
Abstract
Chemical prototypes with broad-spectrum antiviral activity are important toward developing new therapies that can act on both existing and emerging viruses. Binding of the SARS-CoV-2 spike protein to the host angiotensin-converting enzyme 2 (ACE2) receptor is required for cellular entry of SARS-CoV-2. Toward identifying new chemical leads that can disrupt this interaction, including in the presence of SARS-CoV-2 adaptive mutations found in variants like omicron that can circumvent vaccine, immune, and therapeutic antibody responses, we synthesized 5-chloro-3-(2-(2,4-dinitrophenyl)hydrazono)indolin-2-one (H2L) from the condensation reaction of 5-chloroisatin and 2,4-dinitrophenylhydrazine in good yield. H2L was characterised by elemental and spectral (IR, electronic, Mass) analyses. The NMR spectrum of H2L indicated a keto–enol tautomerism, with the keto form being more abundant in solution. H2L was found to selectively interfere with binding of the SARS-CoV-2 spike receptor-binding domain (RBD) to the host angiotensin-converting enzyme 2 receptor with a 50% inhibitory concentration (IC50) of 0.26 μM, compared to an unrelated PD-1/PD-L1 ligand–receptor-binding pair with an IC50 of 2.06 μM in vitro (Selectivity index = 7.9). Molecular docking studies revealed that the synthesized ligand preferentially binds within the ACE2 receptor-binding site in a region distinct from where spike mutations in SARS-CoV-2 variants occur. Consistent with these models, H2L was able to disrupt ACE2 interactions with the RBDs from beta, delta, lambda, and omicron variants with similar activities. These studies indicate that H2L-derived compounds are potential inhibitors of multiple SARS-CoV-2 variants, including those capable of circumventing vaccine and immune responses.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The natural product isatin (Fig. 1) serves as a precursor for many bioactive molecules and it is a versatile substrate that can be modified. Isatin derivatives, mostly those substituted at C-3 such as isatin-3-hydrazones, are generally employed as ligands in coordination Chemistry (El-Sawi et al. 2011; Joshi et al. 1980; Snavely and Un 1981; Radovanović and Andelković 1998; Vine et al. 2007). Synthesis of isatin derivatives have gained attention in recent years due to their biological potential as anticancer (Abadi et al. 2006; Vine et al. 2007; Ashraf et al. 2006; Han et al. 2014; Singh et al. 2012; Solomon et al. 2009; Uddin et al. 2007; Vine et al. 2009), antimalarial (Kumar et al. 2014; Raj et al. 2014), antiviral (Abbas et al. 2013; Zhang et al. 2014; Sin et al. 2009), and antimicrobial agents (Kumar et al. 2010; Nandakumar et al. 2010). For example, it has been reported that halogenation at C-5 produces compounds with increased antimicrobial activity (Gurkok et al. 2008; Nathani et al. 2011; Nain et al. 2023; Patel et al. 2006). The in silico evaluation of some isatin-hydrazone derivatives has also been reported and shown to exhibit diverse properties, including potential interactions with topoisomerase, dihydrofolate reductase, and Chikungunya virus envelope and protease proteins, among others (Bittencourt et al. 2016; Mishra et al. 2016; Velasques et al. 2017).

Isatin
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causes Coronavirus Disease 2019 (COVID-19), that spread worldwide with major effects on human morbidity and mortality (WHO, 2019). While SARS-CoV-2 has likely become endemic since 2023, it continues to cause substantial mortality worldwide, particularly in high-risk populations such as elderly and immunocompromised individuals. SARS-CoV-2 binds to and infects host cells via its trimeric spike glycoprotein, where the receptor-binding domain (RBD) of the S1 segment can directly interact with the host angiotensin-converting enzyme II (ACE2) receptor to gain cellular entry (Xiu et al. 2020). Antagonism of this RBD-ACE2 interaction, for example by therapeutic antibodies such as REGN10933 (Casirivimab) and REGN10987 (Imdevimab), can inhibit multiple variants of SARS-CoV-2 cellular entry and SARS-CoV-2 infection (Starr et al. 2021). Small molecules that can also disrupt this RBD-ACE2 interface may therefore also be developed into lead compounds to disrupt SARS-CoV-2 infection and mitigate COVID-19 progression.
Computer-aided drug design methodologies do not claim to find lead compounds but could accelerate the process of finding a lead compound (Kontoyianni 2017; Lionta et al. 2014; Baig et al. 2016). These often involve structure-based methods when the molecular structure of the drug target is known, e.g., molecular docking to determine the affinity and orientation of a small molecule within a receptor-binding site (Meek and Weaver 2022), molecular dynamics simulations to test for the stability of a small molecule within a receptor site (Esmaielbeiki et al. 2014; Arcon et al. 2021; Kontoyianni 2017; Rogers et al. 2023) and structure-based pharmacophore methods to assist in screening for putative small molecule binders to a receptor based on how well the three-dimensional (3D) structural features of the small molecules correspond to those of known binders (Baig et al. 2016; Wermuth 2006; Urbina et al. 2022). Ligand-based methods often do not require a knowledge of the structural features of a receptor site (Ferreira et al. 2015; Vazquez et al. 2020). These include quantitative structure–activity relationships (QSAR), ligand-based pharmacophore querying methods, and most recently, artificial intelligence/machine learning (AI/ML) models (Selvaraj et al. 2022; Subramanian et al. 2022; Namba-Nzanguim et al. 2022; Turon et al. 2023). Among these methods, the most cite is molecular docking and scoring techniques have been proven to be efficient ways of identifying active compounds from an electronic library of compounds by a what is commonly called virtual screening (Morris & Lim-Wilby 2008; Chen 2015; Lohning et al 2017). Besides, from the docking orientation of ligand poses, there is often a basis for explaining observed biological activities, mostly in vitro activity concentrations and binding affinities based on the structural interactions between the ligand and the target receptor or the drug target (Kontoyianni 2017; Baig et al. 2016; Rogers et al. 2023).
We recently reported on some new hydrazones with biological activity (Majoumo-Mbe et al. 2015, 2019; Nfor et al. 2013; Yong et al. 2016). Based on preliminary docking studies using the spike RBD from the parental SARS-CoV-2 variant (Wuhan), we hypothesized that hydrazone derivatives may be able to disrupt SARS-CoV-2 spike/ACE2 interactions inclusive of mutations in spike that have arisen in subsequent SARS-CoV-2 variants. Based on this, we now report on the synthesis, characterization, and biochemical and computational screening on SARS-CoV-2 spike of a hydrazone derived from 5-chloroisatin and 2,4-dinitrophenylhydrazine.
Experimental
Materials
Chemicals
Reagent grade 5-chloroisatin, and 2,4-dinitrophenylhydrazine were purchased from Sigma-Aldrich. Ethanol as solvent and concentrated acetic acid were used as purchased.
Physical measurements
Elemental analyses were performed with a Thermo Flash EA-1112 series CHNS-O Elemental Analyzer. The melting points were determined with a Stuart SMP11 instrument in sealed capillary and are uncorrected. Infrared spectra were obtained (KBr 400–4000 cm−1) on ALPHA FT-IR Spectrometer from Bruker. UV–visible spectra were carried out with GENESYS 10S UV–Vis spectrophotometer. A Bruker AV 400 MHz Spectrometer was used for the 1H and 13C NMR analysis. Mass spectra were obtained on JEOL Gemate II and Autoflex spectrometers from Bruker.
General procedure for synthesis of 5-chloro-3-[2-(2,4-dinitro- phenyl)hydrazono]indolin-2-one (H2L)
To a 200-mL ethanolic solution of 5-chloro-isatin (1.5 g, 8.28 mmol) and 2,4-dinitrophenylhydrazine (1.64 g, 8.28 mmol) was added a catalytic amount of concentrated glacial acetic acid (three drops) under reflux at 80–85 °C (see Scheme 1). The resulting solution was further stirred for 6 h. After completion of the reaction, the orange-reddish precipitate obtained after cooling overnight was filtered and washed with methanol (100 mL × 2) and dried. Yield 63%; mp > 350 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 11.69 (s, 1H), 11.05 (s, 1H), 8.92 (d, J = 2.6 Hz, 1H), 8.61 (dd, J = 9.4, 2.6 Hz, 1H), 8.14 (d, J = 9.4 Hz, 1H), 7.91 (s, 1H), 8.61 (dd, J = 9.4, 2.6 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ ppm 164.36, 143.99, 143.06, 140.59, 138.08, 133.06, 132.94, 130.99, 126.29, 124.24, 122.98, 117.36, 116.92, 113.07. FTIR (max/cm−1): 3372w, 3336w, 3188br, (OH, NH), 3104w, 3057w, 1729m (C=O), 1692m (C=N), 1612s, 1579s, 1497s (NO2), 1470s, 1449m, 1338s (NO2), 1307s, 1269s, 1230m, 1177s, 1135m, 1109s, 1046m, 847m, 830s, 796m (C–Cl), 719m. Elemental analysis (%): Found: C, 46.40; H, 2.2; N, 19.3 (M+ , 361) C14H8ClN5O5; Calcd (%): C, 46.55; H, 2.2; N, 19.0. UV–vis: max (DMSO/nm) 271, 391, 420sh, 560. ESI (methanol) m/z = 362.1 (M+ , 30%), 360.1 (100, M–2H), 307 (5, M–CO,–HCN).

Synthesis of target compound
AlphaScreen binding assays
AlphaScreen assays were performed as described previously (Tietjen et al. 2021). For RBD-ACE2 assays, 2 nM of ACE2-Fc (Sino Biological, Chesterbrook, PA, USA) was incubated with 5 nM HIS-tagged SARS-CoV-2 Spike-RBDs representing ancestral (“Wild-type” (WT)), beta, delta, lambda, or omicron sequences (SinoBiological) in the presence of 5 μg/mL nickel chelate donor bead in a total of 10 μL of 20 mM Tris (pH 7.4), 150 mM KCl, and 0.05% CHAPS. Test compounds were diluted to 100 × final concentration in DMSO. 5 μL of ACE2-Fc/Protein A acceptor bead was first added to the reaction, followed by 100 nL test compound and then 5 μL of RBD-HIS/Nickel chelate donor beads. All conditions were performed in duplicate. Following incubation at room temperature for 2 h, luminescence signals were measured using a ClarioStar plate reader (BMC Labtech, Cary, NC, USA). Data were then normalised to percent inhibition, where 100% equaled the AlphaScreen signal in the absence of RBD-HIS, and 0% denoted AlphaScreen signal in the presence of both protein and DMSO vehicle control. To measure PD-1-PD-L1 binding, 0.5 nM of human PD-L1-Fc (Sino Biological) was incubated with 5 nM HIS-tagged human PD-1 (Sino Biological) in the presence of 5 μg/mL protein A and 5 μg/mL nickel chelate donor beads in a total volume of 10 μL of 20 mM HEPES (pH 7.4), 150 mM NaCl, and 0.005% Tween. Proteins and test agents were then added, incubated, and analysed as described above.
Selection of crystal structure of spike/ACE2 receptor
At the time of this study, four three dimensional (3D) structures of spike/ACE2 complex of SARS-CoV-2 were available from Protein Data Bank (PDB) (Berman et al. 2000; Burley et al. 2017, 2018) and had been solved via X-ray crystallography (PDB codes: 6M0J, 6VW1, 6M17 and 6LZG). The crystal structure 6M0J (Lan et al. 2020) was chosen due to high-resolution and domain completeness. The crystal structure of the Spike RBD/ACE2 complex has 832 amino acid residues divided into two chains (A and E). Chain A is the N-terminal peptidase domain of ACE2 which has 603 residues, while Chain E is the receptor-binding domain of the Spike protein from SARS-CoV-2 and has 229 amino acids residues. The structure also had bound metallic cofactors (Zn2+ and Cl−), N-Acetyl glucosamine (NAG), and water molecules.
Molecular docking procedures
Generally, molecular docking procedures were performed using similar methods as reported in our previous published papers (Simoben et al. 2018, 2021; Divsalar et al. 2020).
Ligand preparation
The 3D structure of H2L was generated using Molecular Operating Environment (MOE, Chemical Computing Group 2017). The ligand was prepared for docking using the LigPrep tool, as implemented in the Schrödinger’s software (Schrödinger 2017), where all possible tautomeric forms were generated. They were subsequently energy-minimised using the integrated Optimised Potentials for Liquid Simulations (OPLS_2005) force field (Banks et al. 2005). Finally, 60 conformers were calculated with ConfGen using the default settings and allowing minimisation of the output conformations (Watts et al. 2010).
Protein preparation
The crystal structures of spike/ACE2 complex of SARS-CoV-2 (PDB ID: 6M0J) which is the Wuhan variant, along with the human PD-1/PD_L1 (PDB ID: 4ZQK) were downloaded from the Protein Data Bank (PDB; www.rcsb.org) (Berman et al. 2000; Burley et al. 2017, 2018). All water molecules were deleted using MOE software (Chemical Computing Group 2017). Further preparations of the protein structures preparation were done using the Protein Preparation Wizard of Schrödinger software (Schrödinger 2017; Sastry et al. 2013). At this stage, bond orders were assigned and hydrogen atoms added, missing side chains were filled using PRIME, and the H-bond network was subsequently optimised. The protonation states at pH 7.0 were predicted using the Epik-tool in the Maestro package commercialized by Schrödinger (Schrödinger 2017; Shelley et al. 2007). The structures were finally subjected to a restrained energy minimization step (rmsd of the atom displacement for terminating the minimization was 0.3 Å) using the OPLS2005 force field (Banks et al. 2005). Furthermore, the different variants/mutants of the spike/ACE complex of SARS-CoV were obtained from the Wuhan 6M0J structure (as mentioned above) by mutation (manual replacement of the residues of interest around the spike receptor-binding domain (spike-RBD), using the protein builder module in MOE in the spike protein sequence. Table 1 shows the various mutations carried out on the Wuhan strain or the wild type (WT) spike RBD/ACE2 to derive the various mutants (β, δ, λ and ο).
Docking towards the SARS-CoV-2 Spike RBD/ACE2 and the human PD_1/PD_L1
Docking procedures were performed using the Glide program in a similar way as previously demonstrated (Simoben et al. 2018, 2021; Divsalar et al. 2020). In this work three grid boxes for the SARS-CoV-2 viral protein RBD/ACE2 human receptor (PDB ID: 6M0J) and one grid box for the human protein complex PD_1/PD_L1 (PDB ID: 4ZQK, Zak et al. 2015) were generated and using specific protein residues. For the ACE2/SARS-CoV-2 protein (PDB ID: 6M0J), the first grid box of interest constituted of the following amino acid residues D93, Q80, Q68, D277, N272, L125, Y32, K523, F494 and N560 around the ACE2 binding site (further shown and discussed in the Results Section). The second choice was around the spike RBD-ACE2 binding site and was generated using the centroid of the following the residues Q771, Y718, N752, P744, M365, A3769, E05, E311. The last avenue to investigate was where the compounds will preferably bind when the whole structure is explored for the generation of a grid. For this purpose, the following amino acids D597, T598, K516, V321, Q121, K578, A283, S91, N746, Q68, P744, E518 and T610 were used to generate a third grid around the ACE2/SARS-CoV-2 protein. On the other hand, the grid box for the PD_1/PD_L1 structure was generated using the residues F63, V63, N66, Y68, E84, L122, E136, I134 and I126; as reported in literature (Horita et al. 2016; Tang and Kim 2019). For all the generated grid boxes, the sides were set to 36 Å. The generated 3D conformers of the prepared ligand were docked into the different receptor grid files. For the docking process, default settings were used with exception of input ring conformation as well as writing a total of 10 poses per ligand conformer from the 20 poses that were included for each ligand conformer. The GlideScore Standard Precision (SP) mode was used as the scoring function (Halgren et al. 2004).
Results and discussion
Synthesis and characterisation of H2L
The ligand H2L was synthesized and characterised for biochemical activities. In the NMR spectrum in DMSO (Fig. 2) H2L shows trace of the enol form of the ligand (Fig. 3) in solution.

1H NMR of H2L showing keto–enol forms in DMSO

Proposed structure of H2L
In the 1H NMR, the chemical shifts of the N–H groups of the isatin ring and the dinitrophenylhydrazone moieties for the keto form were assigned at 11.05 and 11.7 ppm, respectively, while in the enol form, the OH group of the 5-chloroisatin ring and the N–H group of the dinitrophenylhydrazone moieties were identified at 14.4 and 11.4 ppm, respectively. In the enol form, C12, C9, C7, C5 and C4 signals are downfield shifted with respect to the keto form, while C13 and C15 result in an inverse effect. Amongst the observed signals for both forms, C9 and C4 are the most influenced by the keto–enol equilibrium (shifted about 2.3 and 1.2 ppm, respectively). Similarly, the most affected H-signals are attributed to N–NH, HC15 and HC4 (shifted about 0.3 and 0.2 ppm). The mass spectrum of H2L revealed a molecular ions peak at m/z 360.1 and 362.1 which is closer to the formula weight (361.7) of the ligand and supported the identity of the proposed structure. In the mass fragmentation of the ligand, a peak corresponding to the loss of CO and HCN can also be observed.
Electronic spectral analysis of H2L ligand exhibited three major absorption bands with a shoulder at 271, 391, 420sh and 560 nm. The first observed absorptions can be attributed to the π–π* transitions of the aromatic system (Seleem 2011), the π–π* transitions of C = O and C = N can be attributed to the second absorption, and the n–π* transition due to the lone pairs electron of the oxygen and nitrogen can be attributed to the third absorption. The longest UV-band reflects the charge transfer nature (Seleem et al. 2010, 2011) that gives H2L its strong reddish colour. In the IR spectrum of H2L, the C=O, C=N and NH vibrations were identified at 1729, 1692, 3336, and 3188 cm−1, respectively. Vibrations in the range of 1713–1737, 1685, 3273, and 3193 cm−1 were reported for other compounds of isatin hydrazone derivatives with similar environment (Hussein et al. 2019; Jabbar 2018). Additional spectral data are available in the supplementary data (Figs S7–S10).
In vitro activities of H2L against ligand–receptor interactions
To determine whether H2L may disrupt SARS-CoV-2 entry, we employed a previously-described AlphaScreen technology-based assay (Tietjen et al. 2021; Lan et al. 2020), which uses a SARS-CoV-2 RBD protein containing a C-terminal His tag, bound to an nickel chelate acceptor bead, in addition to a full-length ACE2 peptide with a C-terminal Fc tag bound to a donor protein A bead. When an RBD-ACE2 binding event occurs, the two beads are brought into proximity of each other, at which point excitation at 680 nm results in a singlet oxygen transfer and luminescence at 615 nm. Using this assay, we first asked whether H2L could disrupt interactions with RBD from the parental Wuhan variant of SARS-CoV-2, as this sequence provided a useful baseline for understanding H2L interactions across multiple variants and was best characterised in this assay (Tietjen et al. 2021). In this approach, we found that H2L could disrupt RBD-ACE2 binding with dose dependence and with an IC50 of 0.26 μM (Fig. 4; Table 2), in contrast to an IC50 of 0.0013 μM for the control therapeutic antibody REGN10933. To assess the selectivity of this interaction, we next determined whether H2L could interfere with the unrelated host PD-1/PD-L1 ligand–receptor pair, which we previously observed could be disrupted by the control inhibitor BMS-116611 with an IC50 of 0.0040 μM. Using this assay, we also observed dose-dependent inhibition with H2L but with a much higher IC50 of 2.06 μM (Fig. 4). These results corresponded to a selectivity index [(IC50 PD-1-PD-L1)/(IC50 RBD-ACE2)] of 7.9 (Table 2), indicating selectivity of H2L to disrupt the SARS-CoV-2 RBD-ACE2 interaction. Derivatives of H2L with improved cellular tolerance should therefore be assessed for antiviral activity in vitro using pseudovirus-based or replication competent virus-based cellular assays (Tietjen et al. 2021).

Dose–response curves denoting ability of H2L to disrupt luminescence due to SARS-CoV-2 spike RBD–host ACE2 protein-binding (circles) and PD-1-PD-L1-binding (triangles) AlphaScreen assay. Results denote the mean ± S.D. from 3 independent experiments
In silico analysis of H2L binding to PD-1-PD-L1 ligand–receptor pair
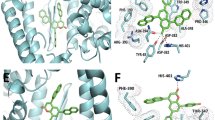
AlphaScreen showed that the H2L ligand was selective towards the inhibition of the spike RBD-ACE2 binding, when compared with PD-1-PD-L1 binding inhibition (Table 2). Interestingly, our work confirmed the comparative studies performed with the unrelated PD-1-PD-L1 ligand–receptor-binding pair. Docking studies showed that, unlike the observed binding of the ligand within the ACE2 binding site for the ACE2-Spike RBD, the ligand was observed to bind between the PD-1 and PD-L1 protein–protein complex as shown in Fig. 5C. The proposed docking pose of the synthesized ligand showed that it had interactions with only two residues (D44 and R96) of the PD-1 surface, thus explaining their non-preference of this complex.

A View of the ACE2-Spike RBD complex. The ACE2 protein backbone is shown as green cartoon, while the spike RBD is shown as orange ribbon. Mutation residues are depicted as lincorice-sticks. The ACE2-Spike RBD domain interface and the preferential ACE2 binding site are shown as purple egg sphere and red rectangle, respectively. B Close view of the proposed binding mode of the ligand (brown) within the ACE2 binding site. Key residues within the site are shown as green sticks. C Proposed binding mode of the ligand (cyan) docked at the interface between PD-1 (light blue) and PD-L1 (grey). For all figures, H-bond interactions are shown as magenta dashed-lines
In silico analysis of H2L binding to RBD-ACE2 ligand–receptor pairs across SARS-CoV-2 variants
To further explain the observed biological activities, computational studies were performed on the spike sequence of the ancestral SARS-CoV-2 variant (i.e., Wuhan variant or “wild-type”, (WT)) as well as SARS-CoV-2 beta, delta, lambda, and omicron variants. Figure 5A depicts the different mutations (as summarized in Table 1 above) that were made to perform this study. The docking studies revealed that the ligand preferentially binds within the ACE2 binding/active site as depicted in Figure S2. This agrees with other studies stipulating that ligands bind within the ACE2 binding site to elicit conformational changes that influence how well the spike RBD would subsequently bind and interact with ACE2 (García-Iriepa et al. 2020; Williams-Noonan et al. 2021). This, therefore, postulates how the synthesised ligand might bind and interact with ACE2 to inhibit the ACE2-spike protein complex formation. Figure 5B exemplifies the binding mode of the ligand within the ACE2 binding for the Wuhan variant. Like the in the other variants, the ligand binds within the ACE2 binding site, interacting with residues on the alpha 1 and alpha 2 (α1 and α2) N-terminal helices of ACE2, and causing conformational changes on the alpha-(α−) and beta-(β−) interfaces of the ACE2 protein (García-Iriepa et al. 2020; Williams-Noonan et al. 2021). These conformational changes (as observed in Fig. 5B) are in proximity with the spike RBD. This could imply that the proposed inhibitory mechanism of ACE2 by H2L would be expected to occur regardless of sequence changes that occur in the assessed SARS-CoV-2 variants.
In vitro activities of H2L against RBD-ACE2 ligand–receptor interactions across SARS-CoV-2 variants
Based on these observations, we hypothesized that the mechanism of inhibition of H2L was unlikely to be perturbed by mutations that are prevalent in variants of concern. To test this, we repeated the AlphaScreen assays for H2L using RBD sequences from beta, delta, lambda, and omicron variants. In these assays, we observed a slightly higher IC50 of 447.5 nM for the WT RBD sequence, while no more than a 1.4-fold difference in IC50 was observed for any variant (maximum IC50 = 628.5 nM using lambda RBD; Table 3). These results for H2L agree with the docking studies and suggest that H2L derivatives may be useful towards antagonising SARS-CoV-2 entry across multiple variants of concern. In contrast, the control therapeutic antibody REGN10933, while inhibiting WT, delta, and lambda RBD-ACE2 interactions with similar activities (IC50s = 0.8–1.4 nM), was ~ 70-fold weaker against the beta RBD (IC50 = 90.9 nM) and had no detected activity against the omicron RBD (IC50 > 700 nM), consistent with previous reports of fluctuating activity of REGN10933 against SARS-CoV-2 variants (Tietjen et al 2021; VanBlargan et al. 2022).
Conclusions
We report the synthesis and characterization of a new compound which showed selective antagonism of the binding of the SARS-CoV-2 viral spike protein RBD to the human angiotensin-converting enzyme 2 at sub-micromolar concentrations and across RBD sequences representing multiple SARS-CoV-2 variants of concern including omicron. This activity of H2L is consistent with binding of ACE2 leading to subsequent disrupting of protein–protein interactions that are required for RBD binding, and thus presumably SARS-CoV-2 cellular entry and replication. The biological activities revealed that, although the compound was less active than the therapeutic antibody REGN10933 (Casirivimab) for the WT, beta, delta and lambda variants, it was more active against the omicron variant. Besides, for spike/ACE2 binding the reported compound was about eightfold selective when compared with binding to the human PD1/PD-L1 protein complex. Molecular modelling of the interaction between the compound and the angiotensin II binding site of the spike/ACE2 complex reveals interactions with key amino acid residues that could prevent recognition with the RBD of the viral spike. Additionally, the binding against the viral spike/ACE2 complexes of all tested variants showed very similar IC50 values, which suggests the design of analogues of H2L that could potentially prevent the transmission of the new variants of the SARS-CoV-2 virus. Molecular docking studies also revealed that the synthesized ligand preferentially binds within the ACE2 receptor-binding site in a region distinct from where spike mutations in SARS-CoV-2 variants occur. As H2L represents a highly-accessible chemical scaffold that disrupts RBD-ACE2 interactions regardless of SARS-CoV-2 variant sequence and with selectivity over unrelated ligand–receptor interactions such as PD-1-PD-L1, additional studies are therefore warranted to assess H2L analogues for their ability to inhibit SARS-CoV-2 variant entry and replication using in vitro cellular infection models. Such leads may be able to support SARS-CoV-2 therapeutic efforts against emerging variants of concern that otherwise circumvent vaccine and host immune responses.
References
Abadi AH, Abou-Seri SM, Abdel-Rahman DE, Klein C, Lozach O, Meijer L (2006) Synthesis of 3-substituted-2-oxoindole analogues and their evaluation as kinase inhibitors, anticancer and antiangiogenic agents. Eur J Med Chem 41(3):296–305. https://doi.org/10.1016/j.ejmech.2005.12.004
Abbas SY, Farag AA, Ammar YA, Atrees AA, Mohamed AF, El-Henawy AA (2013) Synthesis, characterization, and antiviral activity of novel fluorinated isatin derivatives. Monatshefte Fur Chemie 144(11):1725–1733. https://doi.org/10.1007/s00706-013-1034-3S
Arcon JP, Turjanski AG, Martí MA, Forli S (2021) Biased docking for protein–ligand pose prediction. Methods Mol Biol 2266:39–72. https://doi.org/10.1007/978-1-0716-1209-5_3
Baig MH, Ahmad K, Roy S, Ashraf JM, Adil M, Siddiqui MH, Khan S, Kamal MA, Provazník I, Choi I (2016) Computer aided drug design: success and limitations. Curr Pharm Des 22(5):572–581. https://doi.org/10.2174/1381612822666151125000550
Banks JL, Beard HS, Cao Y, Cho AE, Damm W, Farid R, Felts AK, Halgren TA, Mainz DT, Maple JR, Murphy R, Philipp DM, Repasky MP, Zhang LY, Berne BJ, Friesner RA, Gallicchio E, Levy RM (2005) Integrated modeling program, applied chemical theory (IMPACT). J Comput Chem 26(16):1752–1780. https://doi.org/10.1002/jcc.20292
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28(1):235–242. https://doi.org/10.1093/nar/28.1.235
Bittencourt VCD, Almeida RMFC, Bortoluzzi AJ, Gervini VC, de Oliveira AB (2016) (3E)-5-chloro-3-(2-phenylhydrazinylidene)-1Hindol-2(3H)-one. IUCrData 1:x160258. https://doi.org/10.1107/S2414314616002583
Burley SK, Berman HM, Christie C, Duarte JM, Feng Z, Westbrook J, Young J, Zardecki C (2018) RCSB protein data bank: sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci 27(1):316–330. https://doi.org/10.1002/pro.3331
Burley SK, Berman HM, Kleywegt GJ, Markley JL, Nakamura H, Velankar S (2017) Protein data bank (PDB): the single global macromolecular structure archive. Methods Mol Biol 1607:627–641. https://doi.org/10.1007/978-1-4939-7000-1_26
Chemical Computing Group (2017) Molecular Operating Environment (MOE) version 2016.08.39
Chen YC (2015) Beware of docking! Trends Pharmacol Sci 36(2):78–95. https://doi.org/10.1016/j.tips.2014.12.001
Divsalar DN, Simoben CV, Schonhofer C, Richard K, Sippl W, Ntie-Kang F, Tietjen I (2020) Novel histone deacetylase inhibitors and HIV-1 latency-reversing agents identified by large-scale virtual screening. Front Pharmacol 11:905. https://doi.org/10.3389/fphar.2020.00905
El-Sawi EA, Mostafa TB, Radwan HA (2011) Synthesis and biological activity of functionalized phosphorus derivatives of isatin imines. Eur J Med Chem 2(4):539–543. https://doi.org/10.5155/eurjchem.2.4.539-543.55
Esmaielbeiki R, Nebel JC (2014) Scoring docking conformations using predicted protein interfaces. BMC Bioinform 15:171. https://doi.org/10.1186/1471-2105-15-171
Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD (2015) Molecular docking and structure-based drug design strategies. Molecules 20(7):13384–13421. https://doi.org/10.3390/molecules200713384
García-Iriepa C, Hognon C, Francés-Monerris A, Iriepa I, Miclot T, Barone G, Monari A, Marazzi M (2020) Thermodynamics of the interaction between the spike protein of severe acute respiratory syndrome coronavirus-2 and the receptor of human angiotensin-converting enzyme 2. Effects of possible ligands. J Phys Chem Lett 11(21):9272–9281. https://doi.org/10.1021/acs.jpclett.0c02203
Gurkok G, Altanlar N, Suzen S (2008) Investigation of antimicrobial activities of indole-3-aldehyde hydrazide/hydrazone derivatives. Chemotherapy 55(1):15–19. https://doi.org/10.1159/000166999
Han K, Zhou Y, Liu F, Guo Q, Wang P, Yang Y, Song B, Liu W, Yao Q, Teng Y, Yu P (2014) Design, synthesis and in vitro cytotoxicity evaluation of 5-(2-carboxyethenyl)isatin derivatives as anticancer agents. Bioorg Med Chem Lett 24(2):591–594. https://doi.org/10.1016/j.bmcl.2013.12.001
Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47(7):1750–1759. https://doi.org/10.1021/jm030644s
Horita S, Nomura Y, Sato Y, Shimamura T, Iwata S, Nomura N (2016) High-resolution crystal structure of the therapeutic antibody pembrolizumab bound to the human PD-1. Sci Rep 6:35297. https://doi.org/10.1038/srep35297
Hussain I, Ullah A, Khan AU, Khan WU, Ullah R (2019) Synthesis, characterization and biological activities of hydrazone schiff base and its novel metals complexes. Sains Malaysiana 48(7):1439–1446. https://doi.org/10.17576/jsm-2019-4807-13
Jabbar SS (2018) Synthesis, characterization and antibacterial activity of carbamate derivatives of isatin. Orient J Chem 34(4):2026–2030. https://doi.org/10.13005/ojc/3404041
Joshi KC, Pathak VN, Jain SK (1980) Studies of potential organo-fluorine antibacterial agents. Part 5: Synthesis and antibacterial activity of some new fluorine-containing indole-2,3-dione derivatives. Pharmazie 35(11):677–679
Kontoyianni M (2017) Docking and evirtual screening in drug discovery. Methods Mol Biol 1647:255–266. https://doi.org/10.1007/978-1-4939-7201-2_18
Kumar RS, Rajesh SM, Perumal S, Banerjee D, Yogeeswari P, Sriram D (2010) Novel three-component domino reactions of ketones, isatin and amino acids: synthesis and discovery of antimycobacterial activity of highly functionalised novel dispiropyrrolidines. Eur J Med Chem 45(1):411–422. https://doi.org/10.1016/j.ejmech.2009.09.044
Kumar K, Pradines B, Madamet M, Amalvict R, Benoit N, Kumar V (2014) 1H–1,2,3-triazole tethered isatin-ferrocene conjugates: synthesis and in vitro antimalarial evaluation. Eur J Med Chem 87:801–804. https://doi.org/10.1016/j.ejmech.2014.10.024
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X (2020) Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581(7807):215–220. https://doi.org/10.1038/s41586-020-2180-5
Lionta E, Spyrou G, Vassilatis DK, Cournia Z (2014) Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr Top Med Chem 14(16):1923–1938. https://doi.org/10.2174/1568026614666140929124445
Lohning AE, Levonis SM, Williams-Noonan B, Schweiker SS (2017) A practical guide to molecular docking and homology modelling for medicinal chemists. Curr Top Med Chem 17(18):2023–2040. https://doi.org/10.2174/1568026617666170130110827
Majoumo-Mbe F, Nfor EN, Sengeh EB, Njong RN, Offiong EO (2015) Synthesis, crystal structure andbiological activity of 1-(phthalazin-1(2h)-one)[(pyridin-2-yl)ethylidene]hydrazine. Commun Inorg Synth 3:40–46. https://doi.org/10.21060/cis.2015.332
Majoumo-Mbe F, Ngwang Nfor E, Kenfack Tsobnang P, Nguepmeni Eloundou VB, Ngwain Yong J, Iris Efeti I (2019) Synthesis, molecular and crystal structure of 1-(1,2-di-hydro-phthalazin-1-yl-idene)-2-[1-(thio-phen-2-yl)ethyl-idene]hydrazine. Acta Crystallogr E Crystallogr Commun 75(Pt 2):251–254. https://doi.org/10.1107/S2056989019000732
Meek A, Weaver DF (2022) Drug–receptor interactions: it is a numbers game. Can J Neurol Sci 49(4):589–590. https://doi.org/10.1017/cjn.2021.152
Mishra P, Kumar A, Mamidi P, Kumar S, Basantray I, Saswat T, Das I, Nayak TK, Chattopadhyay S, Subudhi BB, Chattopadhyay S (2016) Inhibition of chikungunya virus replication by 1-[(2-methylbenzimidazol-1-yl) methyl]-2-oxo-indolin-3-ylidene] amino] thiourea(MBZM-N-IBT). Sci Rep 6:20122. https://doi.org/10.1038/srep20122
Morris GM, Lim-Wilby M (2008) Molecular docking. Methods Mol Biol 443:365–382. https://doi.org/10.1007/978-1-59745-177-2_19
Nain S, Mathur G, Anthwal T, Sharma S, Paliwal S (2023) Synthesis, characterization, and antibacterial activity of new isatin derivatives. Pharmaceut Chem J 57(2):196–203. https://doi.org/10.1007/s11094-023-02867-4
Namba-Nzanguim CT, Turon G, Simoben CV, Tietjen I, Montaner LJ, Efange SMN, Duran-Frigola M, Ntie-Kang F (2022) Artificial intelligence for antiviral drug discovery in low resourced settings: a perspective. Front Drug Discov 2:1013285. https://doi.org/10.3389/fddsv.2022.1013285
Nandakumar A, Thirumurugan P, Perumal PT, Vembu P, Ponnuswamy M, Ramesh P (2010) One-pot multicomponent synthesis and anti-microbial evaluation of 2′-(indol-3-yl)-2-oxospiro(indoline-3,4′-pyran) derivatives. Bioorg Med Chem Lett 20(14):4252–4258. https://doi.org/10.1016/j.bmcl.2010.05.025
Nathani BR, Pandya KS, Jeni MM, Patel MR (2011) Synthesis and antimicrobial activity of some new isatins derivatives. Der Pharma Chemica 3:367–372
Nfor EN, Husian A, Majoumo-Mbe F, Njah IN, Offiong OE, Bourne SA (2013) Synthesis, crystal structure and antifungal activity of a Ni (II) complex of a new hydrazone derived from antihypertensive drug hydralazine hydrochloride. Polyhedron 63:207–213. https://doi.org/10.1016/j.poly.2013.07.028
Patel A, Bari S, Talele G, Patel J, Sarangapani M (2006) Synthesis and antimicrobial activity of some new isatin derivatives. Iran J Pharm Res. 5(4):e128295. https://doi.org/10.22037/ijpr.2010.685
Raj R, Gut J, Rosenthal PJ, Kumar V (2014) 1H–1,2,3-Triazole-tethered isatin-7-chloroquinoline and 3-hydroxy-indole-7-chloroquinoline conjugates: synthesis and antimalarial evaluation. Bioorg Med Chem Lett 24(3):756–759. https://doi.org/10.1016/j.bmcl.2013.12.109
Rogers DM, Agarwal R, Vermaas JV, Smith MD, Rajeshwar RT, Cooper C, Sedova A, Boehm S, Baker M, Glaser J, Smith JC (2023) SARS-CoV2 billion-compound docking. Sci Data 10(1):173. https://doi.org/10.1038/s41597-023-01984-9
Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27(3):221–234. https://doi.org/10.1007/s10822-013-9644-8
Schrödinger (2017) Maestro Package, Release version 2017-2
Seleem HS, El-Inany GA, Mousa M, Hanafy FI (2010) Spectroscopic and pH-metric studies of the complexation of 3-[2-(4-methylquinolin-2-yl)hydrazono]butan-2-one oxime compound. Spectrochim Acta A Mol Biomol Spectrosc 75(5):1446–1451. https://doi.org/10.1016/j.saa.2010.01.015
Seleem HS (2011) Transition metal complexes of an isatinic quinolyl hydrazone. Chem Cent J 5:35. https://doi.org/10.1186/1752-153X-5-35
Seleem HS, El-Inany GA, El-Shetary BA, Mousa MA, Hanafy FI (2011) The ligational behavior of an isatinic quinolyl hydrazone towards copper(II)-ions. Chem Cent J 5:20. https://doi.org/10.1186/1752-153X-5-20
Selvaraj C, Chandra I, Singh SK (2022) Artificial intelligence and machine learning approaches for drug design: challenges and opportunities for the pharmaceutical industries. Mol Divers 26(3):1893–1913. https://doi.org/10.1007/s11030-021-10326-z
Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M (2007) Epik: a software program for pK(a) prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des 21(12):681–691. https://doi.org/10.1007/s10822-007-9133-z
Simoben CV, Robaa D, Chakrabarti A, Schmidtkunz K, Marek M, Lancelot J, Kannan S, Melesina J, Shaik TB, Pierce RJ, Romier C, Jung M, Sippl W (2018) A novel Class of Schistosoma mansoni histone deacetylase 8 (HDAC8) inhibitors identified by structure-based virtual screening and in vitro testing. Molecules 23(3):566. https://doi.org/10.3390/molecules23030566
Simoben CV, Ghazy E, Zeyen P, Darwish S, Schmidt M, Romier C, Robaa D, Sippl W (2021) Binding free energy (BFE) calculations and quantitative structure-activity relationship (QSAR) analysis of Schistosoma mansoni histone deacetylase 8 (smHDAC8) inhibitors. Molecules 26(9):2584. https://doi.org/10.3390/molecules26092584
Sin N, Venables BL, Combrink KD, Gulgeze HB, Yu KL, Civiello RL, Thuring J, Wang XA, Yang Z, Zadjura L, Marino A, Kadow KF, Cianci CW, Clarke J, Genovesi EV, Medina I, Lamb L, Krystal M, Meanwell NA (2009) Respiratory syncytial virus fusion inhibitors. Part 7: structure-activity relationships associated with a series of isatin oximes that demonstrate antiviral activity in vivo. Bioorg Med Chem Lett 19(16):4857–4862. https://doi.org/10.1016/j.bmcl.2009.06.030
Singh P, Sharma P, Anand A, Bedi PM, Kaur T, Saxena AK, Kumar V (2012) Azide-alkyne cycloaddition en route to novel 1H–1,2,3-triazole tethered isatin conjugates with in vitro cytotoxic evaluation. Eur J Med Chem 55:455–461. https://doi.org/10.1016/j.ejmech.2012.06.057
Snavely FA, Un S (1981) A study of the structure of hydrazones of indole-2,3-dione and 1-methylindole-2,3-dione with nuclear magnetic resonance spectroscopy. J Org Chem 46(13):2764–2766. https://doi.org/10.1021/jo00326a032
Solomon VR, Hu C, Lee H (2009) Hybrid pharmacophore design and synthesis of isatin-benzothiazole analogs for their anti-breast cancer activity. Bioorg Med Chem 17(21):7585–7592. https://doi.org/10.1016/j.bmc.2009.08.068
Starr TN, Greaney AJ, Addetia A, Hannon WW, Choudhary MC, Dingens AS, Li JZ, Bloom JD (2021) Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 371(6531):850–854. https://doi.org/10.1126/science.abf9302
Stojčeva Radovanović BC, Andelković SS (1998) Synthesis and spectral characterization of N-[5-nitro-2-furfurylidene]-N2-[β-isatin]azine and its Zn(II), Cu(II) and Ni(II) complexes. Spectroscop Lett 31(1):63–70. https://doi.org/10.1080/00387019808006761
Subramanian M, Shanmuga Vadivel K, Hatamleh WA, Alnuaim AA, Abdelhady M (2022) The role of contemporary digital tools and technologies in COVID-19 crisis: an exploratory analysis. Expert Syst 39(6):e12834. https://doi.org/10.1111/exsy.12834
Tang S, Kim PS (2019) A high-affinity human PD-1/PD-L2 complex informs avenues for small-molecule immune checkpoint drug discovery. Proc Natl Acad Sci USA 116(49):24500–24506. https://doi.org/10.1073/pnas.1916916116
Tietjen I, Cassel J, Register ET, Zhou XY, Messick TE, Keeney F, Lu LD, Beattie KD, Rali T, Tebas P, Ertl HCJ, Salvino JM, Davis RA, Montaner LJ (2021) The natural stilbenoid (-)-hopeaphenol inhibits cellular entry of SARS-CoV-2 USA-WA1/2020, B.1.1.7, and B.1.351 variants. Antimicrob Agents Chemother 65(12):e0077221. https://doi.org/10.1128/AAC.00772-21
Turon G, Hlozek J, Woodland JG, Kumar A, Chibale K, Duran-Frigola M (2023) First fully-automated AI/ML virtual screening cascade implemented at a drug discovery centre in Africa. Nat Commun 14(1):5736. https://doi.org/10.1038/s41467-023-41512-2.PMID:37714843;PMCID:PMC10504240
Uddin MK, Reignier SG, Coulter T, Montalbetti C, Grånäs C, Butcher S, Krog-Jensen C, Felding J (2007) Syntheses and antiproliferative evaluation of oxyphenisatin derivatives. Bioorg Med Chem Lett 17(10):2854–2857. https://doi.org/10.1016/j.bmcl.2007.02.060
Urbina F, Lowden CT, Culberson JC, Ekins S (2022) MegaSyn: integrating generative molecular design, automated analog designer, and synthetic viability prediction. ACS Omega 7(22):18699–18713. https://doi.org/10.1021/acsomega.2c01404
VanBlargan LA, Errico JM, Halfmann PJ, Zost SJ, Crowe JE Jr, Purcell LA, Kawaoka Y, Corti D, Fremont DH, Diamond MS (2022) An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat Med 28(3):490–495. https://doi.org/10.1038/s41591-021-01678-y
Vázquez J, López M, Gibert E, Herrero E, Luque FJ (2020) Merging ligand-based and structure-based methods in drug discovery: an overview of combined virtual screening approaches. Molecules 25(20):4723. https://doi.org/10.3390/molecules25204723
Velasques JM, Gervini VC, Bortoluzzi AJ, de Farias RL, de Oliveira AB (2017) Crystal structure of (3E)-5-nitro-3-(2-phenyl-hydrazinyl-idene)-1H-indol-2(3H)-one. Acta Crystallogr E Crystallogr Commun 73(Pt 2):168–172. https://doi.org/10.1107/S2056989016020375
Vine KL, Locke JM, Ranson M, Pyne SG, Bremner JB (2007) In vitro cytotoxicity evaluation of some substituted isatin derivatives. Bioorg Med Chem 15(2):931–938. https://doi.org/10.1016/j.bmc.2006.10.035
Wermuth CG (2006) Similarity in drugs: reflections on analogue design. Drug Discov Today 11(7–8):348–354. https://doi.org/10.1016/j.drudis.2006.02.006
Vine KL, Matesic L, Locke JM, Ranson M, Skropeta D (2009) Cytotoxic and anticancer activities of isatin and its derivatives: a comprehensive review from 2000–2008. Anti-Cancer Agents Med Chem 9(4):397–414. https://doi.org/10.2174/1871520610909040397
Xiu S, Dick A, Ju H, Mirzaie S, Abdi F, Cocklin S, Zhan P, Liu X (2020) Inhibitors of SARS-CoV-2 entry: current and future opportunities. J Med Chem 63(21):12256–12274. https://doi.org/10.1021/acs.jmedchem.0c00502
Watts KS, Dalal P, Murphy RB, Sherman W, Friesner RA, Shelley JC (2010) ConfGen: a conformational search method for efficient generation of bioactive conformers. J Chem Inf Model 50(4):534–546. https://doi.org/10.1021/ci100015j
WHO (2019) Coronavirus Disease. https://www.who.int/emergencies/diseases/novel-coronavirus-2019
Williams-Noonan BJ, Todorova N, Kulkarni K, Aguilar MI, Yarovsky I (2021) An active site inhibitor induces conformational penalties for ACE2 recognition by the spike protein of SARS-CoV-2. J Phys Chem B 125(10):2533–2550. https://doi.org/10.1021/acs.jpcb.0c11321
Yong JN, Majoumo-Mbe F, Samje M, Nfor EN (2016) Synthesis, molecular structure and anti-onchocercal studies of 1-(phthalazin-1(2H)-one)[(pyridin-2-yl)ethylidene]hydrazone. Int J Org Chem 6(1):77. https://doi.org/10.4236/ijoc.2016.61008
Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, Dömling A, Dubin G, Holak TA (2015) Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure 23(12):2341–2348. https://doi.org/10.1016/j.str.2015.09.010
Zhang HM, Dai H, Hanson PJ, Li H, Guo H, Ye X, Hemida MG, Wang L, Tong Y, Qiu Y, Liu S, Wang F, Song F, Zhang B, Wang JG, Zhang LX, Yang D (2014) Antiviral activity of an isatin derivative via induction of PERK-Nrf2-mediated suppression of cap-independent translation. ACS Chem Biol 9(4):1015–1024. https://doi.org/10.1021/cb400775z
Acknowledgements
Funding was provided by the Wistar Science Discovery Fund (LJM, JS) and the Canadian Institutes for Health Research (CIHR PJT-153057) (IT). FMM thanks the Cambridge Crystallographic Data Center (CCDC) for their initiative to promote structural studies in Africa and particularly in Cameroon through the FAIRE Programme. This work was also supported by the following grants to LJM: the Robert I. Jacobs Fund of the Philadelphia Foundation and the Herbert Kean, M.D., Family Professorship. FNK acknowledges a Calestous Juma Science Leadership Fellowship from the Bill & Melinda Gates Foundation (award number: INV-036848). FNK also acknowledges joint funding from the Bill Bill & Melinda Gates Foundation and LifeArc (award number: INV-055897) under the African Drug Discovery Accelerator program. FNK acknowledges further funding from the Alexander von Humboldt Foundation for a Research Group Linkage project.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
We declare none.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Majoumo-Mbe, F., Sangbong, N.A., Tadjong Tcho, A. et al. 5-chloro-3-(2-(2,4-dinitrophenyl) hydrazono)indolin-2-one: synthesis, characterization, biochemical and computational screening against SARS-CoV-2. Chem. Pap. 78, 3431–3441 (2024). https://doi.org/10.1007/s11696-023-03274-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-023-03274-5