
Abstract
Propene polymerization kinetic profiles with a diether-based Ziegler–Natta MgCl2-supported catalyst were investigated in a stainless-steel batch reactor. The initial 10 min period characterizes various temperature levels with a constant volume of liquid propene. The lowest temperature level corresponds to the usual prepolymerization temperature (10 °C), and the highest level corresponds to the usual main polymerization temperature (70 °C). The effects of the starting temperature levels were evaluated through polymerization kinetic patterns computed namely from the second polymerization period carried out at 70 °C for the next 90 min. Based on the heat transfer data, the kinetic profiles were fitted to suitable semi-empirical equations derived from fundamental kinetic approaches using the first and second orders of the catalyst active sites decay. Both approaches adequately describe the dependence of the initial activities and deceleration constants on the temperature during the initial period.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Karl Ziegler and Giulio Natta developed the most significant method for synthesizing polyolefins in the mid-1950s (Bárány and Karger-Kocsis 2019). Even 70 years later, the development of the ZN catalyst remains an important topic for both academic and industrial researches, as it is still the most widespread method of isotactic polypropene production (Busico 2009).
There are many ways to influence catalyst behaviour during polymerization, such as modifying its structure by using an internal or external donor, preactivation or prepolymerization of the catalyst (Brambilla et al. 2007). The aim of our work was to describe the dependencies and the influence of prepolymerization on the resulting kinetic profile of polymerization (Liu et al. 2022). This study simply selected multiple initial polymerization temperature levels to demonstrate the significant modification of the catalyst kinetic patterns. These catalysts are widely used for the commercial production of isotactic polypropene (iPP) and stand out through their many unique properties, such as good hydrogen response and high isotacticity without an external donor (Bukatov et al. 2019; Paghadar et al. 2021; Nikolaeva et al. 2018; Milanesi et al. 2023; Yaluma et al. 2006).
Numerous industrial processes used in polypropylene production are based on polymerization in liquid propene employing loop reactors for both the prepolymerization and polymerization steps in the synthesis. In this research, the prepolymerization loop reactor is modelled by a 10 min period during which the polymerization is carried out at various temperature levels in a wide range from 10 °C to 70 °C, while the subsequent main polymerization process is represented namely by 90 min of bulk polymerization in liquid propene at 70 °C.
To broaden the scope of the produced polypropylene grades, the majority of the processes also utilize a gas phase reactor at the end of the production lines, emphasizing the necessity of maintaining catalyst activity also during this period of production by minimizing the catalyst activity deceleration patterns. Therefore, the kinetic profile of the polymerization is one of the crucial features characterizing the behaviour of the catalytic system. Many authors have been involved in the development of various descriptions for the polymerization kinetics in batch reactors, with the aim of describing the polymerization system using mathematical relationships and fundamental dependencies (Tian et al. 2013; Soares and McKenna 2012).
The resulting kinetic profiles can be further utilized in mathematical models of production processes to control the industrial facilities and predict the benefits and disadvantages of any new catalyst system being introduced into the continuous production. Naturally, the kinetic features assessed in the laboratory bench-scale batch reactors have to be processed using complex computations to develop a respective mathematical model applicable to different industrial processes (Touloupides et al. 2010; Reginato et al. 2003; Zacca et al. 1996). These computations, based on statistical evaluation of the catalyst residence time distributions, prioritize the simple approaches of the batch kinetic descriptions minimizing the necessary number of deceleration parameters yet maintain accuracy in the fitting of heat transfer curves obtained during polymerization in the batch reactor.
Experimental part
Chemicals
Polymerization-grade propylene was obtained from the Orlen Unipetrol RPA, s.r.o., Litvinov, Czech Republic. The propylene underwent further purification via six-column filtration. The content of critical impurities (CO, COS) in the propylene after purification was below 10 vol. ppb, while the water and oxygen levels were below 0.1 vol. ppm. Lanxess AG Germany provided the triethylaluminium (TEA) cocatalyst featuring ultra-low aluminium hydride content (< 0.05 wt%, TEA-ULH grade). The TEA was used diluted with n-heptane. The dry catalyst was then transferred into Shell Ondina 933, a white mineral oil. The final catalyst concentration in the mineral oil suspension was approximately 25 wt%.
Polymerization procedures
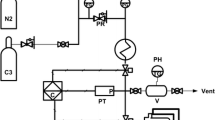
A 4-L stainless-steel reactor designed for batch liquid propene polymerization was used (Fig. 1). After closing the reactor and purging by nitrogen flow at 95 °C, a defined amount of TEA diluted with n-heptane was charged by micropipette (MicromanⓇ, Gilson), TEA/Ti = 120 mol/mol. Then, the reactor was filled with a defined amount of propene leading to a constant liquid phase volume at various prepolymerization temperatures. Concurrently, 32 mmol of hydrogen was added. After stabilization of the starting polymerization temperature, the catalyst suspension was charged by MicromanⓇ micropipette into the catalyst injection device. Subsequently, the catalyst was injected into the reactor through a flush of 40 g of liquid propene. In all the experiments, the final volume of the liquid phase during the prepolymerization period remained constant at 350 cm3.

The scheme of 4-L reactor for propene polymerization in the liquid phase
2.2.1. A short isothermal experiment.
After the injection of the catalyst, the polymerization proceeded at constant temperature and a stirring speed of 630 RPM for 10 min. Afterward, the reactor was vented, and the polymer weight measured.
2.2.2. A sequential two-period experiment.
After charging the catalyst, the 1st polymerization period at the defined temperature level was carried out for 10 min. Next, the reactor was filled with a next propene dose to a total of 1100 g simultaneously with a temperature rise to 70 °C. The 2nd polymerization period began after 98% of the main polymerization temperature (i.e. 68.6 °C of 70 °C) was reached. The 2nd polymerization period proceeded at 70 °C in liquid propene for 90 min. During the whole 2nd polymerization period, additional propene flow of 250 g/h into the reactor was applied to approximately compensate for the consumed propene.
Polymerization data processing
2.3.1. Data collection.
Figure 1 shows that during the experiment, the liquid propylene temperature was monitored using the thermocouple Tr located at the bottom of the reactor, and the temperature of the gas phase using the thermocouple Tr2 placed close to the reactor cover. The temperature of the incoming water reactor jacket was monitored by the thermocouple Tw. During the polymerization, the special double-ribbon stirrer with bottom blades was rotated at 630 RPM to ensure temperature homogeneity throughout the reactor, and all internal walls were in contact with the liquid propene. This statement was supported by the same readings from both internal thermocouples, Tr and Tr2. With such conditions, the technical arrangement of the 4-L reactor allowed the heat flow released by the polymerization reaction to be monitored. The heat caused a slight overheating (up to 2 °C) of the reactor content above the temperature of the water jacket, measured by the thermocouples inside the reactor (Tr and Tr2) and compared with the temperature of the circulating thermostatic bath (Tw). The reactor water jacket encompassed the whole reactor surface, including the reactor cover (Fig. 1).
For better resolution of the temperature difference signals DT = Tr − Tw and dT = Tout − Tin, direct micro-voltage measurement of the thermocouples voltage difference was performed employing suitable high-resolution transmitters (Fig. 1). The signals were recorded every second during the polymerization process and saved in the polymerization data records approximately every 5 s. Simultaneously, any as a basis for signal correction, derivation of temperature Tr was also collected.
2.3.2. Polymerization data evaluation.
In order to maintain a homogeneous temperature in the system, fast cooling water circulation was applied. However, this homogeneity also caused a decrease in the difference between the inlet and outlet temperatures, so it was more precise to assess the kinetic profile using the DT value (Tr − Tw, ΔT ≈ 2.0 °C), rather than dt (Twin − Twout, ΔT ≈ 0.1 °C). Example collected data are depicted in Fig. 2.

Example of a two-step polymerization data record. Polymerization started at 50 °C, after 10 min the temperature, it was gradually increased to 70 °C
The heat signal was automatically corrected for temperature scatter using a calibrated linear function of instant temperature and its derivation. All the collected DT points are depicted in light blue “DT(all)”, while the dark blue points “DT(incl)” correspond to those used for kinetic curve fitting.
2.3.3. Kinetic function.
The polymerization process starts immediately after the activated catalyst comes into contact with propene. This sub-second starting phenomenon is not detectable by the reactor as setup, even if the catalyst is charged at the final 70 °C temperature level, because the charging procedure disturbs the temperature reading for about 1–2 min. On the other hand, if the catalyst is charged at a lower temperature than the final polymerization level, significant activity increase is observed even several minutes after the final polymerization temperature is reached. Thus, the acceleration patterns of the polymerization kinetics are governed not only by the increasing temperature between the periods, but also by the time itself. To include this observation in the polymerization kinetic computation for the whole polymerization period, suitable compensation for the activity increase has to be included in the kinetic functions. To facilitate the subsequent kinetic profile computation deceleration, a simple mathematic approach was chosen to describe the catalyst activity increase between the polymerization periods, based on the linear combination of two non-dimensional transition functions separately describing the temperature increase and also the final polymerization rate increase after the final temperature is reached:
where t is the time since catalyst injection into the reactor (i.e. activation), Ka1, Ka2, P1 and P2 are the process activation parameters.
The function (1) applied in combination with deceleration functions facilitates the transition of the polymerization rate level from zero at zero time to an increased level after which Acc(t) = 1. However, this research is focused namely on the 2nd polymerization period, and zero time is defined as the end of the 1st polymerization period, when catalyst activity is already significant. Thus, additional modification of the acceleration function (1) is necessary:
where t is the time since the end of the first polymerization period, and A1i is a constant dimensionless parameter that allows us to compute the polymerization rate profile of the 2nd polymerization period from the final polymerization rate reached at the end of the 1st polymerization period.
The infinity time limit of the acceleration function (2) is defined as follows:
During the process of polymerization kinetic parameters optimization, the parameter A1i is constant. Its value is derived from the experimentally assessed yield of short 10 min runs performed in the same way as the 1st polymerization period of the sequential runs.
At the beginning of the temperature increase between the 1st and 2nd periods, the experimental DT points are not applicable as input data for optimization of the process activation parameters of the acceleration function due to their large scatter caused by the high power of the thermostat heaters enabling the fast temperature increase. Thus, at the beginning of the temperature increase, the parameters are optimized according to the instant temperature reading and in the context of the experimentally verified dependence of catalyst activity on the instant temperature (Fig. 3). Before 70 °C is reached, the thermostat heaters are automatically switched to a mode in which they maintain the polymerization temperature level. Then, the kinetic profile can be evaluated directly using experimental DT points automatically corrected for the instant reactor temperature and its derivation.

The effect of temperature on polymer yields in the 1st period, T = 10–70 °C, VLC3 = 350 dm3 of liquid propene; mcat = 4 mg, TEA/Ti = 120 mol/mol, nH2 = 32 mmol; t = 10 min
With all these experiments, the highest polymerization activity was typically observed several minutes after the end of the temperature increase to the final polymerization level. Then, a decrease in catalyst activity was observed. Two approaches were studied to describe the polymerization rate decrease:
1: two types of polymerization active site deactivation according to the first order:
2: one type of polymerization active site deactivation according to the second order:
where t is time since the end of the 1st polymerization period, A1f, A2f, A1s and A2s are parameters corresponding to the polymerization rate at zero time of the deceleration functions, from which the active sites deactivate according to their deceleration nature described by the parameters Kd1f, Kd2f and Kd1s.
The zero time in our kinetic description is the end of the 1st polymerization period, when the polymerization process proceeds at various temperature levels rendering to the process various polymerization rates. Thus, to fit the experimental DT points on the kinetic equation FDT over the entire 2nd polymerization period, the simple product of the combination of deceleration functions (3) or (4) and Eq. (2) was used:
and
The experimental DT points profile (exemplified in Fig. 2 for a run with the 1st period at 50 °C) is assumed to be a linear measure of the instant polymerization rate, linearly transferable from their dimension (°C) to the dimension of the polymerization rate—kgPP/(gcat∙h):
where Rp(t) is the polymerization rate profile; DTc is the semi-empirical constant for the polymerization run and FDT(t) is the profile fitted to the experimental DT data according to Eq. (6) or (7).
The product of the acceleration and deceleration functions defined using Eq. (6) or (7) can easily describe polymerization start-up upon constant or increasing temperature. Another advantage of this practical approach is in the easy definition of the kinetic maximum of the polymerization rate (Rp(0)) at zero time of deceleration, defined as the extrapolation of the acceleration function parameters Ka1 and Ka2 to zero (theoretical instant acceleration), which results in the same equation as the extrapolation of the acceleration time to infinity—see Eqs. (2) and (3). Thus, the extrapolated kinetic maximum of the polymerization rate according to the two kinetic approaches is defined as follows:
second order of deceleration:
Fitting the selected experimental points (dark blue) since the end of prepolymerization to the function (4) is depicted in Fig. 2. The fitting procedure is based on a simplex optimization procedure (Nelder and Mead 1965; O’Neill 1971), which computes in several steps all the parameters of the functions (6) and (7), performing the best fit to the experimental DT points also in congruence with the experimentally verified dependence of the yields of short polymerization runs corresponding to 1st periods at different temperatures.
The computation of DTc is the next step after fitting the DT points. According to Eq. (8), each data point DT can be transferred into instant heat flow produced by the polymerization rate. Thus, the time integral of the fitted function FDT(t) (6), including the temperature start-up period, represents the total heat produced by the polymerization process, which corresponds to the total yield of polymer produced during the 2nd period, including the temperature increase part of the 2nd period. Then, the multiplication factor DTc in Eq. (8) valid for the second polymerization period can be computed as a simple ratio of the total yield (corrected for the amount of polymer developed during the 1st period) and the total heat computed as the integral FDT(t) from the end of the 1st period to the end of the 2nd polymerization period:
where YT is the total polymer yield (1st + 2nd periods), as determined by the polymer weighting, Y1st is the polymer yield produced in the first 10 min period calculated from Eq. (12) and the cumulative integral value of the FDT(t) function (6).
Results and discussion
Catalyst activity upon increasing the polymerization temperature (1st period)
Within the scope of this two-polymerization-period research, we separately investigated the effect of different temperatures in the range (10–70 °C) on catalyst activity during the 1st period in the Short Isothermal Experiment mode (Chapter 2.2.1).
The results of this set of runs are depicted in Fig. 3. The experimental polymer yield points corresponding to the temperature levels are fitted by a function constructed according to the transition function (1) described above in the context of kinetic acceleration:
where T is the polymerization temperature, and P1, P2 and P3 are optimized parameters.
The 10 min experiments revealed a steep increase in polymer yield at temperatures above 40 °C. The shape of the yield dependence on polymerization temperature fits well to the function (12), which represents one of the two transition functions used in the expression (1). In principal, this fact rationalizes the use of such a functional expression for the polymerization activity increase between the polymerization periods.
The impact of temperature in the 1st period on catalyst activity in the 2nd period
Research of the two catalyst performance periods using the results of 10 min isothermal polymerizations described in Chapter 3.1, and the subsequent widening of the scope by searching the catalyst performance during the 90 min after the reactor was heated to 70 °C. Naturally, the duration of the temperature increase depends on the temperature difference between the 1st and 2nd periods. To facilitate the definition of the end of the temperature increase phase, the limit of 98% of the targeted 70 °C level (68.6 °C) was defined as the important milestone, after which the duration of the 2nd period was counted until the next 90 min time was reached.
As the temperature levels in the 1st period were the only variable in this research, all the resulting values in the relevant comparisons for all 14 runs performed at seven temperature levels (two runs at each level) were related to the temperature levels. At first, polymer yields between the milestones of the entire process are shown in Fig. 4.

Polymer yields between the milestones of the sequential experiments, mcat = 4 mg, TEA/Ti = 120 mol/mol, nH2 = 32 mmol (red unfilled circle1st period yield…10 min at various temperatures; red unfilled square 2ndT98 yield…temperature rise to 68.6 °C) (Grey filled triangle 2nd net yield…90 min after the temperature increase; blue filled diamond Yield Total…the whole sequential run yield)
The data related to polymer yield formed in the 1st period were already discussed above in Chapter 3.1. The yields were directly determined by weighting the polymer yield obtained from the reference 10 min experiments. Similarly, the total polymer yield obtained after the sequential experiment (1st + 2nd periods) was also determined by simple weighting.
On the other hand, the most important value—the net polymer yield formed during 90 min at a constant temperature of 70 °C—is inherently connected with polymer yield formed during the intermediate temperature increase that lasted over various periods (Yield 2nd T98). The most suitable solution to the intermediate yield evaluation is based on computing the overall polymerization kinetics describing the whole 2nd period, including the temperature increase. Then, the intermediate yield during the temperature increase phase is evaluated as the integral of the kinetic function until the customary limit of 98% of the targeted 70 °C level (i.e. 68.6 °C) is reached. The functional construction for the description of the polymerization rate acceleration during the intermediate period defined by Eq. (2) was already found to be in congruence with the experimentally constructed polymerization yield dependence on the temperature in Eq. (12). Also, further indirect support of the correctness of this approach is the independence of DTc (11) on the temperature differences and also on the duration of the intermediate period. This independence is demonstrated in Fig. 4 by the very low correlation coefficient of presumptive linear dependence.
Computation of the polymerization kinetics as described in Chapter 2.3.3 was performed automatically for both kinetic approaches in relation to the active site ageing process, Eqs. (4) and (5). Computing all 14 runs of this experimental series revealed that the resulting optimized Kd2f values are close to zero, which indicate not realistic statement about their everlasting performance. In other words, it was revealed that the ageing of the more stable active sites according to the first order of their decay is negligible. Thus, Eq. (4) could be modified as follows:
Then, the kinetic computation of all the results of the 14 runs was repeated for Eqs. (7) and (13). A typical comparison of the best fit to the experimental points for both kinetic approaches is exemplified in Fig. 2, while the resulting standard deviations for the experimental DT points for both kinetic approaches are compared in Fig. 5:

Standard deviations between the first-order and second-order kinetic approaches
The relationship between the standard deviations for both kinetic approaches presented in Fig. 5 revealed their congruity and also their correctness, because all of them are well-inside the experimental limits of errors, and both are similar for each run. This resulted in the slope of their linear correlation (≈ 1.02) with negligible intercept. Thus, it is relevant to use both kinetic decay approaches to demonstrate the kinetic profile curves. The resulting comparison of the kinetic profiles of the seven temperature levels for the first-order approach and single selected runs from the duplicates is depicted in Fig. 6 and, for the second order, in Fig. 7.

Polymerization kinetic profiles assuming the first order of deceleration

Polymerization kinetic profiles assuming the second order of deceleration
The almost identical kinetics of the relevant profiles are shown in Figs. 6 and 7, starting at catalyst introduction when the profiles assessed during the 1st polymerization period start. Although the shape of the curves was calculated in the same manner as described above for the 2nd period, their accuracy is limited due to the relatively small volume of the liquid phase inside the reactor. This situation could be a source of error in the interpretation of the heat transfer data because the smaller volume may not fully cover the entire inner surface of the reactor. However, the instant activity at the joint point for both periods (10 min) is assessed with high accuracy because it corresponds to the average activity calculated from the experimental yields of the short (10 min) isothermal runs discussed in Chapter 3.1 and presented in Fig. 3.
After 10 min, the kinetic curves in Figs. 6 and 7 enter into the polymerization temperature increase stage, which corresponds to their sharp increase to maximum, typically occurring several minutes after the temperature level of 70 °C is reached (Fig. 2). Assuming that these maxima are the results of both simultaneously and independently occurring acceleration and deceleration, then with the help of our kinetic approach extrapolated to “immediate” acceleration according to Eqs. (9) and (10), a comparison of both extrapolated maxima is constructed in Fig. 8:

Extrapolated polymerization rate maxima assuming immediate acceleration in both kinetic approach orders
The congruence of both kinetic approaches presented above in Figs. 5, 6, 7 and 8 heralds the suggestion that both mathematic tools—first- and second-order equations—are equivalent for a description of the presented sets of kinetic curves. If so, the optimized parameters for both kinetic approaches might also be in good correlation with the only variable in this research—the polymerization temperature in the 1st period (Figs. 9 and 10):

Comparison of optimized parameters for the first order of deceleration, red filled circle A1f, brown filled square A2f and blue filled diamond Kd1f

Correlation of optimized parameters for the second order of deceleration, red filled circle A1s and blue filled diamond Kd1s
While the three parameters optimized for the first order of kinetic approach do not form any correlation, as presented in Fig. 9, the two parameters of the second-order approach indicate a good ability to be fitted to some subsequent functional expressions, as they both follow smooth monotone trends.
One explanation for these results might be as follows: During the optimization process of the first-order approach, the parameters A1f and A2f both related to the maximum polymerization rate at zero time partially compensate each other. This effect, however, also disturbs the single parameter Kd1f which describes the deceleration features of the polymerization rate.
Moreover, the parameter A2f, here representing stationary active sites in the first-order approach, is naturally regarded as unrealistic. Thus, the second-order kinetic approach is the most appropriate for a mathematical description of the observed kinetic curves. However, further research will be necessary to elucidate an explanation for the chemistry behind this finding. A summary of all the measured and computed results is presented in Table 1.
Hypothesis
A plausible explanation for the described increase in polymerization activity with a decrease in polymerization temperature during the catalyst activation period is related to the monomer–dimer equilibrium of triethylaluminium (Smith 1967; Černý et al. 1988).
When the catalyst is injected into a reactor containing TEA and monomer at a high temperature, active sites are activated immediately and fast polymer chain growth occurs on the active sites within tenths of a second (Mori et al. 2000). Polymer chain growth is an exothermic process, during which the active site surrounding the space can be significantly overheated, and some of the active sites deteriorated and deactivated.
At a lower temperature, TEA occurs in dimer form, unable to create an active site on the catalyst. Most likely, the dimeric form is only adsorbed on the surface of the catalyst particle, including the inner surface of the catalyst particle pores. Thus, after catalyst introduction into the reactor at low temperature, active site formation upon dissociation, the TEA dimer occurs slowly during the temperature ramp between the polymerization periods, and the polymerization heat is easily transferred into the surrounding space of the catalyst particles.
The dissociation is dependent on temperature, thus a relatively slow temperature ramp between the polymerization periods facilitates transfer of the polymerization heat into the space surrounding the catalyst particles and plausible deactivation of part of the active sites on the catalyst due to local overheating is suppressed. The sequential formation of active sites without local overheating leads to increased catalyst activity, as shown in Figs. 6 and 7.
Conclusions
This paper focuses on the influence of temperature in the 1st 10 min polymerization period on the kinetic profiles of the 2nd period. Two-step polymerization based on a Ziegler–Natta catalyst with a diether-based internal donor was carried out in a 4-L stainless-steel reactor. It was shown that increasing the temperature level of the 1st period led to a steep increase in polymer yield within this period but, on the contrary, activity in the 2nd polymerization period and total yield both decreased.
The presented set of two-step polymerization runs in liquid propene revealed significant trend towards stabilization of the polymerization rate deceleration features upon an increase in the starting temperature.
Despite a sufficient description of the kinetic profile by first-order deactivation, no dependence of the A1f, A2f or Kd1f kinetic constants could be found. Based on a similar deviation of fitting by first- or second-order equations, both methods provide congruity and also correctness.
Namely due to the simplicity and conformity of the optimized constants, the deceleration features were better described by the application of the second-order deactivation of active sites. This approach also appeared to be more logical and more suitable for further mathematical processing towards the computation of the catalyst kinetic performance in continuous processes.
References
Brambilla L, Zerbi G, Piemontesi F, Nascetti S, Morini G (2007) Structure of MgCl2–TiCl4 complex in co-milled Ziegler-Natta catalyst precursors with different TiCl4 content: experimental and theoretical vibrational spectra. J Mol Catal a: Chem 263(1–2):103–111. https://doi.org/10.1016/j.molcata.2006.08.001
Bukatov GD, Maslov DK, Sergeev SA, Matsko MA (2019) Effect of internal donors on the performance of Ti–Mg catalysts in propylene polymerization: donor introduction during or after MgCl2 formation. Appl Catal A 577:69–75. https://doi.org/10.1016/j.apcata.2019.03.010
Busico V (2009) Metal-catalysed olefin polymerisation into the new millennium: a perspective outlook. Dalton Trans 41:8794. https://doi.org/10.1039/b911862b
Černý Z, Heřmánek S, Fusek J, Kříž O, Čásenský B (1988) 27Al NMR spectroscopy of triethylaluminium. A direct method to the determination of the proportion of monomer in solution. J Organomet Chem 345:1–9. https://doi.org/10.1016/0022-328X(88)80228-6
Karger-Kocsis J, Bárány T (eds) (2019) Polypropylene handbook: morphology, blends and composites. Springer International Publishing, Cham. https://doi.org/10.1007/978-3-030-12903-3
Liu H, Du G, Du Y, Li D, Chen J (2022) Effects of prepolymerization, temperature, and hydrogen concentration on kinetics of propylene bulk polymerization using a commercial Ziegler-Natta catalyst. Adv Polym Technol 2022:1–9. https://doi.org/10.1155/2022/9980759
Milanesi M, Piovano A, Wada T, Zarupski J, Chammingkwan P, Taniike T, Groppo E (2023) Influence of the synthetic procedure on the properties of three Ziegler-Natta catalysts with the same 1,3-diether internal donor. Catal Today 418:114077. https://doi.org/10.1016/j.cattod.2023.114077
Mori H, Yamahiro M, Terano M, Takahashi M, Matsukawa T (2000) Lifetime of growing polymer chain in stopped-flow propene polymerization using pre-treated Ziegler catalysts. Macromol Chem Phys 201(3):289–295. https://doi.org/10.1002/(SICI)1521-3935(20000201)201:3%3c289::AID-MACP289%3e3.0.CO;2-M
Nelder JA, Mead R (1965) A Simplex Method for Function Minimization. Comput J 7(4):308–313. https://doi.org/10.1093/comjnl/7.4.308
Nikolaeva M, Matsko M, Zakharov V (2018) Propylene polymerization over supported Ziegler-Natta catalysts: effect of internal and external donors on distribution of active sites according to stereospecificity. J Appl Polym Sci 135(23):46291. https://doi.org/10.1002/app.46291
O’Neill R (1971) Algorithm AS 47: function minimization using a simplex procedure. Appl Stat 20(3):338. https://doi.org/10.2307/2346772
Paghadar BR, Sainani JB, Bhagavath P (2021) Internal donors on supported Ziegler Natta catalysts for isotactic polypropylene: a brief tutorial review. J Polym Res 28:1–19. https://doi.org/10.1007/s10965-021-02737-1
Reginato AS, Zacca JJ, Secchi AR (2003) Modeling and simulation of propylene polymerization in nonideal loop reactors. AIChE J 49(10):2642–2654. https://doi.org/10.1002/aic.690491017
Smith MB (1967) Monomer-dimer equilibria of liquid aluminum alkyls. I. Triethylaluminum. J Phys Chem 71(2):364–370. https://doi.org/10.1021/j100861a024
Soares JBP, McKenna TFL (2012) Polyolefin reaction engineering. Wiley-VCH
Tian Z, Xue-Ping G, Feng L-F, Guo-Hua H (2013) A model for the structures of impact polypropylene copolymers produced by an atmosphere-switching polymerization process. Chem Eng Sci 101:686–698. https://doi.org/10.1016/j.ces.2013.07.004
Touloupides V, Kanellopoulos V, Pladis P, Kiparissides C, Mignon D, Van-Grambezen P (2010) Modeling and simulation of an industrial slurry-phase catalytic olefin polymerization reactor series. Chem Eng Sci 65(10):3208–3222. https://doi.org/10.1016/j.ces.2010.02.014
Yaluma AK, Tait PJT, Chadwick JC (2006) Active center determinations on MgCl2 -supported fourth- and fifth-generation Ziegler-Natta catalysts for propylene polymerization: active center determinations. J Polym Sci Part a Polym Chem 44(5):1635–1647. https://doi.org/10.1002/pola.21277
Zacca JJ, Debling JA, Ray WH (1996) Reactor residence time distribution effects on the multistage polymerization of olefins—I. Basic principles and illustrative examples, polypropylene. Chem Eng Sci 51(21):4859–4886. https://doi.org/10.1016/0009-2509(96)00258-8
Acknowledgements
The author would like to thank ORLEN Unipetrol RPA, s.r.o.—Polymer Institute Brno and the University of Technology, Faculty of Chemistry. Many thanks also go to Clariant Produkte (Deutschland) GmbH for their cooperation and support.
Funding
Open access publishing supported by the National Technical Library in Prague.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
We wish to confirm that there are no known conflicts of interest associated with this publication, and there has been no financial support for this work that could have influenced its outcome.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kolomazník, V., Cejpek, I. & Skoumal, M. Temperature effect on the kinetic profile of Ziegler–Natta catalyst in propene polymerization. Chem. Pap. 78, 8397–8408 (2024). https://doi.org/10.1007/s11696-024-03679-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-024-03679-w