumran S, Petsom A, Danielson UH (2006) Inhibition of viral proteases by Zingiberaceae extracts and flavones isolated from Kaempferia parviflora. Pharmazie 61:717–721
Srinivasan A, Giri AP, Harsulkar AM, Ja Gatehouse, Gupta VS (2005) A Kunitz trypsin inhibitor from chickpea (Cicer arietinum L.) that exerts anti-metabolic effect on podborer (Helicoverpa armigera) larvae. Plant Mol Biol 57(3):359–374
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Taylor S, Wakem M, Dijkman G, Alsarraj M, Nguyen M (2010) A practical approach to RT-qPCR-Publishing data that conform to the MIQE guidelines. Methods 50:1–5
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:RESEARCH0034
Xie F, Xiao P, Chen D, Xu L, Zhang B (2012) miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol Biol 80(1):75–84
Zhang S, An S, Li Z, Wu F, Yang Q, Liu Y, Cao J, Zhang H, Zhang Q, Liu X (2015) Identification and validation of reference genes for normalization of gene expression analysis using qRT-PCR in Helicoverpa armigera (Lepidoptera: Noctuidae). Gene 555:393–402
Acknowledgements
This work was supported by eScience grant from the Ministry of Science, Technology and Innovation, Malaysia and RUGS grant from Ministry of Education, Malaysia. Sincere gratitude is expressed to Universiti Putra Malaysia (UPM) and Malaysia Agricultural Research and Development Institute (MARDI) for the facilities.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by W. Wang.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Chan, SN., Abu Bakar, N., Mahmood, M. et al. Identification and expression profiling of a novel Kunitz trypsin inhibitor (KTI) gene from turmeric, Curcuma longa, by real-time quantitative PCR (RT-qPCR). Acta Physiol Plant 39, 12 (2017). https://doi.org/10.1007/s11738-016-2311-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-016-2311-7