
Abstract
A wide amount of existing building materials and consumables contains formaldehyde (HCHO) that may be released as a gas even after installation. The produced gas may concentrate in housing and offices, leading to a long-term exposure of the occupants. Living in such an environment, even at low concentrations less than 0.5 ppm, may lead to carcinogenic effects and damages of the human central nervous system. As a consequence, the development of methods that oxidize and neutralize the formaldehyde at ambient temperature is welcome. The catalytic oxidation method at ambient temperature is preferred over other available techniques due to its ease of operation and high efficiency. In this work, different weight percentages, ranging from 1 up to 5%, of silver-doped manganese oxides (Ag/MnOx) were synthesized by a co-precipitation process followed by a calcination at 450 °C for 4 h. The XRD and BET analysis results show that the sample at 4% wt of Ag doped in the MnOx matrix exhibit highest crystallinity, surface area and highest dispersion values, very near to theoretical ones. The removal performance of HCHO was tested in a silicon wool bed continuous flow reactor characterized by an inner diameter of 4 mm and a length of 12.5 cm. At ambient temperature a removal efficiency and an amount of produced CO2 of 94% and 66%, respectively.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Formaldehyde (HCHO) is a major indoor air pollutant released from decorating wood furniture and paintings that may lead to severe chronic diseases in human beings. To avoid long-term health effects, the WHO recommend short-term and long-term HCHO exposures with a maximum limit of 0.2 mg/m3 and 0.1 mg/m3, respectively (Zhu et al. 2019). Different techniques have been suggested in order to remove indoor HCHO contamination, such as adsorption, microbial decomposition, chemical absorption, photocatalytic decompositions, membrane separation and thermal catalytic oxidation (Ye et al. 2020). Adsorption is one of the most effective removal methods exhibiting highest elimination rates and efficiency values but requires maintenance by replacing the exhausted adsorbent on regular basis which leads to high operating costs and represent a burden to the environment. Microbial decomposition is capable to remove HCHO from industrial gas streams efficiently, but the required equipment is not suitable for home applications since the use of loaded activated carbons with blended biological enzymes may lead to biological hazards (Shao et al. 2020). As a consequence, thermal catalytic formaldehyde removal methods are gaining interest for home applications, and some studies were performed by using a wide variety of catalysts, such as Ag/Co3O4, Ag/SBA-15, Ag/CeO2 and Ag/MnOx-CeO2 (Chen et al. 2009; Zhang et al. 2015; Rong et al. 2018a, b; Alam et al. 2019). These catalysts may be prepared aiming to different structures and morphology to enhance HCHO removal at ambient temperature. Among these catalysts, manganese oxides (MnOx) gain interest due to the work of Sekine et al. (2002) that showed that MnO2 can break the HCHO molecule at room temperature, making this approach up most interesting for commercial use.
Among others, Chen et al. (2009) produced several MnOx catalysts such as pyrolusite, todorokite and cryptomelane observing that the latter one exhibit the best removal efficiency of HCHO. Parallel to this, these researchers produced MnO2 by synthesis such as sol–gel, hydrothermal and co-precipitation leading to different phases labeled δ, β, α and γ- MnO2. They observed that a tunnel-type pattern of MnOx, typical of δ-MnO2, appears to be a key element in obtaining enhanced catalytic activity. In this case, the catalyst is characterized by high efficiency values due to optimized lattice active surface oxygen species capable to complete the removal of HCHO at 80 °C (Zhang et al. 2015). Moreover, it has been noted that addition of K+ and/or a higher Mn4+ to Mn3+ ratio furthermore improves the catalytic activity. Both actions lead to a higher number of Mn vacancies that increases the amount of suspended O− atoms capable to boost the HCHO removal (Rong et al. 2018a). Parallel to this, other researchers observed that the single-crystalline form of α-MnO2 characterized by a 310-facet structure may perform similarly to δ-MnO2 due to the highly active surface activity given by the high number of crystalline defects after hydrothermal treatment in the presence of oxalate ions (Rong et al. 2018c). In this case, the catalyst is able to eliminate almost all of the HCHO at 60 °C.
To enhance the presence of vacancies in the catalyst, noble metals such as platinum (Pt), silver (Ag), palladium (Pd), gold (Au) and rhodium (Rh) can be employed. This kind of doping was observed to be sensibly more efficient if compared to non-noble metal oxide doping methods (Yu et al. 2013). As a consequence, many studies focused on the use of noble metal doped catalysts to investigate about the relationship between the formed structure and the mechanisms underlying catalytic HCHO degradation. As ease of an example, Au- or Ag-loaded CeO2, Co3O4 and MnO2, used as separated or mixed catalytic support, exhibits different performances yet confirming that the dispersion of noble metals is a key element determining the outcome of the removal efficiency. Due to the aggregation of the Au nanoparticles (NPs), the dispersion of the same within the metal oxide appeared to be a difficult task and thus limited in the maximum possible amount (Shen et al. 2008). Moreover, with use of polystyrene (PS) colloidal crystals, it is possible to produce complex structures such as 3D-ordered microporous 3DOM Au-doped CeO2 catalysts (3DOM Au/CeO2) (Zhang et al. 2009).
Recent research work was successful in loading single atoms of Au onto α-MnO2 by means of the adoption of a H2O2 reduction procedure followed by a subsequent heat treatment at 200 °C. Due to surface defects by Au doping that leads to vacancies in the lattice, active oxygen species are attracted to these active sites that are capable to remove HCHO molecules with high efficiency (Chen et al. 2018a). More recently, research started to achieve similar results by adopting Ag since this material is sensibly cheaper than Au; on the other side, it was observed that particular caution has to be used if Ag is adopted as doping material, since the dispersion of the latter in the lattice of the supporting material appears to be a critical point (Ou et al. 2019). If dispersion is controlled with particular care, it was observed that it is possible to obtain catalyst materials that exhibit high efficiency values such as the Ag/MCM catalyst reported elsewhere (Chen et al. 2013). While transition metal oxides (TMO) are sufficient to form pits in the lattice structure to deliver catalytic activity of the supporting material, the addition of Ag leads to the formation of a higher number of vacancies capable to tackle the HCHO decomposition (Yu et al. 2013). The efficiency of the degradation of HCHO appears to be a function of the dispersed mass of the noble metal within the TMO lattice allowing enhanced chemisorption of H2 or CO even at ambient temperature.
Catalytic removal of HCHO appears to be a promising technique due to its ease of use since no external heating source or addition chemicals are required. The here proposed technique may thus qualify as a best available technology (BAT) to an effective and environmentally friendly method for indoor HCHO degradation. In this research work, an Ag-doped manganese dioxide (Ag/MnOx) catalyst was developed and produced at different amounts of Ag by co-precipitation technique; after this, the performances of the latter in terms of catalytic oxidation of HCHO at room temperature was measured and compared to the performances of undoped MnOx. At the end, the best available material used during this work in optimized operating conditions allows to eliminate 94% of HCHO after 2 h. The catalyst was tested for converting HCHO to carbon dioxide (CO2) with time using a lab-scale reactor. The removal of HCHO in the samples was analyzed using the flame ionization detector (FID) of gas chromatography (GC). The effect of noble Ag/MnOx by changing the weight percentage of Ag and optimizing the rate for the best activity of HCHO removal was also studied. This work aimed to synthesize Ag/MnOx catalyst to achieve a strong interaction between the doping Ag and MnOx support by using the simple method of preparation as co-precipitation and optimizing the ratio of the Ag on MnOx for HCHO removal.
Experimental setup
Materials
Manganese nitrate (Mn (NO3)2), HCHO (37% w/w), silver nitrate (AgNO3, 95%), sodium carbonate (Na2CO3) and silicon wool were supplied by Sigma-Aldrich and Mosa Chemical KPK, respectively. All other reagents used in this work were of analytically pure grade.
Synthesis procedure
The adopted procedure is based on the co-precipitation method reported graphically in Fig. 1. The procedure requires to produce two reagent solutions: the first one is a solution of 1 M Mn (NO3)2 and a variable amount of AgNO3 in 100 ml distilled water (solution A); the latter one is a solution of 3 M Na2CO3 in deionized water (solution B). After this, both solutions are added dropwise in the same moment in a beaker containing initially 100 mL of distilled water, such to maintain the pH value in the range 7–8. After some time, a mixed precipitate of MnCO3 and Ag2CO3 was observed and collected on the bottom of the beaker glass, removed by filtering, washed until a constant pH value of the washing solution was observed. The obtained solid was then dried in an oven at 100 °C for 12 h before calcination at 450 °C for 4 h. According to the amount of AgNO3 being used, the obtained products were labeled as 1-Ag/MnOx, 2-Ag/MnOx, 3-Ag/MnOx, 4-Ag/MnOx, and 5-Ag/MnOx when using 0.1 M, 0.2 M, 0.3 M, 0.4 M and 0.5 M, respectively.

Synthesis process of Ag/MnOx
Finally, a sample by adopting the same procedure without any use of AgNO3 was produced to prepare the undoped reference catalyst.
Experimental setup
To allow catalyst testing a conventional tubular fixed bed flow reactor was adopted and provided with an accurate mass flow controller to achieve stable gas flow rates. The bed flow reactor is composed by quartz tubing with an inner diameter and length of 4 mm and 12.5 cm, respectively. The obtained products and all the reactants were analyzed by means of a gas chromatograph equipped with an FID detector. A carbon molecular sieve column was then used for CO2 and CO separation, even if no other carbon based compounds were found except CO2 in the products.
At the beginning a blank test was conducted without catalyst, and as expected no conversion was observed at room temperature. The experiments were designed such to use 0.156 g of the considered Ag/MnOx material for the removal of HCHO in a fixed silicon wool bed. The HCHO gas was produced by bubbling some N2 through a formalin solution at 37% leading to a gas stream containing about 10 ppm of HCHO. The gas mixture, including HCHO gas which is produced by passing the N2 gas from the bottle containing the formalin (37%) solution to convert into a gaseous state at 40 °C leading to input of HCHO of 20 ± 1 ppm and air (1L /min which corresponding to a WHSV 200,000 mL/(gcat h). Mass flow controllers were used for accurate and stable control of gas flow rates. Products and reactants analysis were carried out by a gas chromatograph equipped with an FID. A carbon molecular sieve column was used for CO2 and CO separation. No other carbonaceous compounds were found except CO2 in the products. A blank test was conducted without catalyst and no conversion was observed at room temperature. The manipulating variables for the control of these parameters were the flow rates of the relevant make-up gas streams. The flow diagram of experimental setup is shown in Fig. 2. The following equation was used to calculate the HCHO removal efficiency:
where \({\text{C}}_{{{\text{HCHO}}}} \left( {\text{\% }} \right).\) is the removal. Efficiency, [HCHO]in and [HCHO]out the initial and final concentration of HCHO, respectively. The CO2 percentage production is calculated by the following relationship:
where \(\left[ {{\text{HCHO}}} \right]_{{{\text{in}} \ldots }}\) is the HCHO concentration in the inlet stream and \([{\text{CO}}_{2} ]_{{{\text{out}}}} .\) is the CO2 concentration in the outlet stream.

Adopted experimental setup
An overall scheme of the experimental setup is reported in Fig. 2.
Catalyst testing
Thermal stability was measured by means of thermogravimetric analyzer (TGA) STARe system supplied by Mettler Toledo. The adopted measurement protocol includes the drying of the catalyst for 4 h at 423 K using an oven-style evaporator. Parallel to this, the Ag content as part of the elemental analysis of an inductively coupled plasma-atomic emission spectroscopy (ICP-OES) was used. The crystalline structure of catalysts was determined using X-ray powder diffraction (XRD) (D8-Advance, Bruker, Germany) with Cu K (wavelength = 0.15406 nm) radiation at a tube voltage of 40 kV and current of 40 mA, in the 2θ range 10–80° with a step size of 0.05. The materials’ structural characteristics, specific surface area and pore volume was quantified by means of a quanta chrome physio sorption equipment supplied by Quanta Chrome. In this latter case, the BJH and BET formula was adopted to estimate the average pore size using N2 adsorption isotherms and the specific surface areas of the catalyst, respectively. The morphology of the material was examined with a 30-kV Hitachi JEM-2010F scanning electron microscope (SEM).
Kinetic modeling
Kinetic modeling allows to obtain specific parameter values useful for the subsequent design of reactors and equipment (Xiao et al. 2015). For studying of reaction kinetics of a process, it is important to eliminate the effects of external and internal mass transfer to ensure that surface reaction to be the rate determining step. The surface reaction rate is determined by the surface concentration which is the difference between the logarithmic mean values of inlet and outlet concentration of formaldehyde from the reaction. All other parameters such as temperature, relative humidity and velocity are kept constant. The collected kinetic data of HCHO degradation have been interpreted in light of the bimolecular Langmuir Hinshelwood (LH) model:
K and Ka are the parameters to model and interpret the kinetic data of HCHO degradation. LH model is based on the competitive absorption of the reactant molecules (formaldehyde and oxygen) on the surface of catalyst with best fitting of experimental dataset.
The experimental rate has been obtained by the following equation, according to (Li et al. 2016):
where:
-
C is the initial concentration of formaldehyde (ppm), 10 mg/l
-
X is the conversion of formaldehyde (%),
-
V is the reaction mixture flow rate under normal conditions (m3/h), 0.015 m3/h
-
m (g) is the weight of the catalyst used, 0.156 g
-
Sp (m2/g) is the specific area of the catalyst used, 38.92 m2/g.
With all other operating parameters (i.e., temperature, relative humidity, velocity) fixed, the surface reaction rate was determined by the surface concentration, which was calculated by the logarithmic mean value of inlet and outlet formaldehyde concentrations.
The nonlinear regression of experimental data has been performed by an excel datasheet, adopting as error function the ERRSQ (errors squared) and evaluating the fitness goodness also by the R2 value.
Results and discussion
Calcination temperature effects
TGA was adopted to measure the weight change as a function of temperature and was performed in argon gas environment between 25 and 800 οC. The obtained results from this analysis are reported in Fig. 3. It is possible to observe that the prepared samples include MnCO3 precipitates as a precursor of the MnOx catalyst. In general, TGA provide indication about thermal stability and amount of water of the catalyst precursor, but in particular it may also be affected by the presence of Ag used as a dopant in the catalyst for this study. The weight loss occurs following a typical scheme of three distinct zones dependent of temperature increases (Kumar and Sharma 2017). In a first step, weight loss will occur by removing adsorbed and the chemically bound water of the sample by desorption at the surface of the sample. In this work, only 8% of the weight has been lost operating below 350 °C. The second phase occurred between 350 and 480 °C, characterized by a significant decrease in weight loss and equal to 32%. This observation may be justified by the conversion of metal carbonate to metal oxides and the consequent production of CO2. This effect appears to be completed when temperature rises above 500 °C, and a less extend of additional weight loss is registered due to the occurrence of phase transitions from MnCO3.to Mn3O4, passing through the formation of MnO2 and Mn2O3. Parallel to this active lattice surface oxygen grows and due to oxygen compensation effects produced by surface oxidation reactions of Mn3+.to Mn4+ a small amount of weight gain was registered up to the temperature maximum value, equal to 900 °C (Chen et al. 2014).

TGA curve of different catalyst
Concerning the obtained TGA data on the doped catalyst samples, it generally appears that the Ag doping minimizes MnOx weight losses with some differences in the TGA plot if compared to MnOx. After a common gradual breakdown phase at 60 °C, justified by the presence of volatile contaminants and water dissolution leaving the samples, the Ag-doped MnOx ones loses significant amount of weight up to 350 °C. Between 350 and 500 °C, a dramatic extreme weight loss occurs like the MnOx catalyst. However, in this region, only 29% of the weight is loosed for the reason mentioned above, which is less than 32% weight loss of MnOx catalyst. The thermally stable is shown in the catalyst starting from 350 °C, due to the catalyst precursor converting to its final state phase. Finally, thermal stability of the samples appears to benefit from higher concentrations of Ag doping, stabilizing at higher values of the weight as soon as the temperature is above 500 °C.
Inductively coupled plasma-optical emission spectrometry
Elemental analysis of the samples was performed by means of inductively coupled plasma-optical emission spectrometry (ICP-OES) investigating on the percentage content of Ag in the doped MnOx materials. The obtained results are reported in Table 1. Theoretical and experimental values of Ag doped are both compared with the error percentage in the table. It is possible to observe that there is a little gap between the theoretical and measured values of Ag doped in all the samples. The small error may be due to the sample heterogeneity or human error during the experimental step. But it is worthy to mention that a higher and homogeneous Ag dispersion improve the performances of the catalyst on HCHO removal due to a higher availability of surface-active sites, similar to the observations performed by other researchers using Pt and Au as a dopant (Peng and Wang 2007)(Wang et al. 2021). Ag dispersion in the samples can be seen in the SEM and EDX images as ICP-OES just gives elemental analysis in bulk.
Structural analysis
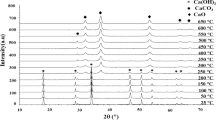
The XRD results of the prepared samples are shown in Fig. 4. It is possible to observe that characteristic peaks of specific facets of tetragonal Hausmannite Mn3O4 (according to JCPDS.008-0017 database) and of pyrolusite MnO2. (according to JCPDS 0.024-0735 database), more detailed in Table 2, appeared in the XRD output.

XRD patterns of different samples
The absence of Ag0 or Ag2O peaks in the analysis but surely present in the samples can be justified by a very fine dispersion of Ag species on the MnOx; indeed, after adding Ag, a peak shift of the XRD pattern is observed and indicate proper dopant incorporation. The high intensity and narrow peak observed in the XRD output plot is a consequence of Ag/MnOx having a high degree of crystallinity and small crystallite size (Li et al. 2019). Among the measured samples, it appears that the 4-Ag/MnOx one has highest crystallinity, and that adding more Ag crystallinity decreases probably due to nanoparticles aggregation phenomena as previously observed in SEM photography. The crystallinity of 5-Ag/MnOx is decreased as further adding the Ag may be due to nanoparticles agglomeration as shown in SEM images. Concerning crystallite size, adding Ag decreases the particle size from 300 to 100 nm as evaluated by using the Debye–Scherrer equation using the highest peak. The same was confirmed previously by BET observations.
Scanning electron microscope (SEM)
It is possible to appreciate from the SEM images (as reported in Fig 5a) that the synthesized catalyst has flower-like morphology and irregularly shaped microparticles in the range between 200 and 400 nm. These nanoparticles may form giant spherical micro clusters ranging in diameter between 1.5 and 2.0 µm.

SEM images of catalysts (a–e) for 1 to 5% Ag doping. (g and h) for MnOx and (f and i) for 4-Ag/MnOx
Surprisingly, increasing the Ag percentage resulted in a complete morphological transition of dense and aggregated nanospheres (Yusuf et al. 2020). From the reported images, it is possible to appreciate the morphological changes occurring to the catalyst and the reduction of limitation of the employed SEM in this work. Exceeding specific Ag values, such of sample 5-Ag/MnOx catalyst, a decrease in surface area can be observed (black spots). Indeed, the agglomeration of the particles into compressed nanospheres as reported in Fig. 5e leads to a decrease in the surface area and results in a decrease in the removal efficiency (Xiao et al. 2015). Moreover, particles without a particular morphology were also observed in the sample, indicating that at the beginning amorphous MnOx coexisted during the synthesis. The figure 5. (f and g) is shown for the 4-Ag/MnOx and MnOx separately to see the dispersion of Ag in the doped samples, respectively. The white spots in the sample in the Left (Fig. 5f) shows that the Ag is well dispersed in the sample. While for Fig. 5g, there is no while spots (Ag) indicating the absence of Ag in the MnO2 sample. From the Fig. 5h it can be revealed that particle size of the undoped MnOx is between 200 to 500 nm. But for the 4-Ag/MnOx samples, the average particle size is around 60 nm as shown in Fig. 5i.
The energy-dispersive X-ray analysis (EDX) exhibit that if no Ag is added during the catalyst formation, MnOx in its simple form is produced; on the contrary, by adding 4% of Ag, EDX is capable to measure this percentage of Ag within the MnOx matrix (Fig. 6) proofing that dispersed doping of Ag was successfully accomplished over the surface of MnOx.

EDX of catalysts a MnOx b 4-Ag/MnOx
Brunauer–Emmett–Teller (BET) analysis
The surface area of the prepared catalysts was determined by using the Brunauer–Emmett–Teller (BET) method; after this, a link between surface area and catalytic activity for the removal of formaldehyde was established. Specific surface area (SBET) and pore volume or porosity of all the samples are measured using N2 adsorption–desorption assays as described in Table 3. SBET values for 1-Ag/MnOx and 2-Ag/MnOx are nearly identical, 26.59 m2 g–1 and 26.73 m2 g–1, respectively. As the amount of Ag in the provided sample is increased, the surface area in the samples sensibly increases: 3-Ag/MnOx and 4-Ag/MnOx exhibit a value of 33.48 m2 g–1 and 38.92 m2 g–1, respectively. The highest surface area was indeed achieved on the 4-Ag/MnOx sample, since 5-Ag/MnOx has an SBET value equal to 34.92 m2 g–1.
The obtained results may be justified by the formation of macropores that arise due to the production of slit-shaped holes in the catalyst (Li et al. 2016). Moreover, the added amount of Ag lead to smaller sized particles, as will be hereafter reported in the XRD analysis section of this paper. An increased specific surface area is generally parallel to a more homogeneous dispersion of Ag in the particles. (Qi et al., 2021). Increasing specific surface area enables more active sites to be exposed and provides a more significant number of reaction sites that leads to higher catalytic efficacy. The average pore size was calculated by means of the BJH formula from desorption of the N2 adsorption-desorption isotherms. All the prepared catalysts from 1-Ag/MnOx to 3-Ag/MnOx, have an average pore diameter from 12.12 to 12.01 nm. On contrary, 4-Ag-MnOx exhibits the largest porosity equal to 12.22 nm. Finally, the pore diameter of the 5-Ag/MnOx sample are similar to that of 3-Ag/MnOx. Adsorption, surface reaction and desorption are responsible of the catalysis by Ag/MnOx, and 4-Ag/MnOx has the highest removal performance because of the largest possible large surface area and pore diameter.
Experimental results on HCHO degradation
An experimental campaign of the prepared catalyst at room temperature for the removal of HCHO as a function of time was performed, and obtained results are reported in Fig. 7. At the beginning, for 20 min, all the used catalysts exhibit their relative best removal rate of HCHO, with exception of 1-Ag/MnOx; parallel to this, the produced amount of CO2 is at maximum levels. The available active sites on the start of the run triggers the adsorption of HCHO and after this the conversion CO2 (Qi et al., 2021).

HCHO% as a function of time at room temperature
With exception of sample 1-Ag/MnOx (34%), the removal efficiency of HCHO is near to 85% for all other samples after 20 min of the run start. After this, deactivation appeared to occur in the following 50 min. The deactivation may be justified by the adsorption of intermediates and water on the catalyst surface and active sites that increase the catalyst saturation levels. Only by adopting sample 4-Ag/MnOx, the removal efficiency of HCHO, equal to around 94%, resulted to hold constant for longer period of time (around 150 min). For other samples, the deactivation of catalyst starts after 50 to 75 min.
The conversion of HCHO to intermediates on the catalyst surface is a crucial intermediary step to increase the catalyst efficiency. Although surface oxygen can rapidly oxidize dioxymethylene (DOM) to format, DOM will accumulate and hold on the surface of the catalyst. The breakdown of HCHO lead to the formation of water (H2O) and carbon dioxide (CO2) as products. As oxygen decreases concentration, efficiency reduction will occur, with the formation of CO but failing complete conversion to CO2 (Wang et al. 2015; Liu et al. 2019). It was observed that the outgoing CO2 concentration values are not the same as evaluated HCHO concentration in the feed stream; most probably more HCHO is adsorbing on the surface without triggering catalytic phenomena and failing to concert to CO2.
The percentage of the maximum CO2 production for each catalyst is reported in Fig. 8 and equal to 31% for 1-Ag/MnOx, 59% for both 2-Ag/MnOx and 3-Ag/MnOx, respectively.

Maximum CO2% generation with time
Again, the 4-Ag/MnOx sample was able to produce the highest values of elimination and equal to 66%; the 5-Ag/MnOx sample generated CO2 for only 61%. The key to the process is the intermediates formation from the oxidized HCHO given by the equilibrium between DOM adsorption and the capability of the active species to trigger. Probably, the 5-Ag/MnOx catalysts allow active species to alter, reducing the efficiency of the process (Li et al. 2016).
At the end of this study, it was observed that all performed analysis as well as the experimental campaign indicates the catalyst 4-Ag/MnOx results to be optimized. In this latter case, the maximum HCHO removal efficiency and CO2 generation of 4-Ag/MnOx is 94% and 66%, respectively. 4-Ag/MnOx has a larger specific surface area which can be facilitated in formaldehyde adsorption (Qi et al., 2021).
The catalytic activity of a deactivated catalyst can be renewed by heating to 400 °C. At this temperature which gasifies the carbonaceous intermediates that previously were formed the reactivate oxygen and other active species on the catalyst surface, as also observed for other catalyst such as Ag/Al2O3,Ag/TiO23, and δ-MnO2 (Fig. 9). (Chen et al. 2018b, 2021; Ji et al. 2020)

HCHO % Removal in 100 min
The kinetic data have been fitted adopting the LH model, as reported previously (Pei et al. 2015; Li et al. 2016). Figure 10 displays the results of kinetic modeling and fitting of experimental data with the LH model.

Regression plots of kinetic data and fittings
The experimental data used for fitting model is in the range of 10–60 min. In the first 10 min, the conversion of formaldehyde was not stable because the catalyst needs some time to be activated therefore. The regressed values of K and Ka are 1.5 ∙10–4 mol/m2s and 2.002∙10–4, respectively. The R-squared value determined is 0.9996.
Conclusion
Ag-doped manganese oxides (Ag/MnOx) with different weight percent of Ag were synthesized by using a co-precipitation method maintaining a pH value in the range of 7–8, followed by a calcination at 450 °C. Increasing Ag doping leads to higher values of thermal stability, crystallinity, surface area, dispersion of Ag and porosity, all increasing factors to removal efficiency. The activity of the 4-Ag-MnOx catalyst exhibit highest performances, mainly due to highest crystallinity (XRD), a homogeneous distribution of Ag within the catalyst matrix (ICP-OES) and surface area (BET). In this case, a removal value of 94% is obtained. This allows to remove HCHO at high efficiency with ease and no need of external heating devises, making the proposed process attractive for large scale use.
References
Alam K, Ali S, Saboor A, Salman M, Maoz HM, Sadiq M, Arif M (2019) Antireflection, superhydrophilic nano-porous SiO2 coating based on aerosol impact spray deposition technique for solar PV module. Coatings 9(8):497
Chen T, Dou H, Li X, Tang X, Li J, Hao J (2009) Tunnel structure effect of manganese oxides in complete oxidation of formaldehyde. Microporous Mesoporous Mater 122:270–274
Chen D, Qu Z, Sun Y, Gao K, Wang Y (2013) Identification of reaction intermediates and mechanism responsible for highly active HCHO oxidation on Ag/MCM-41 catalysts. Appl Catal B 142–143:838–848
Chen Y, He J, Tian H, Wang D, Yang Q (2014) Enhanced formaldehyde oxidation on Pt/MnO2 catalysts modified with alkali metal salts. J Colloid Interface Sci 428:1–7
Chen J, Yan D, Xu Z, Chen X, Chen X, Xu W, Jia H, Chen J (2018a) A novel redox precipitation to synthesize Au-doped α-MnO2 with high dispersion toward low-temperature oxidation of formaldehyde. Environ Sci Technol 52(8):4728–4737
Chen X, Chen M, He G, Wang F, Xu G, Li Y, Zhang C, He H (2018b) Specific role of potassium in promoting Ag/Al2O3 for catalytic oxidation of formaldehyde at low temperature. J Phys Chem C 122:27331–27339
Chen X, Wang H, Chen M, Qin X, He H, Zhang C (2021) Co-function mechanism of multiple active sites over Ag/TiO2 for formaldehyde oxidation. Appl Catal B Environ 282:119543
Ji J, Lu X, Chen C, He M, Huang H (2020) Potassium-modulated δ-MnO2 as robust catalysts for formaldehyde oxidation at room temperature. Appl Catal B Environ 260:118210
Kumar A, Sharma Y (2017) In situ conversion of manganese carbonate to manganese oxide/hydroxide and its supercapacitive analysis in aqueous KOH solution. Ionics 23:3409–3418
Li D, Yang G, Li P, Wang J, Zhang P (2016) Promotion of formaldehyde oxidation over Ag catalyst by Fe doped MnOx support at room temperature. Catal Today 277:257–265
Li X, Ma J, Zhang C, Zhang R, He H (2019) Facile synthesis of Ag-modified manganese oxide for effective catalytic ozone decomposition. J Environ Sci (china) 80:159–168
Liu P, Wei G, He H, Liang X, Chen H, Xi Y, Zhu J (2019) The catalytic oxidation of formaldehyde over palygorskite-supported copper and manganese oxides: catalytic deactivation and regeneration. Appl Surf Sci 464:287–293
Ou CC, Chen CH, Chan TS, Chen CS, Cheng S (2019) Influence of pretreatment on the catalytic performance of Ag/CeO2 for formaldehyde removal at low temperature. J Catal 380:43–54
Pei J, Han X, Lu Y (2015) Performance and kinetics of catalytic oxidation of formaldehyde over copper manganese oxide catalyst. Build Environ 84:134–141
Peng J, Wang S (2007) Performance and characterization of supported metal catalysts for complete oxidation of formaldehyde at low temperatures. Appl Catal B 73:282–291
Qi Q, Zhang W, Zhang Y, Bai G, Wang S, Liang P (2021) Formaldehyde oxidation at room temperature over layered MnO2. Catal Commun 153:106293
Rong S, Li K, Zhang P, Liu F, Zhang J (2018a) Potassium associated manganese vacancy in birnessite-type manganese dioxide for airborne formaldehyde oxidation. Catal Sci Technol 8:1799–1812
Rong S, Zhang P, Liu F, Yang Y (2018b) Engineering crystal facet of α-MnO2 nanowire for highly efficient catalytic oxidation of carcinogenic airborne formaldehyde. ACS Catal 8:3435–3446
Sekine Y (2002) Oxidative decomposition of formaldehyde by metal oxides at room temperature. Atmos Environ 36:5543–5547
Shao Y, Wang Y, Zhao R, Chen J, Zhang F, Linhardt RJ, Zhong W (2020) Biotechnology progress for removal of indoor gaseous formaldehyde. Appl Microbiol Biotechnol 104:3715–3727
Shen Y, Yang X, Wang Y, Zhang Y, Zhu H, Gao L, Jia M (2008) The states of gold species in CeO2 supported gold catalyst for formaldehyde oxidation. Appl Catal B 79:142–148
Wang J, Li D, Li P, Zhang P, Xu Q, Yu J (2015) Layered manganese oxides for formaldehyde-oxidation at room temperature: the effect of interlayer cations. RSC Adv 5:100434–100442
Wang C, Li Y, Zhang C, Chen X, Liu C, Weng W, Shan W, He H (2021) A simple strategy to improve Pd dispersion and enhance Pd/TiO2 catalytic activity for formaldehyde oxidation: the roles of surface defects. Appl Catal B Environ 282:119540
Xiao W, Zhou P, Mao X, Wang D (2015) Ultrahigh aniline-removal capacity of hierarchically structured layered manganese oxides: trapping aniline between interlayers. J Mater Chem A 3:8676–8682
Ye J, Yu Y, Fan J, Cheng B, Yu J, Ho W (2020) Room-temperature formaldehyde catalytic decomposition. Environ Sci Nano 7:3655–3709
Yu F, Qu Z, Zhang X, Fu Q, Wang Y (2013) Investigation of CO and formaldehyde oxidation over mesoporous Ag/Co3O4 catalysts. J Energy Chem 22:845–852
Yusuf A, Sun Y, Snape C, He J, Wang C, Ren Y, Jia H (2020) Low-temperature formaldehyde oxidation over manganese oxide catalysts: potassium mediated lattice oxygen mobility. Mol Catal 497:111204
Zhang J, Jin Y, Li C, Shen Y, Han L, Hu Z, Di X, Liu Z (2009) Creation of three-dimensionally ordered macroporous Au/CeO2 catalysts with controlled pore sizes and their enhanced catalytic performance for formaldehyde oxidation. Appl Catal B 91:11–20
Zhang J, Li Y, Wang L, Zhang C, He H (2015) Catalytic oxidation of formaldehyde over manganese oxides with different crystal structures. Catal Sci Technol 5:2305–2313
Zhu S, Wang J, Nie L (2019) Progress of catalytic oxidation of formaldehyde over manganese oxides. Chem Sel 4:12085–12098
Acknowledgements
The authors wish to thank the researchers whose literature has been cited in this article and acknowledge Sapienza University of Rome and GIK Institute of Pakistan for providing materials, experimental setups and material characterization equipments used in this study.
Funding
Open access funding provided by Università degli Studi di Roma La Sapienza within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Badshah, M., Mehdi, S., Alam, K. et al. Room temperature oxidation of gaseous formaldehyde over silver-doped manganese oxide catalyst. Chem. Pap. 78, 4383–4393 (2024). https://doi.org/10.1007/s11696-024-03397-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-024-03397-3