
Abstract
Oxidation of cyclopentanone (CPO) was studied over metal-free carbon catalysts in solvent-free conditions. Norit SX plus (900 m2 g−1) and Vulcan XC72 (240 m2 g−1) were used as pristine materials. Catalysts were prepared by activation in nitrogen and by treatment with urea, calcined in nitrogen at 500 °C and 950 °C. SEM, EDX, XPS, X-ray powder diffraction, nitrogen adsorption–desorption isotherms, acid–base titration, FTIR and Raman techniques were used for characterization. Catalytic tests carried out at 0.45 MPa and 80–110 °C enabled to find the best results: 30% selectivity to glutaric acid (GA) and 3% to succinic acid at 25% conversion of CPO, over Vulcan type catalysts. A low activity, selectivity and strong deactivation of Norit type catalysts can be ascribed to a higher specific surface in a disordered arrangement providing a high concentration of unselective catalytic sites. A higher extent of side reactions can yield in higher molar mass products, which stick on the surface and block catalytic sites. Optimum amount of accessible planar domains, represented by graphene and/or graphitic plates with some disorders present in Vulcan type catalysts classified them superior to Norit catalysts. A redox reaction scheme of oxidation of CPO to GA is also proposed.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glutaric acid (GA) and its derivatives started to be important chemicals to produce bioplastics and chemicals as surfactants, additives to dietary products and biologically active substances. About 5% increase in production of GA is supposed by the 2032 (Glutaric acid market 2022).
Industrially, GA was produced in the past as a side product in the production of adipic acid (Johnson et al. 2010). Other possibility is the oxidation of CPO and cyclopentanol (CPL) by concentrated nitric acid, or by the carbonylation of butyrolactone (Johnson et al. 2010). A few reports (Lisicki and Orlińska 2018; Vennat and Brégeault 2010) describe research on the oxidation of cyclopentanone or its methyl derivatives with oxygen or air. Lisicki and Orlińska (2018) reported the use Mn(II) salt as a catalyst for the oxidation of cyclopentanone using oxygen in acetic acid. Under relatively mild conditions (100 °C, 6 h, 0.1 MPa), a conversion above 98% and selectivity to GA up to 68% were obtained (Lisicki and Orlińska 2018). α-substituted cyclopentanones were oxidized to oxo-acids in the presence of polyoxometalates or Cu(NO3)2 (in systems involving homogeneous catalysis) (Brégeault et al. 2001) or by homogeneous systems with ruthenium acetate complexes and [Ru(H2O)6)](tosylate)2 (Vennat and Brégeault 2010). In recent years, biotechnological procedures are intensively developed to produce GA, or generally C5 platform chemicals (Park et al. 2013; Rohles et al. 2018; Kim et al. 2019; Han et al. 2020). In this paper, we focus on the possibility to prepare GA from the bio-CPO what stems from the previous research in our laboratories (Hronec and Fulajtarová 2012; Hronec et al. 2014, 2016). Recently, bio-furfural was shown as one of the most promising platform molecule in CPO production (Hronec et al. 2016; Zhou et al. 2017; Shen et al. 2018). The deactivation of copper phosphate catalysts in one-step transformation of bio-furfural to dicarboxylic acid (Soták et al. 2017) was ascribed to the dissolution of metals in the reaction mixture under oxidative conditions. This directed our attention on processes with non-metal containing catalysts, e.g. activated carbon (AC) (Iwanow et al. 2020).
Reported applications of AC include liquid phase oxidations (Besson et al. 2000; Pigamo et al. 2002; Yu et al. 2011; Sun et al. 2013), H2S oxidation (Adib et al. 2000; Duong-Viet et al. 2016), oxidative dehydrogenation reactions (Qi and Su 2014), formic acid dehydrogenation (Barlocco et al. 2020), selective decomposition of hydrazine (Barlocco et al. 2022), etc. Active sites on activated carbon and graphene are of two basic types: (a) carbon domains—bonds and defects, and (b) heteroatoms. As for carbon domains edges and planes are possible active sites, where vacances in sp2 graphitic structure create pentane, and heptane structures (Harris et al. 2008; Navalon et al. 2017). Oxygen, nitrogen, sulphur, phosphorus and boron are considered as the most frequent dopants.
Besson’s research group (Besson et al. 2000) reported that metal-free synthetic carbons catalyze the oxidation of cyclic ketones to dicarboxylic acids. Pigamo found that activity in the cyclohexanone oxidation is mainly dependent on the surface area and microporosity, while the surface oxygen groups improved the selectivity to dicarboxylic acids (Pigamo et al. 2002). Yu et al. (2011) reported a negative effect of oxygen functionalities on carbon nanotubes on the activity of cyclohexane oxidation. In addition, (Figueiredo and Pereira 2010; Sun et al. 2013) basic sites were reported as crucial for oxidations by H2O2, catalytic ozonation and catalytic wet air oxidation catalyzed by carbon. The basic character of activated carbons can be primarily associated with the delocalized electrons of aromatic rings, which could act as Lewis bases (Shafeeyan et al. 2010; Pérez-Cadenas et al. 2003). The basicity of AC is also associated with the presence of chromene and pyrone moieties (Boehm 1966; Pigamo et al. 2002). It is widely reported that introduction of nitrogen can improve the basic property due to the strong electron donor behaviour of nitrogen. Moreover, nitrogen atoms can enhance the electron transfer ability, conductivity, oxidation stability and catalytic performance of carbon materials (Bagreev et al. 2004; Shao et al. 2008; Sun et al. 2013; Lv et al. 2018). However, not only surface functional groups are important in the performance of carbon materials as catalysts. Intrinsic defects (obtained also by removing heteroatoms) are known to be active for several metal-free reactions (Barlocco et al. 2020, 2022).
Carbon surface can be modified by various procedures introducing following moieties:
-
Oxygen (carboxylic, lactone, hydroxyl, carbonyl surface groups) usually introduced by oxidation with nitric acid (Szymański et al. 2002; Shafeeyan et al. 2010; Kampouraki et al. 2019)
-
Nitrogen (aromatic amine, amide, imide, lactame, pyridine, pyrrol, quaternary nitrogen), e.g. by calcination in flow of nitrogen (Seredych et al. 2008), or in the flow of ammonia (Shafeeyan et al. 2010; Watanabe et al. 2015; Fujita et al. 2017) or using urea (Bashkova et al. 2007; Seredych et al. 2008; Hulicova-Jurcakova et al. 2009; Sousa et al. 2013; Ren et al. 2018); Lepre et al. (2023) reports high nitrogen carbonaceous materials synthesized from cytosine
-
Sulphonic groups by a direct sulphonation (Konwar et al. 2015) or by incorporation or impregnation with sodium dodecylbenzenesulphonate (Shao et al. 2010)
-
Nitrogen and sulphonic groups, e.g. by arylation with 4-sulfobenzenediazonium chloride (Malins et al. 2015).
A comprehensive review about characterization of the surface of activated carbon was prepared by Bandosz and Ania (2006). A recent paper (Iwanow et al. 2020) lists SEM, FTIR, nitrogen adsorption, Boehm titrations, adsorption of phenol, methylene blue and iodine, TPD, CHNS/O elemental analysis, EDX, XPS, XRD and TGA as the main characterization methods. Details about Raman spectroscopy as a powerful tool for investigation of carbon materials has been described by Merlen et al. (2017); including occurrence of C(sp2) and C(sp3) atoms. From catalytic point of view, graphitic and non-graphitic carbons were shown to differ significantly (Schuepfer et al. 2020; Scardamaglia et al. 2017). Bokobza et al. (2015) deconvoluted Raman spectra of carbon materials using Gauss–Lorentz band functions and reported an identification of typical G (graphitic) and D (disorder) peaks. A Raman band at 1620 cm−1 in natural graphite was ascribed to defects in the graphite structure (Nakamizo et al. 1978). Fullerene-related structures with pentagonal carbon rings were observed upon treatment at 2000 °C, indicating a profound rearrangement of the carbon structure during high-temperature treatment (Harris et al. 2008). Morphology and crystallographic changes of charcoals prepared from wood and lignin proved a positive effect of temperature on graphitizing (ID/IG decrease), i.e. increase of the C(sp2) carbons (Ishimaru et al. 2007). Similar observation was reported by Schüpfer et al. (2021), who proved a dominant C(sp2) arrangement in commercially available nanodiamond and adamantane (originally bearing sp3-hybridization) treated at 3000 °C.
In our work, we have focused on the nitrogen activated metal-free carbon catalysts in a batch oxidation of pure, undiluted CPO to GA. Our choice of the pristine carbon materials, activated carbon and carbon black, was based on their different properties, origin, surface area, conductivity and structure. Compared to other nanocarbon catalysts, they have the advantage of low cost. The choice of Norit AC (specific surface area about 900 m2/g) stemmed from recent successful applications of Norit-supported metal catalysts (Pikna et al. 2014; Turáková et al. 2014; Hronec et al. 2016) in our laboratories. For comparison, carbon black Vulcan XC72 with a lower specific surface area (about 250 m2/g) and a good applicability in redox reactions, illustrated, e.g. by successful applications in fuel cells (Lázaro et al. 2011), was taken.
Experimental
Catalyst preparation
Two types of carbons were used for the preparation of metal-free catalysts: activated carbon Norit SX plus—powder (Sigma Aldrich) and carbon black Vulcan XC72 (Cabot Carbon Corporation, Cabot 2022).
The catalyst CNH was prepared from a suspension of Norit in 1 M HCl (4 g of carbon in 100 ml of 1 M HCl), stirred for 2 h, then filtered and washed with distilled water. This procedure was repeated twice. Afterwards, the solid was washed with distilled water until the total elimination of free chloride ions (checked with the water solution of AgNO3) and dried in a vacuum oven for 5 h at 105 °C. Next series of catalysts were prepared by activation of CNH in nitrogen at 500 °C or 950 °C. The sample was heated (10 °C/min) in nitrogen flow to the desired temperature and kept at this temperature for another hour. Then, the sample was cooled to the ambient temperature in the nitrogen flow. The resulting catalysts were denoted as CNN500 and CNN950, respectively.
Nitrogen modified catalysts based on Vulcan type carbon were prepared by a similar route. Samples were designed as CVH, CVN500 and CVN950, respectively.
Urea modified catalysts were prepared from the CNH or CVH sample by the impregnation method as reported by Seredych (2008). 3 g of demineralised carbon was mixed with urea solution (2 g of urea in 25 ml ethanol) and stirred at room temperature for 5 h. Then, alcohol was evaporated and the carbon sample was dried at 105 °C. The urea-impregnated carbon sample was heated in nitrogen flow at 10 °C/min rate to 500 °C or 950 °C and kept at this temperature for 1 h, then cooled to room temperature in the flow of nitrogen. Afterwards, the urea treated carbon samples were washed with boiling water to remove any urea decomposition products and dried at 105 °C. According to carbon used and calcination temperature, the samples were designated as CNU500, CNU950 and CVU500, CVU950, respectively.
Catalyst characterization
A scanning electron microscope JEOL 7500F with a COLD FEG cathode at the accelerating voltage of 15 kV with EDX detector was used to study the texture and chemical elements present on the surface of modified carbons. Samples were prepared by deposition of a gold layer in a Balzers SCD 050 apparatus.
Particle sizes were determined by a CILAS 930 liquid device in the range of 0.2 – 500 μm with SW SizeExpert V9.08.
XRD patterns of all the catalysts were recorded on a Philips PW 1830 device with a Cu Kα radiation (λ = 1.5405 Ǻ) source in the scan range of 5 – 140° 2 Theta at 40 kV and 30 mA.
Nitrogen adsorption–desorption isotherms were obtained using Quantachrome NOVA 1000e instrument. The obtained data were treated with the built in SW ASAP 2020 software and by calculation procedures (linear and un-linear fitting) in Excel.
IR spectra were collected by using a IRAffinity-1 spectrophotometer (Shimadzu). The spectra were obtained in the range of 600–4000 cm−1. Samples were prepared by uniformly dispersing the carbon black catalysts in KBr powder and compressing into pellets. The background signal was subtracted.
Raman spectra were recorded from 100 to 3500 cm−1 on a Thermo Scientific Raman DXR 3 spectrophotometer using 532 nm laser source and 50 μm slit aperture. The laser power was set to 28 mW. The collection exposure time was 0.2 s and the number of scans 128. The obtained patterns were deconvoluted using the Origin peak fitting programme (OriginLab 2003).
The surface pH of activated carbon samples was determined by mixing 0.4 g of dry carbon with 20 ml of distilled water, stirring the suspension overnight to reach equilibrium and measuring the pH of the suspension the next day (Bashkova et al. 2007).
X-ray photoelectron spectra were recorded using a Thermo Scientific K-Alpha XPS system equipped with a micro focused monochromatic Al Kα1 X-ray source (1486.6 eV).
Contents of acidic and basic surface groups were determined by the Boehm titration procedure (Boehm 1966), which was performed as follows: one gram of carbon sample was placed in 25 mL of 0.05 M solution of either sodium hydroxide or hydrochloric acid. The vials were sealed, and the carbon suspensions were stirred for 24 h, after which the slurry was filtered. 10 ml of NaOH filtrate was added to 15 ml of 0.05 M HCl, and the solution was then back-titrated with standardized NaOH solution. The number of acidic oxygen sites on the carbon surface was estimated from the amount of base neutralized, under the assumption that NaOH neutralizes all acidic groups (carboxylic, lactone and phenolic groups). The number of surface basic groups was calculated from the amount of hydrochloric acid reacted with the AC sample. 10 ml of HCl filtrate was pipetted, and the excess of acid was titrated with NaOH.
Catalytic tests
The oxidation experiments were carried out in a thermostatic stirred glass lined autoclave equipped with two input/output valves and equipment to monitor oxygen consumption. Typically, 3 ml of CPO and 150 mg of catalyst were added into the reactor. The autoclave was purged with O2 twice and charged with 0.45 MPa of O2 at room temperature. The reaction temperature was then set to the desired value (80 – 110 °C). The zero-reaction time corresponds to the start of reactor stirring. Reaction was usually performed for 210 min, or after ceasing the consumption of oxygen. After the appropriate reaction time, the reactor was cooled in the mixture of ice and water, and reached temperature close to 10 °C, the output tube was led to the solution of Ba(OH)2 in aim to detect CO2, and the pressure was slowly released during 5 min. The liquid phase was separated from the catalyst by centrifugation. The reaction mixture was analysed by gas chromatography (Carbowax column). Dicarboxylic acids were derivatized with a large excess of methanol in the presence of concentrated sulfuric acid. The resulting mixture was analysed by means of GC (SE-30 column), and the yields of methyl esters of dicarboxylic acids were calculated. Each catalytic experiment (except recycling) was repeated twice, and average values were taken for assessment. In the case of experiments with the recycled catalyst, the liquid phase was first removed from reactor and then the catalyst was washed with methanol. Finally, the reactor with dry catalyst was charged with 3 mL of CPO and the oxidation started again.
GC–MS was performed using Shimadzu GC–MS-QP2010 apparatus (capillary column HP-1, 50 m × 0.2 mm).
The rate of oxidation was estimated according to the progress of oxygen consumption. The volume of oxygen was converted to moles using the van der Walls equation, where the temperature of the oxygen reservoir (laboratory temperature) was used as parameter T (Engineersedge 2023).
From quantitative point of view the following variables were assessed:
Conversion of CPO was calculated from the GC data:
where tr is the reaction time.
The selectivity to the formed products was calculated as:
The selectivity defined as (2) in fact indicates how many molecules of a product are formed by the conversion of one molecule of the reactant. This means that the loss of the reactant by other reactions (e.g. by total oxidation of CPO or decarboxylation) is not taken into account in terms of the carbon balance. CO2 formation was monitored based on the formation of BaCO3 precipitate in Ba(OH)2 solution at the end of the reaction.
Results and discussion
Catalyst properties
As described in experimental, five catalysts were prepared from activated carbon Norit SX plus and five catalysts were prepared from the carbon black Vulcan XC72; the carbon activated only with hydrochloric acid and dried at 105 °C was prepared twice. Accordingly, the samples were denoted as: CNH, CNN500, CNN950, CNU500 and CNU950 (generally denoted as CN catalysts), CVHa, CVHb, CVN500, CVN950, CVU500 and CVU950 (generally denoted as CV catalysts).
The morphology of the Norit and Vulcan samples is evident from Fig. 1. Norit is typical with rather large particles about 10–50 µm. Vulcan consists of agglomerates of 2–3 µm. The size of different particles was monitored by laser diffraction measurements as well (Table 1).

SEM images of the Norit sample (CNH, left) and Vulcan sample (CVHa, right) treated with HCl and dried at 105 °C. Magnification 500×
Textural parameters of selected catalysts prepared from Norit and Vulcan carbons are summarized in the Supplement (Table S1). Nitrogen adsorption–desorption isotherms for the samples CNH and CVHa are depicted in Fig. 2. Isotherms for other samples are shown in Fig. S7 (Supplement).

Adsorption–desorption isotherms for the samples CNH and CVHa
At first glance all the adsorption–desorption isotherms are rather alike. However, one should acknowledge the complexity of the structure of the catalyst, i.e. the presence of microporosity (Dp < 2 nm), mesoporosity (2 < Dp < 50) and macropores (Dp > 50 nm). To have a more realistic/practical imagination about the structure of the catalyst, one should take in consideration, among others, the cylindrical type of pores, Kelvin equation for capillary condensation and Halsey equation for the thickness of N2 layer on the wall (Lowell et al. 2004). Here, the contributions of individual pore types appear at x < 0.15 for micropores; 0.15 < x < 0.95 for mesopores and 0.95 < x < 0.999 for macropores, where x is the relative pressure (p/p0).
With the aim to identify latent differences in the texture of the CV catalysts, in addition to the standard data treatment methods (Micromeritics 2020), we also treated our data by other methods previously reported (Horniakova et al. 2001; Kralik et al. 2008; Králik 2014).
Altogether, the following models were used:
-
1.
Standard BET isotherm
-
2.
One parameter BET isotherm
-
3.
t-line model: the model uses the standard t-isotherm according to Harkins Jura equation (Jura and Harkins 1944)
-
4.
BET isotherm with the assumption of micropores being filled before the BET regime of adsorption starts
-
5.
n-layer BET isotherm.
A more detailed description of individual methods can be found in Supplementary. Only peculiarities, which are often overlooked in routine textural analysis, are discussed here.
All the results from textural evaluation are summarized in Table 1. More complicated models with a higher number of parameters enabled much better description of adsorption data what led to better values of the adjusted correlation coefficient R2adj.
Adjusted correlation coefficient of determination was calculated according to the formula:
It is worthy to note that results for the 1-parameter BET and t-line models are very close to those calculated by the ASAP 2020 software (Table S1 in the Supplement, entries for S1par and St.ext). Hence, the commercial data treatment software seems to be eligible here. However, at closer look, the unsuitability of the BET isotherm is well demonstrated, particularly for the CNH sample (see Supplement), what is also expressed by the negative value of R2adj (Table 1). The negative value of R2adj means that the model is even worse than an average value considered for data in given region (0.05 < x < 0.35). This is much worse value than for the CV catalysts (see Table 1). The t-line model is also slightly less suitable for the CNH catalyst in comparison with the CV catalysts. The isotherm with the consideration of micropores (entry Vmicro + BET) proved to be fairly valid both for the CNH and CV catalysts. However, the correlation was better for the CV catalysts. One should note the manifestation of microporosity in CNH and CV catalysts: The number of layers in the n-layer BET isotherm model was about 2 for the CNH catalyst and close to 3 for the CV catalysts, whereas VK,micro for the CNH catalyst and the CV catalysts was 0.4 and 0.1 cm3 g−1, respectively. Due to fact that catalysts based on Norit (CN) exhibited low yields (both the conversion and selectivity) of GA in the oxidation of CPO we did not pay such further detailed attention to textural characterization as for the CV catalysts.
As for the influence of the preparation method no significant difference in texture has been found. The linearized BET model, as well as the treatment of data by the nonlinear regression (see Supplementary data) led to high values of the interaction constant CBET (more than 1.000.000), what is physically unrealistic. Therefore, the one parameter BET model offers higher credibility. The model indicated a very little increase in the specific surface area with the increased temperature of calcination (CVN500: 238; CVN950: 245; CVU500: 225; CVU950: 232, m2 g−1). A slight increase in microporosity was registered for samples calcined at 950 °C (see Vt,micro). These changes in microporosity correlate with the decreasing values of number of layers in the n-layer isotherm (CVN500: 3.06; CVN950: 2.88; CVU500: 2.99; CVU950: 2.77) and by a moderate increase of the specific surface area (CVN500: 249; CVN950: 257; CVU500: 235; CVU950: 243, m2 g−1). This trend can be observed in both models, i.e. at 1-parameter BET and also at n-layer isotherm.
As shown in Supplementary material (Figs. S4 and S6), the sample treated with urea and calcined at 950 °C (CVU950) has a morphology similar to the CVHa catalyst. EDX data confirmed the occurrence of carbon and oxygen on the surface. The presence of nitrogen species was just at the limit of detectability without a possibility for quantitative evaluation. (When working with carbons the N signal is usually hidden by C signal (Bagreev et al. 2004)).
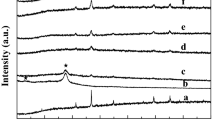
X-ray diffraction patterns (Fig. 3 and Fig. S13 in Supplement) match well with the standard JCPDS file (41-1487). The obtained diffraction peaks for (002), (100) and (101) are assigned to the corresponding crystal planes of graphitic carbon. The presence of a well-developed diffraction around 26° points to the crystalline structure of activated carbon samples, which indicates the superior alignment of otherwise disordered graphitic carbon layers to form the crystalline hexagonal/turbostratic structure (Li et al. 2007; Shao et al. 2010; Xie et al. 2018; Wang et al. 2010). The similarity of XRD patterns (Fig. S13 in Supplement) revealed the identical internal structure (crystal domains) for all the prepared Vulcan catalysts. In the case of the Norit catalyst (CNH), the broadening of diffraction peaks at 26.4° and 42.5° indicates smaller crystal domains compared to the Vulcan catalysts.

XRD pattern for CNH and CVHa samples. Numbers in brackets indicate Miller indices for the hexagonal carbon structure
XPS data for pure nitrogen activated catalysts (CVN500 and CVN950) did not offer any evidence of the presence of nitrogen. In urea activated materials (CVU500 and CVU950) (Fig. S14 in Supplement), it was possible to identify oxygen and nitrogen species. The peak for oxygen at about 530 eV can be assigned to superposition of peaks for the (–CO–O–) (530.35 eV) and C–O (531.58 eV) (Xie et al. 2018). The sample treated with urea and activated at 500 °C under nitrogen presents a large amount of nitrogen. In XPS curve fitting, the N 1 s signal with binding energy of 400.1 eV, indicates that N–H (amine) moieties are formed during this treatment (Boehm 1994).
Figure 4 holds the IR transmittance spectra of individual catalysts. All samples have a broad band at 3040–3400 cm−1, which is attributed to the O–H stretching vibration of hydroxyl groups, carboxylic groups and adsorbed water (Alves et al. 2013; Tazibet et al. 2013; Wang et al. 2011). A broadening of this band to lower wave numbers indicates the presence of hydrogen bonds (Puziy et al. 2002). This region is also associated with the N–H stretching vibration in amine and amide groups (Allwar 2012). The broad peak around 1700 cm−1 can be attributed to C=O stretching vibration of carbonyl groups (Tazibet et al. 2013), which can be conjugated with the carbon black aromatic structures (Peña et al. 2001). As reported in the literature (Tazibet et al. 2013), the peak around 1400 – 1460 cm−1 can be assigned to either aromatic ring vibrations or C–O–H vibrations of carboxylic groups. The small peak occurring at 1367 cm−1 can be ascribed to oxygen functionalities such as highly conjugated C–O stretching, C–O stretching in carboxylic groups and carboxylate moieties (Deng et al. 2010). The band between 1300 and 1000 cm−1 with maximum at 1100 cm−1 is assigned to the C–O stretching in carboxylic acids, alcohols, phenols, ethers and esters (Alves et al. 2013; Tazibet et al. 2013; Wang et al. 2011). The intensities of bands at 3230 cm−1 and 1440 cm−1 increase after modification with HCl (the CVH catalyst). The intensity of these peaks showed that partial removal of carboxylic groups as a result of the increase of temperature during carbonization treatment from 500 to 950 °C takes place. It correlates with the acidic group concentration obtained by Boehm titration (Tazibet et al. 2013).

Transmittance IR spectra of catalysts prepared from the carbon black Vulcan XC72
Raman spectra of the investigated samples are shown in Fig. 5. For comparison, typical frequencies for well-defined materials are indicated; (values in cm−1) graphene: 520, 972, 1580, 2670, adamantane 760, 970, 1230, 2930, diamond: 1330; graphite: 1580, 2722. The inclusion of adamantane is for illustration: its C(sp3) skeleton is comparable to that of diamond (Banfalvi 2012).

Raman spectra for prepared activated carbon Norit (CNH) and carbon black Vulcan (CVH, CVN500, CVN950, CVU500, CVU950) catalysts,  graphene,
graphene,  adamantane,
adamantane,  diamond,
diamond,  graphite
graphite
A comprehensive list of first order Raman bands and vibration modes of activated carbon was reported by Sadezky et al. (2005), where the Lorentz function was used for peak fitting.
~1200 cm−1, D4 (I), weak, Disordered graphitic lattice (A1g-symmetry), polyenes, ionic impurities
~1350 cm−1, D1 (D), very strong, Disordered graphitic lattice (graphene layer edges, A1g- symmetry)
~1500 cm−1, D3 (D’’), medium, Amorphous carbon (Gaussian or Lorentzian)
~1580 cm−1, G, strong, Ideal graphitic lattice (E2g-symmetry)
~1620 cm−1, D2 (D’), strong, Disordered graphitic lattice (surface graphene layers, E2g-symmetry)
The second order Raman spectra bands (rather weak) are at 2450, 2700, 2900 and 3100 cm−1.
Treatment of Raman spectra were carried out by deconvolution method using OriginLab 2003 SW. Some problems connected with this deconvolution procedure are discussed in Supplement.
We started to fit the data according to procedure suggested by Sadezky et al. (2005) (Fig. 6); however, one can be satisfied with results only for the Norit sample (Fig. 6). The Vulcan samples treated by the strict unambiguous assignment of type of peak fitting functions (Lorentzian for the D peak and Gaussian for the G peak) did not allow to obtain a good quality fitting. Therefore, we used a combination of these two functions. To have comparison of individual samples we used the weights on Loretz function 0.5, 0.4, 0.3, 0.4 for the peaks D4, D1, D3 and G, respectively. The peak D2 was not considered. The weight 0.5 was also used for an amorphous part at ca. 825 cm−1. Values of ID/IG (D notation is used for the D1 band) were taken into account (the last entry in Table 1) similarly to the research of Holade et al. (2014). The quality of fitting is illustrated by Fig. 7 and Fig. S15. As illustrated by Fig. 5 and values in Table 1, all the Vulcan samples are similar. There is, however, a significant difference between Vulcan and Norit samples. The ID/IG ratio obtained for our Vulcan sample was 2.19 which is in good agreement with data of Holade et al. (2014) – about 2.14. Samples after thermal treatment exhibited lower values (1.69, 1.63, 2.07, 1.83 for the CVN500, CVN950, CVU500, CVU950, respectively). The increase in the G-fraction; i.e. lowering of the ID/IG ratio; can be the consequence of an increase in graphitizing what is stressed by lower values of the ID/IG ratio for materials treated at 950 °C.

Raman analysis of the CNH catalyst with consideration of D1, D2, D3, D4 and G Raman bands

Raman analysis of the CVH catalyst
The effect of modification was reliably proven by Boehm titration (Figs. 8, 9). Figure 9 compare pristine carbon material Vulcan XC72 as delivered, before and after treatment with HCl. After treatment with HCl, an increase in acidity is noticeable (samples CNH and CVH). The highest content of acidic groups was found in catalysts treated with HCl, whereas due to the higher specific surface area this acidity was significantly higher for the CNH catalyst in comparison with the CVH catalysts (1.17 and 0.45 mmol g−1, respectively). Treatment with nitrogen decreased the content of acidic groups and the surface pH was increased (the entry for CVN500). A higher calcination temperature (950 °C) in the nitrogen atmosphere led to a further increase of the basicity (the entry for the CNN950—Fig. 8 and CVN950—Fig. 9). This behaviour is attributed to the formation of γ-pyrone like structures (Boehm 1966).

Surface pH (orange square, right axis), content of acidic (red) and basic (blue) groups (mmol g−1) in catalysts prepared from the activated carbon Norit SX plus

Surface pH (orange square, right axis), content of acidic (red) and basic (blue) groups (mmol g−1) in catalysts prepared from the carbon black Vulcan XC72
The urea treatment established a significantly higher content of acidic and basic groups (according to Boehm titration methodology) and the surface pH was increased, as well. However, calcination at 950 °C led to a removal of acidic groups and mainly a removal of basic groups. Subsequently, the surface pH decreased (see entry CVU950).
Catalytic performance
As seen in Figs. 11, 13, 15, 16) selectivity to SA was in all catalytic tests very low (< 5%), what was also accompanied by a low formation of CO2. The solution of Ba(OH)2 was virtually clear after bubbling through the off-gas from the reactor, or it exhibited only a slight white opalescence. The solid phase was not possible to isolate, even after ageing. Therefore, we concluded that the amount of BaCO3 was negligible.
In a blank experiment, without catalyst, after 210 min no dicarboxylic acids were detected under given experimental conditions. All catalysts prepared from the activated carbon Norit SX (high surface area and volume of micropores) exhibited low activity in the oxidation of cyclopentanone (Fig. 10). Negative effect of a thermal treatment and urea modification of demineralized Norit on conversion of cyclopentanone and formation of glutaric acid was observed (Fig. 11).

Consumption of oxygen for the Norit-based catalysts in the oxidation of cyclopentanone (90 °C, 0.45 MPa, 150 mg of catalyst, 3 mL of CPO)

Conversion and selectivities to GA and SA for the same catalysts as referred on Fig. 10 (90 °C, 0.45 MPa, 150 mg of catalyst, 3 mL of CPO): black—conversion of CPO, green—selectivity to GA, red—selectivity to SA
Despite rather low conversion, the CN catalysts were quickly deactivated, making the recycling of the catalyst unfavourable.
A high deactivation of the CN catalysts probably stems from the high specific surface area (a high surface concentration of catalytic sites) and the reaction enthalpy of oxidation reactions. The desired reaction releases more than 700 kJ mol−1:
The reaction enthalpy (4) was calculated from formation enthalpies (kJ mol−1) for the 298.15 K: −197.4 (CPO, NIST, liquid phase), 0.0 (O2, NIST), -940.3 (GA, liquid phase) (Steele et al. 2002). This high value of the reaction enthalpy increases a local temperature on the catalysts surface. This increase leads to the formation of heavy secondary condensation/polymerisation products from the beginning of the experiment. Consequently, the reaction rate is decreased. A high extent of side reactions is the reason why the selectivities to GA (10% at maximum) and SA (3.5%) are low.
Due to the low activity of the CN catalysts (see Fig. 10), no further experiments were carried out over them. CNH catalyst as a representative of Norit catalysts was further characterized.
The results obtained using Vulcan-based catalysts (CV) are summarized in Figs. 12 and 13. In the case of these catalysts, the surface area and the pores volumes are significantly smaller compared to the Norit catalysts (Table 1). Boehm titration indicated that the treatment of Vulcan with HCl increased the amounts of acidic functional groups to 0.45 mmol.g−1 (Fig. 9). The activity of CVH catalyst in the oxidation cyclopentanone was higher than that of very acidic demineralised Norit catalyst (CNH), with 1.17 mmol of acidic groups/g of carbon. It means that the catalytic activity in cyclopentanone oxidation depends on factors other than the surface area of the catalyst. Chen (2011) reported that the surface area does not play a major role in catalysis with activated carbon.

Consumption of oxygen for the CV and CNH catalysts in the oxidation of cyclopentanone (90 °C, 0.45 MPa, 150 mg of catalyst, 3 mL of CPO)

Conversion and selectivities to GA and SA for catalysts as referred on Fig. 12 (90 °C, 0.45 MPa, 150 mg of catalyst, 3 mL of CPO): black—conversion of CPO, green—selectivity to GA, red—selectivity to SA
The amounts of acidic functional groups decreased to 0.07 and 0.05 mmol.g−1 for the Vulcan samples treated in nitrogen (activation or modification) at 500 °C and 950 °C, respectively. At lower temperatures (500 °C) strong acid groups, such as carboxylic, anhydride, lactone are decomposed, while higher temperatures (950 °C) eliminate weakly acid groups, such as carboxyl, phenol and quinone (Shafeeyan et al. 2010; Pigamo et al. 2002; Figueiredo and Pereira 2010). Contrary to the literature (Pigamo et al. 2002), no positive effect of increased basicity of prepared CVN500 and CVN950 catalysts (Fig. 13) on the selectivity of glutaric acid formation was observed.
Activities and selectivities were enhanced by modification with urea and following treatment in N2 at 500 °C, which, according to Adib (Adib et al. 2000), creates nitrogen-containing sites. After modification, the contents of surface acids and bases are higher than for demineralized Vulcan activated in nitrogen at the same temperature, yielding surface pH = 8 (Fig. 9). At lower temperature, 500 °C, nitrogen is likely to be bound in the form of -NH, -NH2 or NH4+ (Adib et al. 2000; Unglaube et al. 2023). Formation of N–H (amine) moieties in our case was confirmed by XPS measurement and a broad peak at 3040 − 3400 cm−1 in FTIR spectrum (Fig. 4) is also associated with the N–H stretching vibration in amine and amide groups (Allwar 2012). Nitrogen atoms in the CVU500 catalyst showed a positive effect on the catalytic activity on the oxidation of cyclopentanone into GA with molecular oxygen.
After thermal treatment of the urea modified catalyst at 950 °C (CVU950), nitrogen content determined by XPS and EDX decreased, but the conversion of CPO was inverse to the nitrogen content (Fig. 13). Upon heating to 950 °C most of the nitrogen is incorporated into the carbon matrix, mainly in pyridinic, pyrrolic and quaternary nitrogen form (Adib et al. 2000; Unglaube et al. 2023). The band around 1500–1600 cm−1 in the FTIR spectrum of the CVU950 catalyst (Fig. 4) can be assigned to pyridines (Adib et al. 2000; Unglaube et al. 2023).
An effect of the temperature on the reaction rate is demonstrated in Figs. 14 and 15. Up to 105 °C the reaction rate increases with the increased temperature. A further increase in temperature by only 5 °C no longer increases the reaction rate, on the contrary, it sharply decreases it.

Consumption of oxygen for the CVU950 catalyst in the oxidation of cyclopentanone—for various temperatures (0.45 MPa, 150 mg of catalyst, 3 mL of CPO, temperature indicated in figure)

Conversion and selectivities to GA and SA for the CVU950 catalyst (0.45 MPa, 150 mg of catalyst, 3 mL of CPO, temperature indicated in Fig.): black—conversion of CPO, green—selectivity to GA, red—selectivity to SA
Furthermore, the lifespan and the possibility of recycling of CV catalysts were tested. The initial screening showed that the CVU950 catalyst rendered the best recyclability at a temperature of 90 °C. The duration of the experiment was then extended from original 210 min to 300 min. It was found that a higher conversion of CPO was achieved in this way and the selectivity to GA increased as well (see Fig. 16). However, a problem with the separation of the catalyst appears after such a duration of the experiment, since the reaction mixture at the end of the reaction was thick and after washing with methanol the catalyst did not regain its original activity.

Time development of the conversion and selectivity over CVU950 (90 °C, 0.45 MPa, 150 mg of catalyst, 3 mL of CPO): black—conversion of CPO, green—selectivity to GA, red—selectivity to SA
Therefore, further experiments were conducted with a duration of 210 min. Figures 17 and 18 demonstrate the fair recyclability and lifetime of the CVU950 catalyst under the given conditions.

Consumption of oxygen in experiments with the CVU950 catalyst in the oxidation of cyclopentanone (90 °C, 0.45 MPa, 150 mg of catalyst, 3 mL of CPO) with the fresh (0) and recycled catalyst in three consecutive runs (1-blue, 2-light green, 3-Gy)

Conversion and selectivities to GA and SA for the same catalysts as referred on Fig. 17 (90 °C, 0.45 MPa, 150 mg of catalyst, 3 mL of CPO): black—conversion of CPO, green—selectivity to GA, red—selectivity to SA
As demonstrated in Figs. S16 and S17 in the Supplement (GC–MS analysis), the oxidation of CPO gives a wide range of reaction products. The most important reaction products were:
-
2-Cyclopenten-1-one and 3-Cyclopenten-1-one (oxodehydrogenation—removal of two hydrogen atoms from CPO)
-
Pentanoic acid
-
δ-Valerolactone
-
5-Hydroxypentanoic acid
-
5-Oxopentanoic acid (Glutaraldehydic acid)
-
Pentanedioic acid (GA, desired product)
-
Butanedioic acid (SA, succinic acid)
-
Decanedioic acid
Based on the occurrence of detected intermediates in the course of the oxidation of cyclopentanone and taking into account recent data from literature (Bhanja et al. 2018), we proposed Scheme shown in Fig. 19.

Proposed interactions and reactions during the catalytic oxidation of cyclopentanone by the modified carbon Vulcan catalysts. (Yellow and blue colours indicate the liquid and gas–vapour phases, respectively.)
A very important initial step (1) is the tautomerization of cyclopentanone to pentene-1-ol. Literature (Al-Msiedeen et al. 2016) reports the equilibrium constant for this reaction 7.2 × 10–10, what is a significantly lower value than, e.g. for cyclohexanone (1.0 × 10–8). A lower willingness to form enol form is also underlined by (endothermic) reaction enthalpy; 14.8 and 12.1 kcal/mol for the cyclopentanone and cyclohexanone, respectively. Since protons stabilise the enol form (Attia et al. 2018), and no acid is added at the start of the experiment, an autocatalytic effect of the formed carboxylic acids may be supposed. This elucidates the induction period of the reaction. In most of the reactions catalyzed by AC, the activation of molecular oxygen, i.e. its adsorption with a subsequent transfer of electrons from the basal space of graphitic species (denoted as *), while forming *-O–Oδ− (some portion splitted to *-Oδ−), plays a crucial role (Klepel et al. 2021). In the next step (3), a complex similar to that of proposed by Bhanja et al. (2018) is considered; however, with bonds including electrons from the AC. Here, a relatively high electric conductivity of Vulcan type carbon plays an important role. (For illustration, this conductivity was successfully utilized in fuel cells, Lázaro et al. 2011). The most important step (4) is the transfer of electrons from the C(-O-) atom to the chemisorbed organic oxygenate moiety and a formation of δ-valerolactone. The rate of the hydrolysis of the lactone (step 6 in Scheme) is proportional to the water content and to the activity of hydroxonium ions (Gómez-Bombarelli et al. 2013). The latter factor contributes to the elucidation of the low rate of the oxidation at the start of the experiment, when virtually no hydrogen protons and water are present.
The next step is the oxidation of 5-hydroxypentanoic acid. At first glutaraldehydic acid is formed (step 7, also proved by GC–MS) and finally (step 8) the glutaric acid is created.
The chemisorbed molecules in Scheme are depicted as to be oriented perpendicular to the surface for the sake of graphical simplicity. However, one should keep in mind that p-π interactions between graphite planes with the double bond of pentene-1-ol or oxygen necessitates a planar arrangement. Therefore, a planar, or close to planar geometry in chemisorption should be suggested. This assumption underlines the importance of sufficient amount of accessible planar domains in AC, represented by graphene, or graphitic plates with some disorders on the surface. In this sense, according to Raman analysis CV catalysts are superior to the CN catalysts. The importance of the p-π interactions for the rate and selectivity in the CPO oxidation is in the accordance with Wei et al. (2015), who emphasized this interaction in the cases of selective hydrogenations. In addition to these p-π interactions, activation of chemisorbed molecules may be increased by heteroatoms.
In the case of CPO oxidation over the activated Norit catalysts, the time profiles of oxygen consumptions differ from that of Vulcan type catalysts; the induction period is shorter, the conversion of CPO expressed by oxygen consumption and chemical analysis, as well as selectivities to glutaric acid are much lower. We can assume that a higher specific surface area of CN catalysts and a higher exposure of edges, as indicated by the ID/IG ratio from Raman spectra, allows a higher extent of reactions on edges. One can assume that reactions do not proceed on catalytic sites of “one edge” but catalytic sites from adjacent edges can be employed. The initial rate of reactions on CN is higher than that on CV catalysts and, therefore, an increase in the local temperature on the surface occurs, which leads to the formation of the heavier intermediates, which can irreversibly occupy the catalyst´s surface. The regeneration by calcination of the used CN catalysts led to lower conversion and selectivity compared with the fresh ones.
Conclusions
According to our survey, the topic of solvent-free oxidation of CPO over non-metal-containing activated carbon is not sufficiently described in the literature. This fact underlines the novelty of the presented research. An advantage of catalysts not containing transition metals is no-leaching, what allows good recyclability of the used catalysts under proper reaction conditions and after treatment of the catalysts (washing with methanol and drying). Norit-based catalysts (a significantly higher specific surface area) showed significantly lower activity than Vulcan XC72. In Norit, Raman analysis showed a higher portion of the disordered carbon than in Vulcan type catalysts. We hypothesize that a higher specific surface area of Norit (about 900 m2 g−1, external surface area about 350 m2 g−1) offers more catalytic “edge-type” centres and consequently the higher extent of oxidation, together with the formation of undesired heavy by-products than Vulcan. Vulcan offers a lower specific surface area (240 m2 g−1 and 115 m2 g−1 for the total specific surface area and external surface area, respectively). The majority of catalytic centres on the CN catalysts is quickly occupied by by-products and the course of the reaction is muffled. The lower specific surface area of the CV catalysts enables less local heat generation than in case of CN and, thus, such heat can be removed by conduction and convection from the surface. Based on the occurrence of detected reaction intermediates, on the analysis of morphology of catalysts, a redox reaction scheme has been proposed. This scheme involves steps of formation of enol form of CPO, oxidation to δ-valerolactone, hydrolysis of lactone to 5-hydroxypentanoic acid, oxidation to 5-oxopentanoic acid and its oxidation to glutaric acid. Formation of side products was not considered. Catalysts CV exhibited good catalytic activity, however selectivity to GA did not exceed 35%. To recycle unreacted CPO and to exploit side products from the reaction mixture continues to be a challenge. A great advantage of the CV catalysts is their easy recyclability.
Abbreviations
- AA:
-
Adipic acid
- AC:
-
Activated carbon
- BET:
-
Brunauer–Emmett–Teller adsorption isotherm
- BJH:
-
Barrett–Joyner–Halenda method
- C:
-
Constant “C” in the BET isotherm
- CN:
-
The group of CNH, CNN500, CNN950, CNU500 and CNU950 catalysts
- CNH:
-
Activated carbon Norit SX plus treated with HCl, washed and dried
- CNN500:
-
Activated carbon Norit SX plus treated with HCl, calcined in N2 at 500 °C
- CNN950:
-
Activated carbon Norit SX plus treated with HCl, calcined in N2 at 950 °C
- CNU500:
-
AC Norit SX plus treated with HCl and urea, calcined in N2 at 500 °C
- CNU950:
-
AC Norit SX plus treated with HCl and urea, calcined in N2 at 950 °C
- CPO:
-
Cyclopentanone
- CPL:
-
Cyclopentanol
- CV:
-
The group of CVHa, CVHb, CVN500, CVN950, CVU500 and CVU950 catalysts
- CVHa:
-
Carbon black Vulcan XC72 treated with HCl, washed and dried, sample “a”
- CVHb:
-
Carbon black Vulcan XC72 treated with HCl, washed and dried, sample “b”
- CVN500:
-
Carbon black Vulcan XC72 treated with HCl, calcined in N2 at 500 °C
- CVN950:
-
Carbon black Vulcan XC72 treated with HCl, calcined in N2 at 950 °C
- CVU500:
-
Carbon black Vulcan XC72 treated with HCl and urea, calcined in N2 at 500 °C
- CVU950:
-
Carbon black Vulcan XC72 treated with HCl and urea, calcined in N2 at 950 °C
- D BJH,meso :
-
BJH average diameter of a mesopore, nm
- D BJH,micro :
-
BJH average diameter of a micropore, nm
- D K,meso :
-
Size of a mesopore—Kelvin equation, nm
- D p :
-
Diameter of a pore, nm
- D par :
-
Size of a particle, µm
- EDX:
-
Energy-dispersive spectroscopy
- FTIR:
-
Fourier transform infrared spectroscopy
- GA:
-
Glutaric acid
- GC:
-
Gas chromatography
- GC-MS:
-
Gas chromatography mass spectrometry
- IR:
-
Infrared spectra
- L:
-
Liquid
- n CPO :
-
Number of moles of cyclopentanone
- n L :
-
Number of layers in the n-layer BET isotherm
- \(N_{{\text{m}}}\) :
-
Number of measured points in an experiment
- \(N_{{{\text{par}}}}\) :
-
Number of parameters in a regression model
- p :
-
Pressure, Pa
- p 0 :
-
Pressure of saturated vapours, Pa
- \(R_{{{\text{adj}}}}^{2}\) :
-
Adjusted coefficient of determination
- S :
-
Specific surface area, m2 g−1
- SA:
-
Succinic acid
- S B,ext :
-
External surface area—BET isotherm with the consideration of micropores, m2 g−1
- S GA :
-
Selectivity to glutaric acid, %
- SEM:
-
Scanning electron microscopy
- S K,meso :
-
Surface area of mesopores—Kelvin equation, m2 g−1
- S 1par :
-
Single point surface area at p/p° ≅ 0.3, m2 g−1
- S SA :
-
Selectivity to succinic acid, %
- S t,ext :
-
t-Plot external surface area, m2 g−1
- TGA:
-
Thermogravimetric analysis
- TPD:
-
Temperature programmed desorption
- tr :
-
Reaction time, min
- v a,N2 :
-
Volume of adsorbed nitrogen, cm3 g−1
- V :
-
Volume, cm3 g−1
- V B,micro :
-
Volume of micropores—BET isotherm with the consideration of micropores, cm3 g−1
- V K,meso :
-
Volume of mesopores—Kelvin equation, cm3 g−1
- V K,micro :
-
Volume of micropores—Kelvin equation, cm3 g−1
- V t,micro :
-
t-Plot micropore volume, cm3 g−1
- x :
-
Independent variable
- x :
-
Relative pressure
- X CPO :
-
Conversion of cyclopentanone, %
- XPS:
-
X-ray photoelectron spectroscopy
- XRD:
-
X-ray powder diffraction
- \(y_{i}\) :
-
Value of a variable in the point i
- \(\overline{y}\) :
-
Average value of all the values in points 1,…, Nm
- \(\overset{\lower0.5em\hbox{$\smash{\scriptscriptstyle\frown}$}}{y}_{i}\) :
-
Value predicted by the regression model
- *:
-
Catalytic centre
References
Adib F, Bagreev A, Bandosz TJ (2000) Adsorption/oxidation of hydrogen sulfide on nitrogen-containing activated carbons. Langmuir 16(4):1980–1986. https://doi.org/10.1021/la990926o
Al-Msiedeen AM, Al-Mazaideh GM, Khalil SM (2016) A theoretical study of the enol contents of cyclohexanone, cyclopentanone and acetone. Am Chem Sci J 13(4):1–8. https://doi.org/10.9734/ACSJ/2016/25048
Alves CCO, Franca AS, Oliveira LS (2013) Evaluation of an adsorbent based on agricultural waste (corn cobs) for removal of tyrosine and phenylalanine from aqueous solutions. BioMed Res Int 2013:978256. https://doi.org/10.1155/2013/978256
Allwar A (2012) Characteristics of pore structures and surface chemistry of activated carbons by physisorption, FTIR and Boehm methods. IOSR J Appl Chem 2(1):9–15. https://doi.org/10.9790/5736-0210915
Attia S, Schmidt MC, Schröder C, Pessier P, Schauermann S (2018) Surface-driven Keto–Enol tautomerization: atomistic insights into enol formation and stabilization mechanisms. Angew Chem 130(51):16901–16906. https://doi.org/10.1002/anie.201808453
Bagreev A, Menendez JA, Dukhno I, Tarasenko Y, Bandosz TJ (2004) Bituminous coal-based activated carbons modified with nitrogen as adsorbents of hydrogen sulfide. Carbon 42(3):469–476. https://doi.org/10.1016/j.carbon.2003.10.042
Bandosz TJ, Ania CO (2006) Surface chemistry of activated carbons and its characterization. Interf Sci Technol 7:159–229. https://doi.org/10.1016/S1573-4285(06)80013-X
Banfalvi G (2012) C12: the building block of hexagonal diamond. Open Chem 10(5):1676–1680. https://doi.org/10.2478/s11532-012-0085-3
Barlocco I, Bellomi S, Tumiati S, Fumagalli P, Dimitratos N, Roldan A, Villa A (2022) Selective decomposition of hydrazine over metal free carbonaceous materials. Phys Chem Chem Phys 24(5):3017–3029. https://doi.org/10.1039/D1CP05179B
Barlocco I, Capelli S, Lu X, Tumiati S, Dimitratos N, Roldan A, Villa A (2020) Role of defects in carbon materials during metal-free formic acid dehydrogenation. Nanoscale 12(44):22768–22777. https://doi.org/10.1039/D0NR05774F
Bashkova S, Baker FS, Wu X, Armstrong TR, Schwartz V (2007) Activated carbon catalyst for selective oxidation of hydrogen sulphide: on the influence of pore structure, surface characteristics, and catalytically-active nitrogen. Carbon 45(6):1354–1363. https://doi.org/10.1016/j.carbon.2007.01.005
Besson M, Blackburn A, Gallezot P, Kozynchenko O, Pigamo A, Tennison S (2000) Oxidation with air of cyclohexanone to carboxylic diacids on carbon catalysts. Top Catal 13:253–257. https://doi.org/10.1023/A:1009090518341
Bhanja P, Chatterjee S, Patra AK, Bhaumik A (2018) A new microporous oxyfluorinated titanium (IV) phosphate as an efficient heterogeneous catalyst for the selective oxidation of cyclohexanone. J Colloid Interface Sci 511:92–100. https://doi.org/10.1016/j.jcis.2017.09.115
Bokobza L, Bruneel JL, Couzi M (2015) Raman spectra of carbon-based materials (from graphite to carbon black) and of some silicone composites. C 1(1):77–94. https://doi.org/10.3390/c1010077
Boehm HP (1966) Chemical identification of surface groups. Adv Catal 16:179–274. https://doi.org/10.1016/S0360-0564(08)60354-5
Boehm HP (1994) Some aspects of the surface-chemistry of carbon blacks and other carbons. Carbon 32(5):759–769. https://doi.org/10.1016/0008-6223(94)90031-0
Brégeault JM, Launay F, Atlamsani A (2001) Catalytic oxidative carbon-carbon bond cleavage of ketones with dioxygen: assessment of some metal complexes. Some alternatives for preparing α, ω-dicarboxylic acids. C R Acad Sci Paris Ser IIC-Chem 4(1):11–26. https://doi.org/10.1016/S1387-1609(00)01228-2
Cabot (2022) Datasheet-VULCAN-XC72%20carbon%20black.pdf
Chen Y, Chen W, Tang Q, Guo Z, Yang Y, Su F (2011) Aerobic oxidation of benzyl alcohol over activated carbon supported manganese and vanadium catalysts: effect of surface oxygen-containing groups. Catal Lett 141:149–157. https://doi.org/10.1007/s10562-010-0447-4
Deng H, Zhang G, Xu X, Tao G, Dai J (2010) Optimization of preparation of activated carbon from cotton stalk by microwave assisted phosphoric acid-chemical activation. J Hazard Mater 182(1–3):217–224. https://doi.org/10.1016/j.jhazmat.2010.06.018
Duong-Viet C, Liu Y, Ba H, Truong-Phuoc L, Baaziz W, Nguyen-Dinh L, Nhut JM, Pham-Huu C (2016) Carbon nanotubes containing oxygenated decorating defects as metal-free catalyst for selective oxidation of H2S. Appl Catal B Environ 191:29–41. https://doi.org/10.1016/j.apcatb.2016.03.018
Engineersedge (2023) https://www.engineersedge.com/thermodynamics/van_der_waals_constants_for_gases_15688.htm. Accessed 22 May 2023
Figueiredo JL, Pereira MFR (2010) The role of surface chemistry in catalysis with carbons. Catal Today 150(1–2):2–7. https://doi.org/10.1016/j.cattod.2009.04.010
Fujita SI, Yoshida H, Arai M (2017) Nitrogen-doped activated carbon as metal-free catalysts having various functions. C. https://doi.org/10.3390/c3040031
Glutaric acid market (2022) https://www.futuremarketinsights.com/reports/glutaric-acid-during-market
Gómez-Bombarelli R, Calle E, Casado J (2013) Mechanisms of lactone hydrolysis in acidic conditions. J Org Chem 78(14):6880–6889. https://doi.org/10.1021/jo4002596
Han T, Kim GB, Lee SY (2020) Glutaric acid production by systems metabolic engineering of an L-lysine–overproducing Corynebacterium glutamicum. Proc Natl Acad Sci USA 117(48):30328–30334. https://doi.org/10.1073/pnas.2017483117
Harris PJ, Liu Z, Suenaga K (2008) Imaging the atomic structure of activated carbon. J Phys Condens Matter 20(36):362201. https://doi.org/10.1088/0953-8984/20/36/362201
Holade Y, Morais C, Servat K, Napporn TW, Kokoh KB (2014) Enhancing the available specific surface area of carbon supports to boost the electroactivity of nanostructured Pt catalysts. Phys Chem Chem Phys 16(46):25609–25620. https://doi.org/10.1039/c4cp03851g
Horniakova J, Králik M, Kaszonyi A, Mravec D (2001) A practical approach to the treatment of adsorption–desorption isotherms, acidity and catalytic behaviour of zeolite catalysts. Microporous Mesoporous Mater 46(2–3):287–298. https://doi.org/10.1016/S1387-1811(01)00309-2
Hronec M, Fulajtarová K (2012) Selective transformation of furfural to cyclopentanone. Catal Commun 24:100–104. https://doi.org/10.1016/j.catcom.2012.03.020
Hronec M, Fulajtárova K, Soták T (2014) Highly selective rearrangement of furfuryl alcohol to cyclopentanone. Appl Catal B Environ 154–155:294–300. https://doi.org/10.1016/j.apcatb.2014.02.029
Hronec M, Fulajtárová K, Vávra I, Soták T, Dobročka E, Mičušík M (2016) Carbon supported Pd–Cu catalysts for highly selective rearrangement of furfural to cyclopentanone. Appl Catal B Environ 181:210–219. https://doi.org/10.1016/j.apcatb.2015.07.046
Hulicova-Jurcakova D, Seredych M, Lu GQ, Bandosz TJ (2009) Combined effect of nitrogen-and oxygen-containing functional group of microporous activated carbon on its electrochemical performance in supercapacitors. Adv Funct Mater 19(3):438–447. https://doi.org/10.1002/adfm.200801236
Ishimaru K, Hata T, Bronsveld P, Nishizawa T, Imamura Y (2007) Characterization of sp2- and sp3-bonded carbon in wood charcoal. J Wood Sci 53:442–448. https://doi.org/10.1007/s10086-007-0879-7
Iwanow M, Gärtner T, Sieber V, König B (2020) Activated carbon as catalyst support: precursors, preparation, modification and characterization. Beilstein J Org Chem 16(1):1188–1202. https://doi.org/10.3762/bjoc.16.104
Johnson RW, Pollock CM, Cantrell RR (2010) Dicarboxylic acids. In: Kirk-Othmer Encyclopedia of Chemical Technology. https://doi.org/10.1002/0471238961.0409030110150814.a01.pub2
Jura G, Harkins WD (1944) Surfaces of solids. XI. Determination of the decrease (π) of free surface energy of a solid by an adsorbed film. J Am Chem Soc 66(8):1356–1362. https://doi.org/10.1021/ja01236a046
Kampouraki ZC, Giannakoudakis DA, Triantafyllidis KS, Deliyanni EA (2019) Catalytic oxidative desulfurization of a 4, 6-DMDBT containing model fuel by metal-free activated carbons: the key role of surface chemistry. Green Chem 21(24):6685–6698. https://doi.org/10.1039/C9GC03234G
Kim HT, Khang TU, Baritugo KA, Hyun SM, Kang KH, Jung SH, Song BK, Park K, Oh MK, Kim GB, Kim HU, Lee SY, Park SJ, Joo JC (2019) Metabolic engineering of Corynebacterium glutamicum for the production of glutaric acid, a C5 dicarboxylic acid platform chemical. Metab Eng 51:99–109. https://doi.org/10.1016/j.ymben.2018.08.007
Klepel O, Utgenannt S, Vormelchert C, König M, Meißner A, Hansen F, Bölte JH, Sieber T, Heinemann R, Bron M, Rokicińska A, Jarczewski S, Kuśtrowski P (2021) Redox catalysts based on amorphous porous carbons. Microporous Mesoporous Mater 323:111257. https://doi.org/10.1016/j.micromeso.2021.111257
Konwar LJ, Mäki-Arvela P, Salminen E, Kumar N, Thakur AJ, Mikkola JP, Deka D (2015) Towards carbon efficient biorefining: multifunctional mesoporous solid acids obtained from biodiesel production wastes for biomass conversion. Appl Catal B Environ 176:20–35. https://doi.org/10.1016/j.apcatb.2015.03.005
Kralik M, Vallusova Z, Laluch J, Mikulec J, Macho V (2008) Comparison of ruthenium catalysts supported on beta and mordenite in the hydrocycloalkylation of benzene. Pet Coal 50(1):44–51
Králik M (2014) Adsorption, chemisorption, and catalysis. Chem Pap 68(12):1625–1638. https://doi.org/10.2478/s11696-014-0624-9
Lázaro MJ, Calvillo L, Celorrio V, Pardo JI, Perathoner S, Moliner R (2011) Study and application of carbon black Vulcan XC-72R in polymeric electrolyte fuel cells. In: Sanders IJ, Peeten TL (eds) Carbon black: production, properties and uses. Nova Science Publishers, Inc, p 41. ISBN:978-1-61209-535-6
Lepre E, Rat S, Cavedon C, Seeberger PH, Pieber B, Antonietti M, López-Salas N (2023) Catalytic properties of high nitrogen content carbonaceous materials. Angew Chem 135(2):e202211663. https://doi.org/10.1002/ange.202211663
Li ZQ, Lu CJ, Xia ZP, Zhou Y, Luo Z (2007) X-ray diffraction patterns of graphite and turbostratic carbon. Carbon 45:1686–1695. https://doi.org/10.1016/j.carbon.2007.03.038
Lisicki D, Orlińska B (2018) Oxidation of cyclic ketones to dicarboxylic acids. Pol J Chem Tech 20:102–107. https://doi.org/10.2478/pjct-2018-0061
Lowell S, Shields JE, Thomas MA, Thommes M (2004) Characterization of porous solids and powders: surface area, pore size and density. Springer, Netherlands. https://doi.org/10.1007/978-1-4020-2303-3
Lv Q, Si W, He J, Sun L, Zhang C, Wang N, Yang Z, Li X, Wang X, Deng W, Long Y, Huang C, Li Y (2018) Selectively nitrogen-doped carbon materials as superior metal-free catalysts for oxygen reduction. Nat Commun 9(1):3376. https://doi.org/10.1038/s41467-018-05878-y
Malins K, Kampars V, Brinks J, Neibolte I, Murnieks R (2015) Synthesis of activated carbon based heterogenous acid catalyst for biodiesel preparation. Appl Catal B Environ 176:553–558. https://doi.org/10.1016/j.apcatb.2015.04.043
Merlen A, Buijnsters JG, Pardanaud C (2017) A guide to and review of the use of multiwavelength Raman spectroscopy for characterizing defective aromatic carbon solids: from graphene to amorphous carbons. Coatings 7(10):153. https://doi.org/10.3390/coatings7100153
Micromeritics (2020) Microactive ASAP®2020 accelerated surface area and porosimetry system. Copyright © 2019. Micromeritics Instrument Corporation
Nakamizo M, Honda H, Inagaki M (1978) Raman spectra of ground natural graphite. Carbon 16(4):281–283. https://doi.org/10.1016/0008-6223(78)90043-X
Navalon S, Dhakshinamoorthy A, Alvaro M, Antonietti M, García H (2017) Active sites on graphene-based materials as metal-free catalysts. Chem Soc Rev 46(15):4501–4529. https://doi.org/10.1039/c7cs00156h
OriginLab (2003) Peak fitting module. USA. www.OriginLab.com
Park SJ, Kim EY, Noh W, Park HM, Oh YH, Lee SH, Song BK, Jegal J, Lee SY (2013) Metabolic engineering of Escherichia coli for the production of 5-aminovalerate and glutarate as C5 platform chemicals. Metab Eng 16:42–47. https://doi.org/10.1016/j.ymben.2012.11.011
Peña JM, Allen NS, Edge M, Liauw CM, Valange B, Santamaria F (2001) The use of microwave and FTIR spectroscopy for the characterisation of carbon blacks modified with stabilisers. Polym Degrad Stab 74(1):1–24. https://doi.org/10.1016/S0141-3910(01)00009-X
Pérez-Cadenas AF, Maldonado-Hodar FJ, Moreno-Castilla C (2003) On the nature of surface acid sites of chlorinated activated carbons. Carbon 41(3):473–478. https://doi.org/10.1016/S0008-6223(02)00353-02
Pigamo A, Besson M, Blanc B, Gallezot P, Blackburn A, Kozynchenko O, Tennison S, Creeze E, Kapteijn F (2002) Effect of oxygen functional groups on synthetic carbons on liquid phase oxidation of cyclohexanone. Carbon 40(8):1267–1278. https://doi.org/10.1016/S0008-6223(01)00284-6
Pikna Ľ, Heželová M, Demčáková S, Smrčová M, Plešingerová B, Štefanko M, Turáková M, Králik M, Puliš P, Lehocký P (2014) Effect of support on activity of palladium catalysts in nitrobenzene hydrogenation. Chem Pap 68:591–598. https://doi.org/10.2478/s11696-013-0497-3
Puziy AM, Poddubnaya OI, Martínez-Alonso A, Suárez-García F, Tascón JMD (2002) Synthetic carbons activated with phosphoric acid: I. Surface chemistry and ion binding properties. Carbon 40(9):1493–1505. https://doi.org/10.1016/S0008-6223(01)00317-7
Qi W, Su D (2014) Metal-free carbon catalysts for oxidative dehydrogenation reactions. ACS Catal 4(9):3212–3218. https://doi.org/10.1021/cs500723v
Ren Y, Yuan Z, Lv K, Sun J, Zhang Z, Chi Q (2018) Selective and metal-free oxidation of biomass-derived 5-hydroxymethylfurfural to 2, 5-diformylfuran over nitrogen-doped carbon materials. Green Chem 20(21):4946–4956. https://doi.org/10.1039/C8GC02286K
Rohles CM, Gläser L, Kohlstedt M, Gießelmann G, Pearson S, del Campo A, Becker J, Wittmann C (2018) A bio-based route to the carbon-5 chemical glutaric acid and to bionylon-6,5 using metabolically engineered Corynebacterium glutamicum. Green Chem 20(20):4662–4674. https://doi.org/10.1039/C8GC01901K
Sadezky A, Muckenhuber H, Grothe H, Niessner R, Pöschl U (2005) Raman microspectroscopy of soot and related carbonaceous materials: spectral analysis and structural information. Carbon 43(8):1731–1742. https://doi.org/10.1016/j.carbon.2005.02.018
Scardamaglia M, Susi T, Struzzi C, Snyders R, Di Santo G, Petaccia L, Bittencourt C (2017) Spectroscopic observation of oxygen dissociation on nitrogen-doped graphene. Sci Rep 7(1):7960. https://doi.org/10.1038/s41598-017-08651-1
Seredych M, Hulicova-Jurcakova D, Lu GQ, Bandosz TJ (2008) Surface functional groups of carbons and the effects of their chemical character, density and accessibility to ions on electrochemical performance. Carbon 46(11):1475–1488. https://doi.org/10.1016/j.carbon.2008.06.027
Shafeeyan MS, Daud WMAW, Houshmand A, Shamiri A (2010) A review on surface modification of activated carbon for carbon dioxide adsorption. J Anal Appl Pyrolysis 89(2):143–151. https://doi.org/10.1016/j.jaap.2010.07.006
Shao D, Jiang Z, Wang X (2010) SDBS modified XC-72 carbon for the removal of Pb (II) from aqueous solutions. Plasma Process Polym 7(7):552–560. https://doi.org/10.1002/ppap.201000005
Shao Y, Sui J, Yin G, Gao Y (2008) Nitrogen-doped carbon nanostructures and their composites as catalytic materials for proton exchange membrane fuel cell. Appl Catal B Environ 79(1):89–99. https://doi.org/10.1016/j.apcatb.2007.09.047
Shen T, Hu R, Zhu C, Li M, Zhuang W, Tang C, Ying H (2018) Production of cyclopentanone from furfural over Ru/C with Al11.6PO23.7 and application in the synthesis of diesel range alkanes. RSC Adv 8(66):37993–38001. https://doi.org/10.1039/C8RA08757A
Schuepfer DB, Badaczewski F, Guerra-Castro JM, Hofmann DM, Heiliger C, Smarsly B, Klar PJ (2020) Assessing the structural properties of graphitic and non-graphitic carbons by Raman spectroscopy. Carbon 161:359–372. https://doi.org/10.1016/j.carbon.2019.12.094
Schüpfer DB, Badaczewski F, Peilstöcker J, Guerra-Castro JM, Shim H, Firoozabadi S, Beyer A, Volz K, Presser V, Heiliger C, Smarsly B, Klar PJ (2021) Monitoring the thermally induced transition from sp3-hybridized into sp2-hybridized carbons. Carbon 172:214–227. https://doi.org/10.1016/j.carbon.2020.09.063
Soták T, Hronec M, Gál M, Dobročka E, Škriniarová J (2017) Aqueous-phase oxidation of furfural to maleic acid catalyzed by copper phosphate catalysts. Catal Lett 147:2714–2723. https://doi.org/10.1007/s10562-017-2191-5
Sousa JP, Pereira MF, Figueiredo JL (2013) Modified activated carbon as catalyst for NO oxidation. Fuel Process Technol 106:727–733. https://doi.org/10.1016/j.fuproc.2012.10.008
Steele WV, Chirico RD, Cowell AB, Knipmeyer SE, Nguyen A (2002) Thermodynamic properties and ideal-gas enthalpies of formation for 1, 4-diisopropylbenzene, 1, 2, 4, 5-tetraisopropylbenzene, cyclohexanone oxime, dimethyl malonate, glutaric acid, and pimelic acid. J Chem Eng Data 47(4):725–739. https://doi.org/10.1021/je010088b
Sun X, Wang R, Dangsheng SU (2013) Research progress in metal-free carbon-based catalysts. Chin J Catal 34(3):508–523. https://doi.org/10.1016/S1872-2067(11)60515-9
Szymański GS, Karpiński Z, Biniak S, Świa̧tkowski A (2002) The effect of the gradual thermal decomposition of surface oxygen species on the chemical and catalytic properties of oxidized activated carbon. Carbon 40(14):2627–2639. https://doi.org/10.1016/S0008-6223(02)00188-4
Tazibet S, Boucheffa Y, Lodewyckx P (2013) Heat treatment effect on the textural, hydrophobic and adsorptive properties of activated carbons obtained from olive waste. Microporous Mesoporous Mater 170:293–298. https://doi.org/10.1016/j.micromeso.2012.12.008
Turáková M, Králik M, Lehocký P, Pikna Ľ, Smrčová M, Remeteiová D, Hudák A (2014) Influence of preparation method and palladium content on Pd/C catalysts activity in the liquid phase hydrogenation of nitrobenzene to aniline. Appl Catal A Gen 476:103–112. https://doi.org/10.1016/j.apcata.2014.02.025
Unglaube N, Florent M, Otto T, Gutiérrez-Serpa A, Stötzer M, Grothe J, Kaskel S, Bandosz TJ (2023) Weighting the role of specific nitrogen and oxygen functionalities on porous carbons in formaldehyde removal: combining surface chemistry analysis and factorial dimensionally reduction method. Carbon 215:118454. https://doi.org/10.1016/j.carbon.2023.118454
Vennat M, Brégeault JM (2010) Oxidative cleavage of cycloalkanones with dioxygen catalyzed by supported catalysts or homogeneous systems: evidence for novel active ruthenium (II) and/or (III) species. Appl Catal A Gen 386(1–2):9–15. https://doi.org/10.1016/j.apcata.2010.07.016
Wang L, Wang X, Zou B, Ma X, Qu Y, Rong C, Li Y, Su Y, Wang Z (2011) Preparation of carbon black from rice husk by hydrolysis, carbonization and pyrolysis. Bioresour Technol 102(17):8220–8224. https://doi.org/10.1016/j.biortech.2011.05.079
Wang M, Liu Q, Sun H, Ogbeifun N, Xu F, Stach EA, Xie J (2010) Investigation of carbon corrosion in polymer electrolyte fuel cells using steam etching. Mater Chem Phys 123:761–766. https://doi.org/10.1016/j.matchemphys.2010.05.055
Watanabe H, Asano S, Fujita SI, Yoshida H, Arai M (2015) Nitrogen-doped, metal-free activated carbon catalysts for aerobic oxidation of alcohols. ASC Catal 5(5):2886–2894. https://doi.org/10.1021/acscatal.5b00375
Wei Z, Pan R, Hou Y, Yang Y, Liu Y (2015) Graphene-supported Pd catalyst for highly selective hydrogenation of resorcinol to 1, 3-cyclohexanedione through giant π-conjugate interactions. Sci Rep 5(1):15664. https://doi.org/10.1038/srep15664
Xie R, Zhou L, Lan C, Fan F, Xie R, Tan H, Xie T, Zhao L (2018) Nanostructured carbon black for simultaneous electrochemical determination of trace lead and cadmium by differential pulse stripping voltammetry. R Soc Open Sci 5(7):180282. https://doi.org/10.1098/rsos.180282
Yu H, Peng F, Tan J, Hu X, Wang H, Yang J, Zheng W (2011) Selective catalysis of the aerobic oxidation of cyclohexane in the liquid phase by carbon nanotubes. Angew Chem 17(123):4064–4068. https://doi.org/10.1002/anie.201007932
Zhou M, Li J, Wang K, Xia H, Xu J, Jiang J (2017) Selective conversion of furfural to cyclopentanone over CNT-supported Cu based catalysts: model reaction for upgrading of bio-oil. Fuel 202:1–11. https://doi.org/10.1016/j.fuel.2017.03.046
Acknowledgements
Authors thank Ing. Simona Klempová from the Institute of Natural and Synthetic Polymers, FCHPT STU, for the Raman spectra measurements. This work was created thanks to the generous support under the Operational Programme Integrated Infrastructure for the project: "Support of research activities of Excellence laboratories STU in Bratislava", Project no. 313021BXZ1, co-financed by the European Regional Development Fund (50 % support) and the authors gratefully thank to the Slovak Research and Development Agency under the Contract no. APVV-17-0302 (50 % support). This work was supported also by the Ministry of Education, Research, Development and Youth of the Slovak Republic (project VEGA 1/0374/23).
Funding
Open access funding provided by The Ministry of Education, Science, Research and Sport of the Slovak Republic in cooperation with Centre for Scientific and Technical Information of the Slovak Republic.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gašparovičová, D., Králik, M., Horváth, B. et al. Liquid phase oxidation of cyclopentanone over metal-free carbon catalysts. Chem. Pap. 78, 5943–5960 (2024). https://doi.org/10.1007/s11696-024-03517-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-024-03517-z