
Abstract
Novel oral anticoagulants are frequently used for the pharmacotherapy of thromboembolic disorders but still have drawbacks and side effects. While numerous synthetic and semisynthetic derivatives of nontoxic isosteviol possess potential therapeutic properties, including anticoagulant activity. Besides, thiourea is recognized in medicinal chemistry research as a component of a common framework of many drugs or bioactive compounds. The present work combines molecular modeling and docking approach for searching and designing novel thiourea isosteviol-based compounds as potential FXa inhibitors. Elaborated regression model well reflects the relationships between experimentally determined anticoagulant activity and molecular descriptors and may be used for the prediction of FXa inhibitory activity of novel thiourea isosteviol compounds. Among 20 descriptors incorporated into the ANN model, 60% are 2D topological descriptors, 25% describe three-dimensional molecular structure, and remaining 15% belong to constitutional descriptors. Additionally, docking simulation confirms the prominent binding of the newly in silico designed molecules with the active sites of the protein, which may be the lead molecules and can be further optimized for the efficient pharmacodynamic and pharmacokinetic profiles. Based on the results obtained, thiourea derivatives of isosteviol with 3-chloro-4-fluorophenyl, 3-fluoro-4-chlorophenyl or 4-(oxazol-5-yl)phenyl substituent may be promising FXa inhibitors. Findings reported in the present work can be used as valuable information for the development of anticoagulants.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cardiovascular diseases (CVDs) rank ignominiously first among the leading causes of death worldwide, with an estimated nearly 18 million deaths annually (World Health Organization 2023). More than four out of five deaths are caused by heart attacks and strokes, and one-third of these deaths occur prematurely in people under 70 years of age. Early identification of patients at high risk of cardiovascular disease and implementation of appropriate pharmacotherapy are the most effective means to prevent premature deaths. Thrombosis is defined as the development of blood clots within a blood vessel. This common causal pathology leads to the development of prevalent cardiovascular disorders such as stroke, acute coronary syndrome, and venous thromboembolism (Zacconi 2018). Antithrombotic drugs, used for the prevention and management of thrombosis, counteract clots from forming and magnifying and include antiplatelet drugs, anticoagulants, and fibrinolytic agents. Antiplatelet agents and anticoagulants prevent blood clot formation in the course of the primary and secondary hemostasis process, while fibrinolytic drugs are capable of stimulating the dissolution of a formed thrombus (Bisacchi 2013). Because of the high thrombosis incidence and its serious implications, effective prophylaxis and treatment are indispensable and connected with widespread oral anticoagulant use. Recently, the efforts of researchers have focused on new classes of anticoagulants targeting specific enzymes or coagulation steps in the coagulation cascade (Mccarty and Robinson 2016). Activated Blood Coagulation Factor X (FXa) is an Arg-specific serine protease related to trypsin which plays a pivotal role in hemostasis an essential part of the blood-clotting cascade by catalyzing the thrombin production and strengthens the initial response of the coagulation process. For this reason, it has become the main drug design target that significantly influenced current effective pharmacological thromboprophylaxis. This resulted in the development of direct FXa inhibitors introduced in the market after 2011 such as apixaban, rivaroxaban, edoxaban, and betrixaban, which are among the most widely used oral anticoagulants for the efficient pharmacotherapy of thromboembolic disorders (Rodríguez et al. 2022). It is worth noting that Eliquis® (apixaban) takes sixth place among the 10 best-selling drugs in the world in 2021 with sales of US$ 10.8 billion, which also exhibited an increase compared to 2020 (Urquhart 2022). As was emphasized, FXa, which contains four principal subsites S1, S2, S3, and S4, plays a crucial role in coagulation (Zacconi 2018). However, recent data revealed some functions beyond blood coagulation of FXa. Activation of FX by the tissue factor-FVIIa complex influences immune reactions in inflammation, cancer, and autoimmunity (Ruf 2021).
The broad spectrum of pharmacological activity of naturally occurring diterpene compounds has attracted huge interest and encouraged medical chemists to synthesize, purify, and analyze more selective and potent isosteviol (ISV) analogs (Ullah et al. 2019). The isosteviol (ent-16-oxobeyran-19-oic acid) is a sweet tetracyclic diterpene containing a beyerane skeleton and a hydrolysis product of stevioside. After the hydrolytic cleavage of glucose fragments from a glycoside molecule, the acidic environment forces the aglycone’s immediate rearrangement from steviol to isosteviol. Due to many possible skeletal rearrangements ISV may be a subject of quantitative structure–activity relationship (QSAR) analyses (Gackowski et al. 2022a). Moreover, its structure incorporates many functional groups as synthetic handles available for rapid chemical iteration. Besides, ISV is commercially available, affordable, nontoxic especially at low doses, and approved by the European Food Safety Authorities and other regulatory agencies worldwide (Wang et al. 2018; Ullah et al. 2019). As the molecular weight (MW) increases during the drug development from a leading structure to a drug candidate, the isosteviol (MW of 318.2) is appropriate for further structural optimization according to the criteria of Lipinski’s rule of five (MW < 500) (Hann and Oprea 2004). Novel isosteviol-like compounds have been therefore synthesized by chemical modification of the isosteviol core (Chen et al. 2019; Shi et al. 2020; Zhang et al. 2021). As a metabolite of stevioside, it is renowned for a wide variety of biological activities including antioxidant, anti-diarrheal, antidiabetic, antibacterial, anticancer, acetylcholine, and DNA topoisomerase inhibition, anti-tuberculosis, and cardiovascular effects (Wang et al. 2018; Ullah et al. 2019). Recently, the antithrombic activity of ISV-based compounds has been postulated by Chen et al. (2019) and Shi et al. (2020). Reported FXa interactions and potent anticoagulant activity encourage researchers to continue the efforts toward searching and designing novel and safer anticoagulant drugs based on the isosteviol core.
Numerous synthetic and semisynthetic ISV-like compounds displayed various biological activities and possess potential therapeutic properties. Moreover, thiourea represents a privileged structure in medicinal chemistry as it is a component of a common scaffold of a variety of drugs or bioactive compounds that exhibit a wide range of pharmacological activity. The above-mentioned marketed direct oral anticoagulants still have drawbacks and side effects such as a significant increase in gastrointestinal bleeding, drug interaction, and skin toxicity. And besides, their synthesis is not only layered involving several steps, but also dangerous reagents and toxic solvents are indispensable (Rodríguez et al. 2022). Hence, the isosteviol-based compounds may be potential lead molecules and alternatives to currently used drugs. In view of the above, efforts toward designing novel ISV compounds and meant to improve the understanding of a molecular basis of the anticoagulant activity are highly desirable.
Experimental
Isosteviol derivatives
A set of twenty isosteviol ((4α,8β,13β)‐13‐‐Methyl‐16‐oxo‐17‐norkauran‐18‐oic acid) analogs bearing thiourea fragments synthesized and evaluated for human activated coagulation factor X (FXa) inhibitory activity by Shi et al. were incorporated into the molecular modeling study and docking simulation (Shi et al. 2020). Their structures and biological activity expressed as IC50 against FXa are shown in Table 1. Molecules were built, and energy minimization was performed with the Polak–Ribiere algorithm to a maximum energy gradient of 0.01 kcal (Å⋅mol) − 1 with molecular mechanics (MM +) and Austin Model 1 (AM1) force fields using HyperChem 8.0 (Hypercube Inc., Gainesville, FL, USA).
Molecular descriptors
Molecular descriptors are generated as mathematical representations of a molecule’s chemical information. The information encoded in the molecular structure, e.g., geometry, shape or atomic properties is converted into numbers that are used to establish quantitative relationships between structures and for example biological activities. More than 5000 descriptors can be calculated employing several software applications (Consonni and Todeschini 2010). In the present study, 4885 0D constitutional, 1D structural, 2D topological, and 3D geometrical descriptors belonging to 29 logical blocks (Talete and List of molecular descriptors calculated by Dragon 2023) were calculated using Dragon 7 (Talete, Milano, Italy) software. Descriptors having constant and near constant values, standard deviation less than 0.0001, pairwise correlation (|r| > 0.95) and at least one missing value were rejected. As a consequence, 1117 molecular descriptors were retained in the descriptor space.
Regression analysis
Selected descriptors were subjected to Artificial Neural Network (ANN) analysis with the use of Statistica 13.3 software (TIBCO Software Inc., Palo Alto, California, USA). It should be underlined that to decrease the network's complexity and increase its predictive performance, the most informative and representative molecular features with respect to the studied activity should be selected and used as input to construct a model (Dobchev and Karelson 2016). For this reason, 20 of the most important descriptors were selected from 1117 candidates with the use of Feature Selection and Variable Screening, as implemented in Statistica 13.3 Data Miner, based on F-values and p-values (Fig. 1 and Table 2).

Importance of preselected descriptors
Due to the data ranges over several orders of magnitude, the values of biological activity were transformed to a negative decimal logarithm. The designed predictive model is dedicated to solving regression problem, and for this reason the response of the network is qualitative, namely inhibitory FXa activity expressed as pIC50.
The ANN approach attempts to simulate the information processing of the brain in a simplified form. The network’s architecture incorporates connected neurons grouped in an input, output, and hidden layer situated between these two latter layers. There is a connection flow from the input to the output direction. The molecular descriptors are entered into the input units by the input layer, and then the hidden and output layer units are executed sequentially in their order. Each of them calculates its activation value by taking the weighted sum of the outputs of the units in the previous layer. The activation value is passed through the activation function to produce the output of the neuron. When the network has been executed, the neurons of the output layer are assigned to a desired feedback (biological activity) of the whole network (TIBCO Statistica® User’s Guide Statistica Automated Neural Networks (SANN) 2023; Dobchev and Karelson 2016). In the present study, the construction of the network began with the division of the entire set of isosteviol derivatives into a training set (subjected to the learning process), a test set (for the final cross-assessment of network quality) and a validation set (to check the effects of the learning algorithm) based on Random Sample Selection in Statistica 13.3 Data Miner. Automated Network Search was used in the procedure for training and testing about two hundred different networks with optimal architecture, differing in the number of neurons in individual layers and activation functions for hidden and output neurons. STATISTICA Automated Neural Networks scale the input and target variables using linear transformations such that the original minimum and maximum of each variable is mapped to the range (0, 1). The BFGS (Broyden-Fletcher-Goldfarb-Shanno) learning algorithm was applied. The best neural network able to determine correct answers after introducing new input data was chosen based on the correlation between the experimental values and the predicted by the network for the training, the test, and the validation set, taking into consideration the mean square error. Sensitivity analysis was used to test whether the variables in the ANN model are significant (error quotient for a particular variable is greater than 1) and provide a good prediction of FXa inhibitory activity. The elaborated model was employed to predict the activity of several newly designed thiourea isosteviol compounds (e1–e11).
Molecular docking simulation
Molecular Docking is an advanced computational tool used for analyzing interactions between the drugs or therapeutically effective molecules and the proteins involved in the pathogenesis of diseases. The drug is called a ligand that interacts with the binding or active site of protein in highly stable conformations with the least energy. These possible conformations in which drug binds are called Docking Poses each having different energy called Binding Affinity or Docking Score expressed in terms of kcal/mol unit. The molecule having conformation with the least docking score is considered to be the most effective and stable in terms of ligand–protein interaction, which can be further used in pharmacophore modeling or QSAR studies to optimize the designed therapeutic molecules (Pappachen et al. 2017).
In this study, human activated coagulation factor X protein complexed with apixaban inhibitor was selected (PDB ID: 2P16) and its X-ray diffraction structure was taken from RCSB Protein Data Bank in PDB format (Fig. 2) (RCSB Protein Data Bank and 2P16: Factor Xa in Complex with the Inhibitor APIXABAN (BMS-56 (2247)) AKA 1-(4-METHOXYPHENYL)-7-OXO-6-(4-(2-OXO-1-PIPERIDINYL)PHENYL)-4, 5, 6, 7-TETRAHYDRO-1H-PYRAZOLO[3, 4-C]PYRIDINE-3-CARBOXAMIDE., 2023).

Structure of human factor Xa protein in complex with apixaban inhibitor taken from RCSB protein data bank (PDB ID: 2P16)
The protein was prepared using AutoDock Vina software (The Center for Computational Structural Biology, La Jolla, California, USA). In this, water molecules, unwanted residues, the same amino acid chains, and another inhibitor (Apixaban) already present in the protein were removed as well as Kollmann and Gasteiger charges were assigned. The protein was then saved in PDB file format (Morsy et al. 2020). The structure of the protein showing the active or binding site of human activated coagulation factor X protein for the molecules to interact is presented in Fig. 3.

The structure of protein showing the active or binding site of human activated coagulation factor X protein for the molecules to interact
The preparation of ligands comprised the set of 23 isosteviol compounds (20 bearing thiourea fragments and tested and validated by the designed ANN model) including newly designed compounds with the most promising FXa inhibitory activity predicted by the elaborated ANN model (e1, e4, and e10) as well as standard drugs, namely apixaban and edoxaban, were chosen for docking and saved in PDB file format using BIOVIA Discovery Studio 2021 Client (Discngine S.A.S., Paris, France). The standard drugs were selected for comparison of docking scores, and their structures were taken from Pubchem and saved in 3D conformer as SDF format (later converted to PDB Format) (Morsy et al. 2020). The binding site of the protein was identified based on the position of the apixaban inhibitor already present, and its dimensions were noted using Discovery Studio 2021 Client. The structure of the protein and twenty-five ligands were loaded in PyRx software (SourceForge, San Diego, CA, USA). The ligands were energy minimized, and then both protein and ligands were converted to PDBQT file format. The grid box was positioned in the protein to define the binding site for the ligands to interact or dock according to the dimensions of the active site of the protein obtained in Discovery Studio 2021 Client (Ren et al. 2020). The size of the grid box was 25 × 25 × 25, and it was centered at coordinates X = 9.54, Y = 43.27, and Z = 63.47. Molecular docking simulation was performed using Vina in PyRx software after assigning grid dimensions. The process was allowed to run, and all the ligands were docked, each giving nine conformations with corresponding docking scores. The docking interactions of all the molecules with protein were visualized in 2D and 3D images using Discovery Studio 2021 Client.
Results
Molecular modeling
To find optimal predictive model, about two hundred different networks were trained and tested. The predictive model for establishing quantitative structure–activity relationships was selected based on the correlation and the mean square error. The input layer consisted of 20 artificial neurons, the hidden one of 11 artificial neurons of the logistic activation function and one neuron formed the output layer of the logistic activation function. Broyden-Fletcher-Goldfrab-Shanno BFGS (Quasi-Newton) learning algorithm was employed for the network development.
This artificial neural network–a multilayer perceptron (MLP) 20-11-1 developed to solve the regression problem was characterized by a high correlation between the actual pIC50 value and the value predicted by the network of 0.976 for training, 0.959 for testing, and 0.994 for validation. The error of the regression model discussed was 0.014 for the training set, 0.065 for the test set, and 0.131 for validation. The results of the correlation between the experimentally determined pIC50 values and the pIC50 values predicted by the MLP 20-11-1 network model are shown graphically in Fig. 4.

Graphical presentation of regression analysis: training set R = 0.976; test set R = 0.959 and validation set R = 0.994
Sensitivity analysis revealed the most important variables for the designed MLP 20-11-1 model. The ranking of the descriptors in descending order of importance along with their dimensionality, classification, and definition is shown in Table 3. An error quotient for a single variable that is greater than 1 indicates the significance of variable. However, in the opposite case, a given variable would have no effect or could even worsen the performance of the network and should therefore be excluded. In the case of the elaborated predictive model, all preselected descriptors proved to be important for the FXa inhibitory activity and were therefore used to design the network.
Among 20 preselected predictors incorporated into the network, 60% are 2D topological descriptors, 25% describe three-dimensional molecular structure, and remaining 15% belong to constitutional descriptors. In the case of the error quotient larger than 2, six of the most prominent descriptors were distinguished belonging to following blocks: 2D Atom Pairs, GETAWAY descriptors, Burden eigenvalues, Constitutional indices and P_VSA-like descriptors. Based on structure–activity relations that have been established in previous (Chen et al. 2019; Gackowski et al. 2022a) and current investigation, eleven new thiourea isosteviol structures were designed and their values of potential FXa inhibitory activity (Table 4) were calculated.
Molecular docking
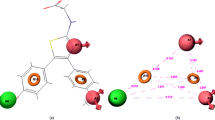
To study the molecular interaction and affinity of binding of studied isosteviol compounds and standard drugs to human activated coagulation factor X protein, a molecular docking simulation was carried out. Among the 23 studied molecules, compound i23 has shown the best binding score of − 9.3 kcal/mol against the target protein compared to edoxaban (− 8.7 kcal/mol) and very close to apixaban (− 10.3 kcal/mol). In addition to this, compounds i32, i33, and i34 showed the same docking score of − 9.0 kcal/mol close to the apixaban standard. Hence, the derivatives designed in the present study showed better docking scores and potential interaction with the selected target protein. The docking scores of all the ligands are given in Table 5 (molecules with binding scores below − 9.0 kcal/mol are marked in Bold) and 2D and 3D interactions of all ligands with the protein are shown in Figs. 5, 6, 7, 8, 9, 10, 11. The interaction energy includes van der Waals energy, electrostatic energy, as well as intermolecular hydrogen bonding for each minimized complex. The residues thus predicted are energetically important for ligand binding inside the active site via hydrophobic, hydrogen bond interactions, etc., in almost all complexes.

3D and 2D interaction of compound i23 with human activated coagulation factor X protein

3D and 2D interaction of compound i20 with human activated coagulation factor X protein

3D and 2D interaction of compound i32 with human activated coagulation factor X protein

3D and 2 D interaction of compound i33 with human activated coagulation factor X protein

3D and 2D interaction of compound i34 with human activated coagulation factor X protein

3D and 2D interaction of Apixaban with human activated coagulation factor X protein

3D and 2D Interaction of Edoxaban with human activated coagulation factor X protein
The fused polycyclic region of all molecules shown in Figs. 5, 6, 7, 8, 9 occupies the S4 pocket with residues Tyr99, Phe174, Lys96, and Glu97. The derivatized aromatic moiety forms van der Waals, conventional H-bond, and Pi-alkyl and Amide-Pi stacked bonds with amino acids in the S1 pocket.
The molecule presented in Fig. 10—Apixaban shows a unique donor-donor interaction between the NH of the benzopyrazole ring with Glu192 at the bottom of the S1 site.
In the case of Edoxaban (Fig. 11), there is Pi-sulfur interaction in the S4 region of the protein with the thiophene ring. Two carbonyl groups of the ligand are involved in conventional H-bond in the S1 and S2 region. Comparing the docking scores of both standard compounds, apixaban shows higher negative value (− 10.3) than edoxaban (− 8.7), which indicates that apixaban has higher affinity to protein and forms more stable complex. This can be easily described by the 2D interactions of both standards with protein whereby apixaban interacts with 20 amino acids through 10 different types of interactions than edoxaban interacting with 19 amino acids through 5 different interactions, namely Conventional Hydrogen Bond, van der Waals, Carbon Hydrogen Bond, Pi-Pi Stacked and Alkyl Bond which are also shown by apixaban. The additional 5 interactions shown by apixaban are Unfavourable Donor-Donor Bond, Pi-Sulfur, Pi-Alkyl, Pi-Pi T-shaped bond and Amide-Pi Stacked bond. Thus, these additional interactions explain higher affinity of apixaban to target protein and more complex stability.
Discussion
Direct oral anticoagulants (DOACs) have been increasingly used in recent years compared with vitamin K antagonists (VKAs) (Capiau et al. 2022). They are widely prescribed for preventing ischemic stroke in patients with non-valvular atrial fibrillation and for the management and prevention of venous thromboembolism (Ferri et al. 2022). On the one hand, DOACs are considered a safer alternative to VKAs due to significant reductions in stroke, intracranial hemorrhage and mortality in comparison to warfarin. But on the one hand, important side effects such as increased gastrointestinal bleeding, skin toxicity, and drug interactions constitute restrictions on their use. As all DOACs are substrates of P-glycoprotein, they may be susceptible to strong inducers or inhibitors of this drug transporter. Moreover, rivaroxaban and apixaban are substrates of cytochrome P450, which underlies possible interactions with inducers or inhibitors of CYP activities. Therefore, there is still a great demand for novel, potent FXa inhibitors with a better safety profile.
Even though since the beginning of the eighties, the factor Xa had already been associated as a pharmacological target for the anticoagulant agents, its first naturally occurring inhibitor antistasin was extracted from the salivary glands of the Mexican leech Haementeria officinalis in 1987 (Zacconi 2018). Drug discovery and medicinal chemistry fields have always been inspired by natural products and their structural analogs. They played a crucial role in finding medications for the pharmacotherapy of for instance infectious diseases, cancer, cardiovascular diseases, or multiple sclerosis. Molecular scaffolds of natural and semisynthetic lead compounds have served as starting points for drug development. Recently, interest in natural products as lead structures for drugs has been revived due to recent technological developments that facilitate the discovery of drugs based on natural products (Rodrigues et al. 2016; Atanasov et al. 2021). Given abundant range of biological activity and other favorable features described above, the isosteviol analogs are promising lead compounds for new drug discovery. Therefore, the present study was designed to investigate relationships between descriptors encoding molecular properties and FXa inhibitory activity, design novel isosteviol-based compounds and predict their potential activity using the elaborated model, and finally analyze the interactions with a target molecule.
Scientific methods applied in established molecular modeling procedure involved geometry optimization using AM1 semiempirical method, descriptor calculation for spatially optimized molecules, and model building using Artificial Neural Network algorithm. The application of semiempirical method assures not only the high level of agreement with ab initio calculations but also makes the whole procedure more economical and time-saving (Ford and Wang 1993). The computational approach toward molecule optimization and descriptor calculation was established based on a considerable set of anthrapyrazoles (Gackowski et al. 2022b) and successfully applied to design a QSAR model for isosteviol compounds (Gackowski et al. 2022a). ANN algorithm applied for the establishment of the structure–activity relations in the current investigation is renowned for the ability to perform nonlinear mapping of the molecular descriptors to the biological activity of analyzed molecules with a remarkable quality of the fit to the training data. It enables producing QSAR models that are superior in predictive performance (Deeb and Hemmateenejad 2007). In particular, one type of ANN, the multilayer perceptron, ensures near-perfect predictions in comparison with linear models (Žuvela et al. 2018). This highly adaptive nonlinear optimization algorithm enables the development of models used in many fields of science. Among other modern approaches, especially in recent years, ANNs have been found wide applications in the process of drug discovery. Despite the criticism for overtraining problems, complexity, black box mechanism, or difficulty of interpretation, it constitutes a great help in drug design (Dobchev and Karelson 2016).
An optimal predictive QSAR model of the highest quality was selected from about two hundred different developed neural networks. The selected network is a system of interconnected neurons arranged in three layers. The input layer consists of 20 neurons, each of which accepts one of the descriptors (Table 2) for further processing. The intermediate layer (hidden) is a key component of the designed network, consisting of eleven neurons and processing the data by applying the sigmoid function (sometimes called logistic function) to them. It captures and reproduces extremely complex input–output relationships. The final layer of the network is the output layer, which consists of one neuro and produces the final result, expressed as a value of pIC50. As presented in Fig. 4, the predicted pIC50 values are in good agreement with experimental ones, which indicates the significant correlation between selected descriptors and FXa inhibitory activity.
Sensitivity analysis of predictive neural network confirmed that all preselected descriptors are important to enter the MLP 20-11-1 model. Nevertheless, the ranking list (Table 3) sorted all descriptors by significance. Six, the most important molecular descriptors can be distinguished if the error quotient is greater than 2. They are representatives of the following classes of descriptors: 2D Atom Pairs, GETAWAY descriptors, Burden eigenvalues, Constitutional indices, and P_VSA-like descriptors. The first two descriptors from the ranking list belong to the 2D Atom Pairs and are two-dimensional descriptors that consider the internal atomic arrangement of compounds (Gozalbes et al. 2002). Both B01[C–Cl] and B07[N–Cl] encode the presence of chlorine atoms at different topological distances. It may be suggested that the presence of chlorine atoms affects the most anticoagulant activity of thiourea isosteviol compounds. It supports previous findings that the introduction of the chlorine atom on the phenyl group led to an improvement of the inhibitory activity (Shi et al. 2020). Moreover, the same descriptor, namely B01[C–Cl], was the first to appear in the QSAR model developed by Gackowski et al. using multivariate adaptive regression splines (MARSplines) (Gackowski et al. 2022a). The third most important descriptor of the ANN network is the representative of the GETAWAY (GEometry, Topology, and Atom-Weights AssemblY) descriptors. This block of 3D descriptors is characterized by a generally good modeling capability, which makes them useful in QSAR studies (Consonni et al. 2002b). GETAWAY descriptors are based on a leverage matrix, the so-called molecular influence matrix, which has been proposed as a new molecular representation simply calculated from the spatial coordinates of the molecular atoms in a chosen conformation. GETAWAY descriptors attempt to match the 3D-molecular geometry and atomic relatedness provided by the molecular influence matrix with chemical information through molecular topology by using different atomic weightings (atomic mass, polarizability, van der Waals volume, and electronegativity, along with unit weights) (Consonni et al. 2002a). The HATS4s descriptor integrated into the QSAR model is based only on the diagonal elements of the leverage matrix, which take into account the relative position of each atom in the 3D molecular space and are related to the intrinsic properties of a single molecule. The next most significant descriptor belongs to Burden eigenvalues. It is SpMax2_Bh(m), i.e., the largest eigenvalue no. 2 of the Burden matrix weighted by mass, which is calculated by Dragon from the Burden matrix Bh(w) based on a hydrogen-filled molecular graph. In this particular case, atomic mass constitutes the diagonal elements of the adjacency matrix. SpMax2_Bh(m) is suitable for a description of chemical similarity/diversity on large databases (Yu and Liu 2021). Mean atomic polarizability (Mp) is the fifth descriptor in the ANN model. This constitutional descriptor is calculated as the mean of all atomic polarizabilities scaled over the carbon atoms. It delineates the deformation of an electron cloud in response to an external field of atoms or molecules (Khan et al. 2018). The last descriptor among the best six ones represents P_VSA descriptors. These indices are labeled as the amount of van der Waals surface area (VSA) having a property P in a certain range. P_VSA_LogP_8 is calculated as the sum of atomic contributions to the van der Waals surface area (VSA), from atoms having the highest LogP values, that is with high hydrophobicity (Casanova-Alvarez et al. 2021).
It should be emphasized that descriptors that entered the designed ANN model represent different logical blocks as well as diverse dimensionality. Constitutional descriptors (0D) are related to atom and bond type counts, and topological descriptors (2D) are obtained from molecular graphs and are conformationally independent, while 3D-descriptors are subjected to the geometrical coordinates of the atoms of molecules (Helguera et al. 2008). The elaborated QSAR model comprising molecular 0D, 2D, and 3D descriptors, in light of the above, comprehensively reflects molecular properties. The joint use of different descriptors containing information on the whole molecular structure provides more predictive models than only indices related to certain features of the molecule (Consonni et al. 2002b). In addition, the elaborated ANN model was successfully employed for the prediction of FXa inhibitory activity of newly designed thiourea isosteviol compounds (Table 4). Although the novel thiourea isosteviol-like compounds designed in this study are not as active as standard drugs, the results indicate clearly that this is the correct direction of research, because the elaborated model may be applied to predict the activity of other possible thiourea isosteviol derivatives.
Virtual screening techniques are widely and frequently used to reduce the duration and cost of the drug development process (da Silva Rocha et al. 2019). The molecular docking approach is used to find new ligands for biological proteins and is important for both structure-based and ligand-based drug design (Agarwal and Mehrotra 2016). To better understand the molecular basis of the biological activity of isosteviol derivatives, the technique has been effectively used for the theoretical prediction of ligand-target interactions. It also provides more insight into the potential mode of action and how agents bind to the FXa protein. When isosteviol compounds were docked using the PyRx tool, highly active molecules were found within the group of 23 isosteviol-like compounds that produced better molecular interactions and lower binding energies (9.0–10.0 kcal/mol) with the chosen protein. The binding affinity of isosteviol compounds to the FXa receptor increases with negative docking score, resulting in more efficient bioactive molecules.
Conclusions
The present paper demonstrates a new approach for the search and design of novel isosteviol-based compounds as potential FXa inhibitors. An elaborated regression model establishes the relationships between experimentally determined biological activity and molecular descriptors and enables the prediction of FXa inhibitory activity for newly designed compounds. The obtained results proved that the Artificial Neural Network algorithm facilitates the search for the most promising isosteviol derivatives containing thiourea fragments as FXa inhibitors. The elaborated QSAR model comprising molecular 0D, 2D, and 3D descriptors comprehensively reflects molecular properties. Moreover, the docking simulation confirms the prominent binding of the compounds to the active sites of the protein, which may be the lead molecules and can be further optimized for efficient pharmacodynamic and pharmacokinetic profiles that yield the potent biological activity discussed in the present work. Based on the results obtained, thiourea derivatives of isosteviol possessing 3-chloro-4-fluorophenyl, 3-fluoro-4-chlorophenyl or 4-(oxazol-5-yl)phenyl substituent may be promising FXa inhibitors. Despite shortcomings of the present study (i.e., biological activity taken from the literature, limited accuracy of AM1 semiempirical method, limitations of descriptors generation and ANN algorithm, and finally the stability of docking complexes justified to a certain extent), the results reported in this paper can be used as valuable information for the development of anticoagulants.
References
Agarwal S, Mehrotra R (2016) An overview of molecular simulation. JSM Chem 4:1024–1028
Atanasov AG, Zotchev SB, Dirsch VM, Orhan IE, Banach M, Rollinger JM, Barreca D, Weckwerth W, Bauer R, Bayer EA, Majeed M, Bishayee A, Bochkov V, Bonn GK, Braidy N, Bucar F, Cifuentes A, D’Onofrio G, Bodkin M, Diederich M, Dinkova-Kostova AT, Efferth T, El Bairi K, Arkells N, Fan TP, Fiebich BL, Freissmuth M, Georgiev MI, Gibbons S, Godfrey KM, Gruber CW, Heer J, Huber LA, Ibanez E, Kijjoa A, Kiss AK, Lu A, Macias FA, Miller MJS, Mocan A, Müller R, Nicoletti F, Perry G, Pittalà V, Rastrelli L, Ristow M, Russo GL, Silva AS, Schuster D, Sheridan H, Skalicka-Woźniak K, Skaltsounis L, Sobarzo-Sánchez E, Bredt DS, Stuppner H, Sureda A, Tzvetkov NT, Vacca RA, Aggarwal BB, Battino M, Giampieri F, Wink M, Wolfender JL, Xiao J, Yeung AWK, Lizard G, Popp MA, Heinrich M, Berindan-Neagoe I, Stadler M, Daglia M, Verpoorte R, Supuran CT (2021) Natural products in drug discovery: advances and opportunities. Nat Rev Drug Discov 20:200–216. https://doi.org/10.1038/s41573-020-00114-z
Bisacchi GS (2013) Anticoagulants, antithrombotics, and hemostatics. In: Abraham DJ (ed) Burger’s medicinal chemistry and drug discovery cardiovascular agents and endocrines, 6th edn. Wiley, Hoboken, pp 283–338
Capiau A, Mehuys E, Dhondt E, De Backer T, Boussery K (2022) Physicians’ and pharmacists’ views and experiences regarding use of direct oral anticoagulants in clinical practice. Br J Clin Pharmacol 88:1856–1865. https://doi.org/10.1111/bcp.15106
Casanova-Alvarez O, Morales-Helguera A, Cabrera-Pérez MÁ, Molina-Ruiz R, Molina C (2021) A novel automated framework for QSAR modeling of highly imbalanced Leishmania high-throughput screening data. J Chem Inf Model 61:3213–3231. https://doi.org/10.1021/acs.jcim.0c01439
Chen P, Zhang D, Li M, Wu Q, Lam YPY, Guo Y, Chen C, Bai N, Malhotra S, Li W, O’Connor PB, Fu H (2019) Discovery of novel, potent, isosteviol-based antithrombotic agents. Eur J Med Chem 183:111722. https://doi.org/10.1016/j.ejmech.2019.111722
Consonni V, Todeschini R, Pavan M (2002a) Structure/response correlations and similarity/diversity analysis by GETAWAY descriptors. 1. Theory of the novel 3D molecular descriptors. J Chem Inf Comput Sci 42:682–692. https://doi.org/10.1021/ci015504a
Consonni V, Todeschini R, Pavan M, Gramatica P (2002b) Structure/response correlations and similarity/diversity analysis by GETAWAY descriptors. 2. Application of the novel 3D molecular descriptors to QSAR/QSPR studies. J Chem Inf Comput Sci 42:693–705. https://doi.org/10.1021/ci0155053
Consonni V, Todeschini R (2010) Molecular descriptors. In: Challenges and advances in computational chemistry and physics. pp 177–208
da Silva Rocha SFL, Olanda CG, Fokoue HH, Sant’Anna CMR (2019) Virtual screening techniques in drug discovery: review and recent applications. Curr Top Med Chem 19:1751–1767. https://doi.org/10.2174/1568026619666190816101948
Deeb O, Hemmateenejad B (2007) ANN-QSAR model of drug-binding to human serum albumin. Chem Biol Drug Des 70:19–29. https://doi.org/10.1111/j.1747-0285.2007.00528.x
Dobchev D, Karelson M (2016) Have artificial neural networks met expectations in drug discovery as implemented in QSAR framework? Expert Opin Drug Discov 11:627–639. https://doi.org/10.1080/17460441.2016.1186876
Ferri N, Colombo E, Tenconi M, Baldessin L, Corsini A (2022) Drug-drug interactions of direct oral anticoagulants (DOACs): from pharmacological to clinical practice. Pharmaceutics 14:1–27
Ford GP, Wang B (1993) New approach to the rapid semiempirical calculation of molecular electrostatic potentials based on the am1 wave function: comparison with ab initio hf/6-31g* results. J Comput Chem 14:1101–1111. https://doi.org/10.1002/jcc.540140911
Gackowski M, Golec KS, Katarzyna M, Pluskota R, Koba M (2022a) Quantitative structure–activity relationship analysis of isosteviol‐related compounds as activated coagulation factor X ( FXa ) Inhibitors
Gackowski M, Szewczyk-Golec K, Pluskota R, Koba M, Madra-Gackowska K, Woźniak A (2022b) Application of multivariate adaptive regression splines (MARSplines) for predicting antitumor activity of anthrapyrazole derivatives. Int J Mol Sci 23:5132. https://doi.org/10.3390/ijms23095132
Gozalbes R, Doucet JP, Derouin F (2002) Application of topological descriptions in QSAR and drug design: history and new trends. Curr Drug Targets Infect Disord 2:93–102. https://doi.org/10.2174/1568005024605909
Hann MM, Oprea TI (2004) Pursuing the leadlikeness concept in pharmaceutical research. Curr Opin Chem Biol 8:255–263. https://doi.org/10.1016/j.cbpa.2004.04.003
Helguera A, Combes R, Gonzalez M, Cordeiro MN (2008) Applications of 2D descriptors in drug design: a DRAGON tale. Curr Top Med Chem 8:1628–1655. https://doi.org/10.2174/156802608786786598
Khan PM, Rasulev B, Roy K (2018) QSPR modeling of the refractive index for diverse polymers using 2D descriptors. ACS Omega 3:13374–13386. https://doi.org/10.1021/acsomega.8b01834
Mccarty D, Robinson A (2016) Factor Xa inhibitors: a novel therapeutic class for the treatment of nonvalvular atrial fibrillation. Ther Adv Cardiovasc Dis 10:37–49. https://doi.org/10.1177/1753944715605011
Morsy MA, Ali EM, Kandeel M, Venugopala KN, Nair AB, Greish K, El-Daly M (2020) Screening and molecular docking of novel benzothiazole derivatives as potential antimicrobial agents. Antibiotics 9:221. https://doi.org/10.3390/antibiotics9050221
Pappachen LK, Zachariah SM, Chandran D (2017) In silico design, synthesis and characterization of some novel benzothiazole derivatives as anticancer agents. Asian J Pharm Clin Res 10:150–155. https://doi.org/10.22159/ajpcr.2017.v10i4.16407
RCSB Protein Data Bank 2P16: Factor Xa in Complex with the Inhibitor APIXABAN (BMS-562247) AKA 1-(4-METHOXYPHENYL)-7-OXO-6-(4-(2-OXO-1-PIPERIDINYL)PHENYL)-4,5,6,7-TETRAHYDRO-1H-PYRAZOLO[3, 4-C]PYRIDINE-3-CARBOXAMIDE. https://www.rcsb.org/structure/2P16. Accessed 14 Mar 2023
Ren L, You T, Li Q, Chen G, Liu Z, Zhao X, Wang Y, Wang L, Wu Y, Tang C, Zhu L (2020) Molecular docking-assisted screening reveals tannic acid as a natural protein disulphide isomerase inhibitor with antiplatelet and antithrombotic activities. J Cell Mol Med 24:14257–14269. https://doi.org/10.1111/jcmm.16043
Rodrigues T, Reker D, Schneider P, Schneider G (2016) Counting on natural products for drug design. Nat Chem 8:531–541. https://doi.org/10.1038/nchem.2479
Rodríguez DF, Durán-Osorio F, Duarte Y, Olivares P, Moglie Y, Dua K, Zacconi FC (2022) Green by design: convergent synthesis, computational analyses, and activity evaluation of new fxa inhibitors bearing peptide triazole linking units. Pharmaceutics 14:33. https://doi.org/10.3390/pharmaceutics14010033
Ruf W (2021) Roles of factor Xa beyond coagulation. J Thromb Thrombolysis 52:391–396. https://doi.org/10.1007/s11239-021-02458-8
Shi Y, Pan BW, Li WC, Wang Q, Wu Q, Pan M, Fu HZ (2020) Synthesis and biological evaluation of isosteviol derivatives as FXa inhibitors. Bioorganic Med Chem Lett 30:126585. https://doi.org/10.1016/j.bmcl.2019.07.044
Talete SRL List of molecular descriptors calculated by Dragon (2023) http://www.talete.mi.it/products/dragon_molecular_descriptor_list.pdf. Accessed 18 Apr 2023
TIBCO Statistica® User’s Guide Statistica Automated Neural Networks (SANN) - Neural Networks Overview. https://docs.tibco.com/pub/stat/14.0.0/doc/html/UsersGuide/GUID-F60C241F-CD88-4714-A8C8-1F28473C52EE.html. Accessed 15 Jan 2023
Ullah A, Munir S, Mabkhot Y, Badshah SL (2019) Bioactivity profile of the diterpene isosteviol and its derivatives. Molecules 24:1–24. https://doi.org/10.3390/molecules24040678
Urquhart L (2022) Top companies and drugs by sales in 2021. Nat Rev Drug Discov 21:251. https://doi.org/10.1038/d41573-022-00047-9
Wang M, Li H, Xu F, Gao X, Li J, Xu S, Zhang D, Wu X, Xu J, Hua H, Li D (2018) Diterpenoid lead stevioside and its hydrolysis products steviol and isosteviol: biological activity and structural modification. Eur J Med Chem 156:885–906. https://doi.org/10.1016/j.ejmech.2018.07.052
World Health Organization (2023) Cardiovascular diseases. https://www.who.int/health-topics/cardiovascular-diseases#tab=tab_1. Accessed 9 Feb 2023
Yu X, Liu J (2021) Prediction of reaction rate constants of hydroxyl radical with chemicals in water. Water Environ Res 93:934–939. https://doi.org/10.1002/wer.1485
Zacconi FC (2018) FXa direct synthetic inhibitors. Anticoagulant Drugs. https://doi.org/10.5772/intechopen.76518
Zhang H, Liu B, Xu G, Xu C, Ou E, Liu J, Sun XO, Zhao Y (2021) Synthesis and in vivo screening of isosteviol derivatives as new cardioprotective agents. Eur J Med Chem 219:113396. https://doi.org/10.1016/j.ejmech.2021.113396
Žuvela P, David J, Wong MW (2018) Interpretation of ANN-based QSAR models for prediction of antioxidant activity of flavonoids. J Comput Chem 39:953–963. https://doi.org/10.1002/jcc.25168
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gackowski, M., Madriwala, B. & Koba, M. In silico design, docking simulation, and ANN-QSAR model for predicting the anticoagulant activity of thiourea isosteviol compounds as FXa inhibitors. Chem. Pap. 77, 7027–7044 (2023). https://doi.org/10.1007/s11696-023-02994-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-023-02994-y