
Abstract
The world is now facing intolerable damage in all sectors of life because of the deadly COVID-19 pandemic caused by the severe acute respiratory syndrome coronavirus 2. The discovery and development of anti-SARS-CoV-2 drugs have become pragmatic in the time needed to fight against this pandemic. The non-structural protein 3 is essential for the replication of transcriptase complex (RTC) and may be regarded as a possible target against SARS-CoV-2. Here, we have used a comprehensive in silico technique to find potent drug molecules against the NSP3 receptor of SARS-CoV-2. Virtual screening of 150 Isatin derivatives taken from PubChem was performed based on their binding affinity estimated by docking simulations, resulting in the selection of 46 ligands having binding energy greater than −7.1 kcal/mol. Moreover, the molecular interactions of the nine best-docked ligands having a binding energy of ≥ −8.5 kcal/mol were analyzed. The molecular interactions showed that the three ligands (S5, S16, and S42) were stabilized by forming hydrogen bonds and other significant interactions. Molecular dynamic simulations were performed to mimic an in vitro protein-like aqueous environment and to check the stability of the best three ligands and NSP3 complexes in an aqueous environment. The binding energy of the S5, S16, and S42 systems obtained from the molecular mechanics Poisson–Boltzmann surface area also favor the system's stability. The MD and MM/PBSA results explore that S5, S16, and S42 are more stable and can be considered more potent drug candidates against COVID-19 disease.
Similar content being viewed by others

Introduction
No one can imagine how a small virus-like SARS-CoV-2 can completely change the lives of the entire human population. What has COVID-19 not destroyed? It has an impact on the economic and social sectors and exposes the medical industry's flaws (McKibbin and Fernando 2020). The globe has been under attack by a pandemic generated by a tiny virus known as Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) that first appeared in late 2019 in Wuhan, China (Bharadwaj 2021). On March 12, 2020, the World Health Organization (WHO) announced a pandemic, World Health Organization (WHO) (Zhang et al. 2020). The worldwide number of COVID-19 deaths and cases has reached 6,287,187and 520,579,701, respectively, as reported on May 14, 2022. It can spread up to 1–2 m via droplets, nasal discharges, or saliva of an infected person and can remain on the surface for days (Kampf et al. 2020). The laboratory test of the COVID-19 patients reveals an increase in neutrophils (a type of white blood cell) count in the blood, which acts as a first-line defense against any disease in the human body (Wang et al. 2020). Coronaviruses (CoVs) are positive-sense, enveloped, single-stranded RNA viruses (Singhal 2020) belonging to the Coronaviridae family and Coronavirinae subfamily of the order Nidovirales (Lima C.M.A.d.O. 2020), which are divided into four genera \(\left( {\alpha ,\beta ,\Upsilon {\text{ and }}\partial } \right)\). The SARS-CoV-2 belongs to the \(\beta\) genus like SARS-CoV-1, and MERS emerged early in 2002 and 2012 (Sen 2020).
The human coronavirus genome has many conserved structural proteins such as enveloped (E) proteins, nucleocapsid (N) protein, membrane (M) protein, and spike (S) glycoprotein (Sen 2020). It also consists of at least four non-structural proteases, such as papain-like protease (PLPRO), RNA-dependent RNA polymerase (RDRP), helicase, and the main protease (MPRO) (Sen 2020). Among NSPs, two important MPROand PLPRO plays a crucial role in forming the replication–transcription complex and regulating various virus replication functions (Naidoo 2020). The PLPRO is an essential component of the replica–transcriptase complexes, which comprise the largest multidomain of all coronaviruses within NSP3 and contain 1945 residues (Rut, et al. 2020). Three proteins, NSP1, NSP2, and NSP3, are released when PLPRO cleaves peptide bonds at three different sites (Rut, et al. 2020). Because of their critical role in viral replication and transcription, the MPRO and PLPRO are considered attractive drug targets among coronaviruses. So, interruption of any replication process may reduce the multiplication process of the virus (Zmudzinski, et al. 2020). In this study, we have taken the NSP3 (PDB 6WOJ) protein released by PLPRO as a potential drug target.
Most researchers are working to find a therapeutic agent for people infected by this disease, but no effective therapy has yet been discovered (Sen 2020). It is pertinent to mention here that drug development is a lengthy process. It may take months to years, or even decades, to discover a new medicine from the initial Stage to the market (Augen 2002). In silico, drug discovery has become a versatile and intensive way to find potent therapeutic agents against any disease. Furthermore, this method is cost-effective and less time-consuming than the wet experimental drug discovery method (Wadood et al. 2013). So, it is worth mentioning here that we do not have enough time to wait for any drug against COVID-19 (Muhammad 2021). The association of computer-based virtual screening and molecular docking approach is one of the best strategies to find potential therapeutic drug targets against SARS-CoV-2. The other strategy is the in silico ADMET prediction of different databases' drug-likeness of potential therapeutic agents (Sepay et al. 2021). Among biologically active compounds, the Isatin derivatives (a class of Schiff bases) group has been gaining popularity among scientists due to their intensive and diverse medicinal applications, which include antiviral (Jarrahpour et al. 2007), anti-inflammatory (Sharma et al. 2016), antifungal (Chohan et al. 2004), antibacterial (Guo 2019), anti-parasitic (Wang et al. 2017), anti-TB (Xu et al. 2017), anti-HIV (Bal et al. 2005), anticancer (Sabet et al. 2010; Khan et al. 2022), anti-mycobacterial (Xu et al. 2017), anticonvulsant (Verma et al. 2004; Ilyas et al. 2022), anti-hemolytic (Melo, et al. 2016) and antioxidant activities (Andreani et al. 2010). As isatin derivatives are Schiff bases that can be synthesized with a high output (yield) by simple condensation of carbonyl and amine compounds (Aslam et al. 2013). This research looked at 150 Isatin derivatives as possible SARS-CoV-2 NSP3 inhibitors. This paper discusses drug-likeness, virtual screening, molecular docking, and molecular dynamics simulation study of isatin derivatives. The information in this paper will allow scientists to investigate a good drug to control the pandemic.
Materials and methods
Retrieval of macromolecule
The protein data bank (www.rcsb.org) was utilized to create the three-dimensional X-ray crystalline structure of non-structural protein 3 (NSP3) in the form of a PDB format (Mishra 2020). The NSP3 protein (PDB ID = 6WOJ) consists of 4 chains (A, B, C, and D), 678 residues, 5432 atoms, 78.22 kDa total structural weight, the sequence length of 176, and complexed with ADP-ribose. Our study used only chain A of the NSP3 receptor for docking analysis. The protein is prepared by using the MGL tools. The co-crystalized ligand (ADP-ribose), other chains, and water molecules detached from the receptor, adding polar hydrogen. The detection of active sites in protein–ligand complexes is critical in the drug development phase (Muhammad 2021). We also studied our receptor through Cavity Plus, online software used to identify active sites within the receptor. The total number of binding pockets found within the receptor was 15. We only selected the three binding pockets based on draggability and surface area of the bag (Xu et al. 2018).
Ligand's structure modeling
By searching the literature, about 95 Isatin derivative compounds based on less toxicity and a molecular weight of less than 500 were chosen from PubChem, an online free access database of compounds (Mahmud 2021). The compounds were downloaded in SDF format. To get ligands ready for docking analysis. The tool Autodock 1.5.6 was used. In the final step of ligand preparation, the resulting structures were transformed into the PDB format (Muhammad 2021; Lim et al. 2021; Bolton et al. 2008).
Molecular docking
The PDB files of receptors and ligands were changed into PDBQT form to perform molecular docking analysis of selected Isatin derivatives with the NSP3 receptor using Autodock Vina. The grid dimensions of the NSP3 receptor were set by getting the binding site amino acid information gathered by using the free online software CASTp 3.0 (Tian, et al. 2018). The grid size dimension of protein is x = 45, y = 60, z = 54 and grid center dimensions x = 23, y = 25, z = 35 and exhaustiveness 8 set as a default. After that, we performed the Docking of selected Isatin derivatives with Autodock Vina (Trott and Olson 2010). The ligands were flexible, having different rotatable bonds, while the receptor was kept as a rigid target. The ligands with the highest binding affinity (more negative value) were considered the most stable for the receptor. All the ligands have a good binding affinity for the receptor. Then we selected 46 ligands having a binding affinity of more than 7.1 kcal/mol. Biova Discovery Studio 2020 client (Biovia 2017), a free version of the software, was used to evaluate the interactions of the nine selected ligands with a binding affinity of greater than 8.5 kcal/mol. Moreover, to make our results more reproducible, we made ten docking replicas of 9 hit ligands and then took the average of their binding energy. The Table S1of replica results are represented in supporting information. The three hits with five or more hydrogen-bonding interactions were selected for further MD study from the interaction analysis. We have also validated our docking protocol.
ADME Study
To check ADMET (Absorption, Digestion, Metabolism, Excretion, and Toxicity) calculations of the top 9 Isatin derivatives, SWISS ADME and PKCSM free online web servers were utilized. These top 9 ligands showed the highest binding affinity against the SARS-CoV-2 NSP3 receptor. These servers have robust data to check high precision physiochemical properties such as drug-likeness, water solubility, lipophilicity, toxicity, and pharmacokinetics (Daina et al. 2017; Pires et al. 2015; Muhammad et al. 2022). Furthermore, none of the ligands violate Lipinski's rule of five. This shows that all the ligands are orally bioavailable (ISMAİL et al. 2018).
Molecular dynamics Study
A molecular dynamics simulation study was carried out to check the stability and conformational behavior of protein alone and upon complex formation with 3 top hit Isatin derivatives. All MD simulations of proteins and complexes were completed in 120 ns (12 × 106 fs). The CHARMM force field (Best et al. 2012) in NAMD (Phillips et al. 2020) was used to measure these simulations. The VMD program completed the trajectories, RMSF, RMSD, the radius of gyration, solvent-accessible surface area, and the number of hydrogen bonds. Three separate replica simulations were used to ensure that the findings were reliable. Results from single simulation tests are frequently not repeatable, and conclusions drawn from several shorter replicas are more trustworthy than those removed from a single, more extensive simulation (Knapp et al. 2018). The whole system was solvated by adding water molecules and neutralized by adding NaCl. The temperature, pressure, cutoff radius, and time step were 310 K, 1 atm, 10 A \(^\circ\), and 2 fs.
MM/PBSA binding free energy calculations
The binding free energy of the selected antiviral compounds (S5, S16, and S42) against NSP3 of SARS-CoV-2 was executed by molecular mechanics Poisson–Boltzmann surface area (MM/PBSA). The MM/PBSA method was first described by (Kollman et al. 2000). The MM/PBSA methodology was used to determine the interaction of ligand (L) and protein (P) in the form of a protein–ligand complex and calculated by Eq. 1
The \(\Delta G_{{\text{bind }}}\) in the equation is dependent on three factors which are \(\Delta G_{{{\text{MM}}}}\) (the gas-phase free energy), \(\Delta G_{{{\text{Sol}}}}\) (solvation free energy) and \(T\Delta S\) (the change in the system). Equation 2 summarized these terms to calculate, MM/PBSA which was given as follows:
where, as
In these equations, \(\Delta G_{{{\text{MM}}}}\) deals with the variation in internal energies such as electrostatic and van dar Waal energies \(\Delta E_{{{\text{ele}}}} {\text{and}}\user2{ }\Delta E_{{{\text{vdW}}}}\) and \(\Delta E_{{{\text{int}}}}\) Represents changes in bond angle and dihedral energy. \(\Delta G_{{{\text{Sol}}}}\) is the sum of polar interactions \(\left( {\Delta G_{{{\text{PB}}}} } \right)\) and nonpolar interactions \(\left( {\Delta G_{{{\text{SA}}}} } \right)\). The \(\Delta G_{{{\text{SA}}}}\) nonpolar energy is determined by solvent-accessible surface area. Here,\(T{\Delta }S\) denote the temperature of the system and the entropy of the solute in a vacuum, respectively.
The \(T\Delta S\) terms justify the change in conformational entropy of the protein–ligand complex upon ligand binding. The calculation of binding free energy for 120 ns time trajectory was accomplished by the CaFE (Calculation free energy) tool (Liu and Hou 2016).
Results and discussion
Selection of binding pockets and isatin derivatives
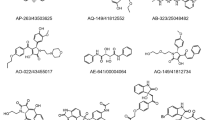
An online software CASTp was utilized to predict the binding pockets within the receptor NSP3 (PDB ID: 6WOJ). This software reveals that the selected chain of receptor NSP3 consists of 15 binging pockets having different volumes and surface areas. The decreasing volume of the first eight (8) important binding pockets is 175.43 > 5.16 > 3.82 > 2.30 > 0.803 > 0.154 > 0.110 > 0.102, respectively. Similarly, the decreasing surface area of these selected pockets is 289.28 > 17.11 > 12.61 > 9.082 > 3.225 > 1.838 > 1.169 > 1.039, respectively. The first one with the highest volume and surface area is selected from these eight pockets. The chosen binding pocket contains 29 residues. These residues are ALA21, ASP22, ILE23, ALA38, ALA39, ASN40, LEU43, LYS44, HIS45, GLY46, GLY47, GLY48, VAL49, ALA50, VAL95, GLY97, PRO125, LEU126, SER127, SER128, ALA129, GLY130, ILE131, PHE132, PRO126, VAL155, PHE156, ASP157, and LEU160. The complete detail of the binding cavities is represented in supporting information in Figures S4 and S5. Isatin derivatives were chosen as potential inhibitors for the COVID-19 NSP3 receptor (Majumder et al. 2020). Further, Isatin is also present naturally in different body organs in different concentrations. It was revealed in a literature study that Isatin derivatives have many biological effects, such as anaptotic, anticancer, antipyretic, anti-inflammatory, antiviral, anti- HIV, and antibiotic (Pandeya et al. 2005). As a result, these were chosen and retrieved from PubChem as antipyretic, anti-inflammatory drugs, antivirals, anti-HIV drugs, and antibiotics (Pandeya et al. 2005). So, these were selected and retrieved from the PubChem online server (Pandeya et al. 2005). The 2D structures of 9 Isatin derivatives are presented in Fig. 1.

Structures of Isatin natural derivatives used in the molecular docking study having good binding affinities
Binding Affinities
Information about the ligand–protein binding interaction mechanism is essential in drug design because it can lead to discovering new drug candidates. As a result, a basic understanding of the essence of molecular interaction is critical, as it can provide vital insights into drug development, design, and discovery (Du et al. 2016). Molecular docking is broad in silico technique for predicting the binding mode of a ligand–protein interaction (Sousa et al. 2013). All the ninety-five (95) Isatin-based ligands were docked successfully against the NSP3 receptor of SARS-CoV-2. The ligand's binding affinities with the receptor were −5.4 kcal/mole to −9.6 kcal/mole.
Further, to make our study more comprehensive, we took 46 ligands with a binding affinity greater than −7.0 kcal/mol, usually used as a benchmark in several docking studies. These forty-six (46) ligands with their binding affinity, PubChem ID, and inhibition constants are presented in Table S4. As the essential structural parts of all the selected Isatin derivatives are similar, the best 9 having a docking score equal to or above −8.5 kcal/mol were selected for interaction analysis. The introduction of an electron-withdrawing group as a substituent on isatin structure leads to a decrease in its binding affinity with the receptor NSP3, i.e., S1, S5, S18, S27, and S36, which have F, acetyl, CF3, and phenyl group as a substituent on either benzene or azole ring of Isatin have lower binding affinity in comparison with Isatin. Removal of electron-withdrawing F from 3-position of substituents in S2 and S3 leads to a bit of rising in binding affinity compared to S1.
Conversely, introducing the electron-donating alkyl group leads to an increase in binding affinity. S42 shows the highest binding relationship because of two such groups, an electron-donating alkyl group on the azole ring and another good electron donor phenoxy as sulfonamide substituent on the benzene ring of Isatin. So, we can conclude that further novel derivatives of Isatin with potent Inhibition against NSP3 of SARS-CoV-2 can be synthesized by introducing influential electron donor groups on either benzene or azole ring of Isatin. The binding affinity of nine top hits of isatin derivatives is shown in Table S7, and their structures are shown in Fig. 2.

The binding affinity (kcal/mol) graph of 9 best docked Isatin derivatives within the active site of NSP3 receptor of SARS-CoV-2
Docking validation
The Discovery studio visualizer of the redocked inhibitor complex visualized the Cartoon view after docking in Autodock vina using the same set of boundary and grid parameters we used in our study. We experimentally determined co-crystallized inhibitor complex downloaded from PDB as shown in Fig. S14 (a) and (b). Then we superimposed both of our inhibitors (redocked and co-crystallized) as shown in Fig. S15 (a) and both of the complexes, i.e., redocked inhibitor complex and co-crystallized inhibitor complex by pymol as shown in Fig. S15 (b) and calculated RMSD which was astonishingly 0. This 0 RMSD shows that ADP-ribose inhibitor interacted in just a similar manner in both the cases as interacting residues exactly matched in both (redocked and co-crystallized complex) the circumstances, as shown in Fig. S15 (a) and (b). In this way, the docking protocol was validated (Tallei 2020; Shivanika et al. 2020).
Ligand and protein Interaction analysis
The interaction results of the nine (9) best Isatin derivatives having a high Binding Affinity/low docking score and their bond distances are presented in Table S5. The surface view poses view and receptor–ligand 2D interactions of our three (3) best ligands, S5, S16, and S42, are shown in Fig. 3. The surface view, pose view, and receptor–ligand 2D interactions of the remaining six ligands are presented in the supporting information in Figs. S1 and S2. The surface view poses an argument, and 2D interactions of the remaining six ligands and three reference drugs are represented in supporting information in Fig. S3.

The non-bonded interactions of the top three docked complexes, Where A, B, C and D represents the interactions analysis diagrams of N-Benzoylisatin (S5), 5-(2-phenylethyl) Isatin (S16) and Isatin sulphonamide 34 (S42) respectively. These figures were generated by using Discovery Studio version
Ligand S42 was the best of the selected 3, having a binding affinity of -9.6 kcal/mol and forming 14 interactions in the binding site of the NSP3 receptor. It was observed to interact with ILE23 (2.93 Å), ILE23 (1.91 Å), Val29 (2.40 Å), GLY48 (3.15 Å), and GLY130 (3.32 Å) amino acid in the binding pocket of the NSP3 receptor through both carbon–hydrogen and conventional hydrogen type interactions. Besides carbon–hydrogen and traditional bond interactions, it was also found to make a bond with VAL49 (3.62 Å), VAL49 (3.86 Å), ILE131 (3.91 Å), PHE156 (3.88 Å), PHE132 (4.89 Å), GLY48 (5.26 Å), GLY48 (4.96 Å), ALA50 (4.77 Å) and ALA38 (5.10 Å) residues through pi–pi-stacked, pi–sigma, amide–pi, and pi–alkyl hydrophobic interactions. Ligand S16 was the second-best among the four-hit ligands, having a binding affinity of −8.9 kcal/mol and forming a total of 15 interactions within the binding pocket of the NSP3 receptor. It was observed to bond with GLY48 (2.52 Å), VAL49 (3.09 Å), ALA50 (2.23 Å), LEU126 (2.84 Å), and PHE132 (2.96 Å) residues in the binding site via conventional and pi–donor hydrogen bond type interaction. Besides hydrogen bond interactions, it was observed to make bonds with ALA38 (3.72 Å), PHE132 (5.93 Å), PHE132 (5.04 Å), ALA38 (4.45 Å), ALA50 (5.42 Å), ILE131 (4.11 Å), ILE131 (5.19 Å), LEU126 (5.04 Å), ALA129 (5.04 Å), and ALA155 (5.36 Å) residues via pi–sigma, pi–pi T-shaped and pi–alkyl type hydrophobic interactions. The last one of the best three ligands, S5, having a binding affinity of −8.8 kcal/mol, also forms six hydrogen bond interactions and eight hydrophobic interactions within the binding pocket of the NSP3 receptor SARS-CoV-2. It was also found to bind to VAL (1.95 Å), SER128 (2.84 Å), SER128 (2.21 Å), ALA129 (2.28 Å), GLY130 (2.26 Å), and GLY48 (3.14 Å) amino acids via conventional and carbon–hydrogen bond interactions. Apart from carbon and traditional bond of hydrogen interactions, it also forms a bond with ALA38 (3.71 Å), PHE132 (4.96 Å), PHE132 (5.32 Å), ILE131 (5.09 Å), ALA38 (4.48 Å), ALA50 (5.20 Å), ILE131 (4.25 Å), and ALA129 (4.57 Å) residues via pi-shaped, pi–pi T-shaped, and pi–alkyl hydrophobic interactions. The three-dimensional interaction analysis diagram of the best three-hit ligands S5, S16, and S42 within the binding cavity of the NSP3 receptor of SARS-CoV-2, along with polar and nonpolar interaction diagrams taken from the discovery studio visualizer and Pymol software are shown in Figs. S11 and 4, respectively. The NSP3, which is a multidomain protein, consists of a macrodomain "ADP-ribose phosphatase domain (ADRP)," a marker domain, an N-terminal Nsp3a domain, an RNA binding domain, a PLpro domain, a transmembrane domain, a SARS-unique domain, and a Y-domain (https://coronavirus3d.org). The ADRP domain (primarily identified as the X domain) was unique and conserved in the genomes of the Hepeviridae, Coronaviridae, and Togaviridae families using bioinformatics techniques (Lee et al. 1991).

Polar and Non-Polar interaction diagrams of the best 3 leads (S5, S16 and S42). Red area represents polar, green region show complete protein surface and light blue region for non-polar
The ADRP domain is involved in several pathways, including ADP-ribose metabolism and post-translational modification of proteins. It is understood that the ADRP domain performs a significant role in modifying natural immunity. Viruses with a mutant macrodomain multiplied poorly in bone marrow-derived macrophages, which are the primary cells involved in establishing an innate immune response, according to studies looking at the role of this domain in enabling the immunological response (Grunewald et al. 2019).
Pretreatment with interferon was also effective against viruses with an inactivated macrodomain (Kuri et al. 2011). These findings support the idea that the ADRP domain is essential in disease pathogenesis and that inhibiting it might lower viral load and speed recovery (Zheng 2020). The authors of this study chose to look for chemicals that interact with the ADRP domain as antiviral medicines. Previous research revealed that the binding site of ADPr coupled to the ADRP domain consists of the following residues: Ala21, Asp22, Ile23, Ala38, Asn40, Lys44, His45, Gly46, Gly47, Gly48, Val49, Leu126, Ser128, Ala129, Gly130, Ile131, Phe132, Ala154, Phe156, and Leu160 (Pandey and Sharma 2021). As shown in Fig. 3, our selected ligands interacted with many of these desired essential residues.
Drug-likeness
Any ligand that forms a good interaction within the receptor's binding site has little or no use if it has poor absorption or if its excretion from the body is too fast. Then, it is essential to anticipate the drug-likeness of the ligands, for which a rule of five, also known as Lipinski's rule, was utilized. This rule states that any ligand can be considered a drug based on some crucial parameters such as the number of H-bond acceptors less than 10, molecular weight < 500 Dalton, number of H-bond donors < 5, and lipophilicity (WlogP) < 5. Our nine best-selected ligands powerfully fulfill this rule and show no violation, as represented in Table 1. It is pertinent to mention here that we can consider these ligands as drugs by seeing the Table 1 results.
Absorption
Numerous parameters affect absorption, i.e., Log P, gastrointestinal absorption, and TPSA. All three ligands, S5, S16, and S42, have comparablegastrointestinal absorption to the reference molecule. Total polar surface area (TPSA) influences absorption and should be below 140A2. As given in Table 1, our chosen compounds meet this requirement. The optimal range for Log P is 1–5, which indicates the hydrophobicity of the therapeutic molecule. As shown in Table S6, our three ligands have the highest Log P-value. All our ligands are within this range.
Distribution
BBB permeability and P-gp efflux are two characteristics that influence medication distribution. All ligands we chose are not P-gp substrates, and BBB permeation is in the range −0.95–0.56, as shown in Table S6.
Metabolism
Improper metabolism might result in various adverse consequences, i.e., toxic effects of reactive metabolites, poor bioavailability, Cytochrome P450 plays an essential role in drug metabolism in the body, and our lead compounds are likely to be efficiently metabolized since they do not block these cytochromes P4502C9 and P4502D6, except for S42, which inhibits P4502C19, as shown in Table S6.
Excretion
An active medicine must be specific to its target and stay in the body long enough to perform the intended activity. Our three ligands had total clearance (TC) values ranging from 0.09 to 0.41 (ml/min/kg). Similarly, except for S16, our lead compounds are non-substrates of the organic cation transporter (OCT2), which is important in excusing drugs (secondary metabolites) from the kidney.
Toxicity
It is advantageous to identify drug toxicity early in the medication development process. To function as an effective medicine, a molecule must be safe, with no adverse effects on the liver (hepatotoxicity) or the skin (skin sensitization). As shown in Table S6, our lead compounds do not produce such toxicity. They also do not cause any hERG: I and II inhibition.
Molecular dynamic study
Docking procedures are considered to be rough and rapid. But due to flexibility loss of protein by Docking, the reliability of the resultant complexes interface. As a result, more detailed molecular dynamics simulations with higher computing costs may better correlate with Docking. In general, MD is utilized to investigate macromolecule behavior, and it employs Newton's equation of motion to calculate the speed and position of every atom in the studied system. As a result, it produces a more realistic representation of protein movement. By utilizing the NAMD package, MD simulations were performed on the protein and ligand–protein complexes to simulate the interaction of these three ligands with the COVID-19 NSP3 for the 120 ns timescale. Each complex was subjected to three separate MD simulations. The data shown are an average of three MD runs.
RMSD analysis
The RMSD of proteins and ligand–protein complexes can be used to evaluate the conformational stability of the system. The variations from the initial positions of protein and ligand–protein complexes atoms are measured using the RMSD analysis. Findings derived from single simulation tests are frequently not repeatable, and conclusions drawn from several shorter replicas are more trustworthy than those removed from a single, more extensive simulation. Here, we form three representations of each complex represented in Fig. 5. The replicas of complexes RMSD and RMSF are illustrated in Figs. 5 and 8, respectively. But the replica of other parameters such as radius of gyration (Rg), SASA, and number of H bonds are represented in supporting information in Figs. S8, S9, and S10.

Three RMSD replica of three best hit ligands of isatin derivatives
The RMSD analysis of replicas of S5, S16, and S42 complexes showed that replica 1 is more stable among these complexes, as shown in Fig. 5. Moreover, we also compared the results of RMSD of the most stable replicas of S3, S16, and S42 complexes with RMSD of Protein, as shown in Fig. 6. To keep the explanation clearer, four colors (maroon (protein), orange–brown (S5), purple (S16), and mustard (S42) were chosen.

The RMSD analysis of protein (maroon) and the most stable replica of the three best ligands complexes of Isatin derivatives
The average, maximum and minimum values of RMSD of protein and ligand–protein complexes are represented in Table 2. Figure 6 showed that the protein (maroon) showed fluctuations from about 10 ns to 16 ns, and after that, the protein gets equilibrated and did not show any severe changes throughout the MD simulations. The maximum, minimum, and average RMSD offered by S5-complex (orange–brown color) is 4.43 Å,3.81 Å, and 4.06 Å. The S5-complex showed slight fluctuation of RMSD from 60 ns and, after that, achieved equilibrium throughout the MD simulation. The maximum, minimum, and average RMSD values S16-complex (purple) are 4.82 Å, 3.20 Å, and 4.12 Å.
.
The S16-complex showed fluctuations at two places, i.e., 16–20 ns and 76–80 ns. After that, the system gets equilibrium. The maximum, minimum, and average RMSD values of S42-complex (mustard) are 4.47 Å, 3.81 Å, and 4.34 Å. All the complexes have higher RMSD values than protein RMSD, which occurred due to combining the ligands with the protein NSP3.
RMSF analysis
Figure 7 depicts the RMSF of the protein (maroon) and S5-complex, S16-complex, and S42-complex as a function of several residues. As shown in Fig. 7, in the case of S5 and S16 ligand's RMSF is below 3 Å, which is considered an optimum value for a globular protein (Kufareva and Abagyan 2011). However, in the case of S42, fluctuations at a few residues, i.e., Gly51, Asn54, Thr71, and Asn101, are observed. At these points, RMSF is more significant than 3 Å. But more importantly, none of these residues is involved in interaction with S42, as shown in Fig. 3, and RMSF is below 3 Å in the remaining parts of the RMSF trajectory. The maximum, minimum, and average RMSD of protein and protein–ligand complexes are given in Table 2. Moreover, the replicas of RMSF of three complexes are represented in Fig. 8.

The RMSF analysis of protein (maroon) and the most stable replica of the three best ligands complexes of Isatin derivatives

Three RMSF of replica of three best hit ligands of Isatin derivatives
Number of H bonds, Rg, and SASA
In protein and ligand–protein complexes and molecular recognition, the number of H bonds is critical for stability. The rigidity of the complexes was demonstrated by the fact that all three ligand–protein complexes remained relatively stable throughout the simulation timeline. The maximum, minimum and average number of H bonds are represented in Table 2. Figure 9 shows the H bonds in S5-complex, S16-complex, and S42-complex, which are reasonably in good numbers. The average number of H bonds are 10, 15, and 15 for S5-complex, S16-complex, and S42-complex, respectively (Sinha and Wang 2020).

The number of hydrogen bonds (H-bond) of protein (maroon) and three best ligand complexes of isatin derivatives
The Rg value significantly impacts the extent of protein and its complexes folding and unfolding. The maximum, minimum, and average Rg values of protein and its complexes (S5-complex, S16-complex, and S42-complex) are presented in Table 2. This Table showed that the protein has low maximum, minimum, and average Rg values than ligand–protein complexes. The S42-complex has the most significant average Rg values compared to two other complexes (S5-complex and S16-complex). Figure 10 shows the Rg of protein and S5-complex, S16-complex, and S42-complex.

The radius of gyration (Rg) of protein (maroon) and three best ligand complexes of Isatin derivatives
To measure the protein and its complex's volume change along the simulation period, the SASA of the protein and its complexes was examined from the MD trajectory. The maximum, minimum, and average SASA values of protein and its complexes (S5-complex, S16-complex, and S42-complex) are presented in Table 2. This Table showed that the protein has low maximum, minimum, and average SASA values compared to ligand–protein complexes. The S5-complex has the smallest average SASA values compared to two other complexes (S16-complex and S42-complex). Figure 11 shows the SASA of protein and S5-complex, S16-complex, and S42-complex. The MD simulations of protein and its complexes confirmed the stability of the complexes with the protein as shown by Rg, number of H bonds, and SASA.

The SASA of protein (maroon) and three best ligand complexes of isatin derivatives
MM/PBSAbinding free energy calculations
The MM/PBSA technique is a more robust and sophisticated model for calculating binding free energy than the MM/PBSA approach (Hubbard and Grothey 2017). Revalidation of docking findings for specified complexes like as S5-NSP3, S16-NSP3, and S42-NSP3, the binding free energy \(\user2{ }\Delta G_{{{\text{bind}}}}\) the calculation was performed by MM/PBSA method (Ahmad 2020). The values of \(\user2{ }\Delta G_{{{\text{bind}}}}\) for selected antiviral complexes (S5, S16, and S42) are summarized in Table 3. The van der Waals interactions contributed the major role in stability of complex and showed higher values of \(\user2{ }\Delta E_{{{\text{vdW}}}}\) such as S5 (−18.460 kcal/mol), S16 (−21.9377 kcal/mol), and S42 (−30.3528 kcal/mol). The electrostatic interaction has a negligible effect on complex stability. The net of solvation energy in terms of polar \(\left( {\Delta G_{{{\text{PB}}}} } \right)\) and nonpolar \(\left( {\Delta G_{{{\text{SA}}}} } \right)\) interactions in the stability of complex also mentioned in table 6. The complex S42 showed high binding free energy against NSP3 of SARS-CoV-2 compared to complexes S5 and S16. The net binding free energy \(\left( {\user2{ }\Delta {\varvec{G}}_{{{\varvec{bind}}}} } \right)\) of complexes (S5, S16, and S42) determined by the MM/PBSAtechnique were −0.8659, −1.6459, and −7.1352 kcal/mol, indicating that these three complexes are the most promising NSP3 inhibitors.
Conclusion
With the constant increase in SARS-CoV-2 infection and the unavailability of any potent antiviral therapeutic agent for its treatment, synthetic compounds are considered a good source of antiviral drug discovery. Herein, ninety-five (95) Isatin derivatives were docked within the binding pocket of the NSP3 receptor of SARS-CoV-2. Moreover, virtual screening based on binding affinity and inhibition constant (Ki) was performed, resulting in a selection of 46 ligands. Molecular interactions of the nine ligands having a critical relationship of ≥ −8.5 kcal/mol were analyzed. All these selected ligands have higher binding affinity than the three reference drugs. The interaction analysis explores those three ligands (S5, S16, S42) that are mainly stabilized by hydrophobic interactions with the receptor's active site. These three ligands were then passed through PKCSM and SWISSADME online servers to anticipate their drug-likeness and ADMET. On the other hand, pharmacological and toxicity analysis of the drug molecules suggests a more robust metabolism and absorption profile and no toxicity risks. Finally, MD simulations were run to determine the stability of the protein alone and in complex with these three ligands. The MD results indicate that S5, S16, and S42 are stable, as shown by RMSD and RMSF values, which could be used as drugs against the SARS-CoV-2 NSP3 receptor. As a result, in vitro tests for drug production against SARS-CoV-2 infection may be used to evaluate these agents as SARS-CoV-2 NSP3 inhibitors further.
References
Ahmad S et al (2020) Molecular docking, simulation and MM-PBSA studies of nigella sativa compounds: a computational quest to identify potential natural antiviral for COVID-19 treatment. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1775129
Andreani A et al (2010) New isatin derivatives with antioxidant activity. Eur J Med Chem 45(4):1374–1378
Aslam M et al (2013) Synthesis, x-ray crystallography, molecular Docking and biological screening of 2-aminophenol based schiff bases. J Chil Chem Soc 58(3):1867–1871
Augen J (2002) The evolving role of information technology in the drug discovery process. Drug Discov Today 7(5):315–323
Bal TR et al (2005) Synthesis and evaluation of anti-HIV activity of Isatin β-thiosemicarbazone derivatives. Bioorg Med Chem Lett 15(20):4451–4455
Best RB et al (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J Chem Theory Comput 8(9):3257–3273
Bharadwaj S et al (2021) Exploration of natural compounds with anti-SARS-CoV-2 activity via Inhibition of SARS-CoV-2 Mpro. Brief Bioinform. https://doi.org/10.1093/bib/bbaa382
Biovia DS (2017) Discovery studio visualizer. S Diego CA USA. https://doi.org/10.1002/cpe.6086
Bolton EE et al (2008) PubChem: integrated platform of small molecules and biological activities. Annual reports in computational chemistry. Elsevier, pp 217–241
Chohan ZH et al (2004) Isatin-derived antibacterial and antifungal compounds and their transition metal complexes. J Enzyme Inhib Med Chem 19(5):417–423
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7(1):1–13
de Lima CMA, O (2020) Information about the new coronavirus disease (COVID-19). Radiol Bras. https://doi.org/10.1590/0100-3984.2020.53.2e1
Du X et al (2016) Insights into protein–ligand interactions: mechanisms, models, and methods. Int J Mol Sci 17(2):144
Grunewald ME et al (2019) The coronavirus macrodomain is required to prevent PARP-mediated Inhibition of virus replication and enhancement of IFN expression. PLoS Pathog 15(5):e1007756
Guo H (2019) Isatin derivatives and their anti-bacterial activities. Eur J Med Chem 164:678–688
Hubbard JM, Grothey A (2017) Napabucasin: an update on the first-in-class cancer stemness inhibitor. Drugs 77(10):1091–1103
Ilyas M et al (2022) A DFT approach for finding therapeutic potential of two dimensional (2D) graphitic carbon nitride (GCN) as a drug delivery carrier for curcumin to treat cardiovascular diseases. J Mol Struct 1257:132547
Ismai̇l S. et al (2018) Insilico molecular docking and pharmacokinetic studies of selected phytochemicals with estrogen and progesterone receptors as anticancer agent for breast cancer. J Turk Chem Soc Sect A Chem 5(3):1337–1350
Jarrahpour A et al (2007) Synthesis, antibacterial, antifungal and antiviral activity evaluation of some new bis-schiff bases of isatin and their derivatives. Molecules 12(8):1720–1730
Kampf G et al (2020) Persistence of coronaviruses on inanimate surfaces and their inactivation with biocidal agents. J Hosp Infect 104(3):246–251
Khan MA et al (2022) Controlled supramolecular interaction to enhance the bioavailability of hesperetin to targeted cancer cells through graphyne: a comprehensive in silico study. RSC Adv 12(10):6336–6346
Knapp B, Ospina L, Deane CM (2018) Avoiding false positive conclusions in molecular simulation: the importance of replicas. J Chem Theory Comput 14(12):6127–6138
Kollman PA et al (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 33(12):889–897
Kufareva I, Abagyan R (2011) Methods of protein structure comparison. Homology modeling. Springer, pp 231–257
Kuri T et al (2011) The ADP-ribose-1’’-monophosphatase domains of SARS-coronavirus and human coronavirus 229E mediate resistance to antiviral interferon responses. J Gen Virol 92:1899–1905
Lee H-J et al (1991) The complete sequence (22 kilobases) of murine coronavirus gene 1 encoding the putative proteases and RNA polymerase. Virology 180(2):567–582
Lim JPL, Mac Kevin EB, Nellas RB (2021) The effect of ligand affinity to the contact dynamics of the ligand binding domain of thyroid hormone receptor-retinoid X receptor. J Mol Gr Model 104:107829
Liu H, Hou T (2016) CaFE: a tool for binding affinity prediction using end-point free energy methods. Bioinformatics 32(14):2216–2218
Mahmud S et al (2021) Virtual screening and molecular dynamics simulation study of plant-derived compounds to identify potential inhibitors of main protease from SARS-CoV-2. Brief Bioinform. https://doi.org/10.1093/bib/bbaa428
Majumder I et al (2020) Chloroform fraction of chaetomorpha brachygona, a marine green alga from indian sundarbans inducing autophagy in cervical cancer cells in vitro. Sci Rep 10(1):1–12
McKibbin W, Fernando R (2020) The global macroeconomic impacts of COVID-19: seven scenarios. Asian Econ Pap. https://doi.org/10.2139/ssrn.3547729
Melo MC et al (2016) Inhibition of the hemolytic activity caused by Staphylococcus aureus alpha-hemolysin through isatin-Schiff copper (II) complexes. FEMS Microbiol lett 363(1):fnv207
Mishra CB et al (2020) Identifying the natural polyphenol catechin as a multi-targeted agent against SARS-CoV-2 for the plausible therapy of COVID-19: an integrated computational approach. Brief Bioinform. https://doi.org/10.1093/bib/bbaa378
Muhammad S et al (2021) Exploring the new potential antiviral constituents of moringa oliefera for SARS-COV-2 pathogenesis: an in silico molecular Docking and dynamic studies. Chem phy lett. https://doi.org/10.1016/j.cplett.2021.138379
Muhammad S et al (2022) Insighting the therapeutic potential of fifty (50) shogaol derivatives against mpro of SARS-CoV-2. J Comput Biophy Chem. https://doi.org/10.1142/S273741652250020X
Naidoo D et al (2020) Cyanobacterial metabolites as promising drug leads against the Mpro and PLpro of SARS-CoV-2: an in silico analysis. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1794972
Pandey A, Sharma M (2021) Reappraisal of trifluperidol against Nsp3 as a potential therapeutic for novel COVID-19: a molecular docking and dynamics study. Futur Virol 16(7):491–506
Pandeya SN et al (2005) Biological activities of isatin and its derivatives. Acta Pharm 55(1):27–46
Phillips JC et al (2020) Scalable molecular dynamics on CPU and GPU architectures with NAMD. J Chem Phys 153(4):044130
Pires DE, Blundell TL, Ascher DB (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58(9):4066–4072
Rut W et al (2020) Activity profiling and structures of inhibitor-bound SARS-CoV-2-PLpro protease provides a framework for anti-COVID-19 drug design. BioRxiv. https://doi.org/10.1101/2020.04.29.068890
Sabet R et al (2010) QSAR study of isatin analogues as in vitro anticancer agents. Eur J Med Chem 45(3):1113–1118
Sen D et al (2020) Identification of potential inhibitors of SARS-CoV-2 main protease and spike receptor from 10 important spices through structure-based virtual screening and molecular dynamic study. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1819883
Sepay N et al (2021) Anti-COVID-19 terpenoid from marine sources: a docking, admet and molecular dynamics study. J Mol Struct 1228:129433
Sharma PK et al (2016) Synthesis and anti-inflammatory activity evaluation of novel triazolyl-isatin hybrids. J Enzyme Inhib Med Chem 31(6):1520–1526
Shivanika C et al (2020) Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1815584
Singhal T (2020) A review of coronavirus disease-2019 (COVID-19). Indian J Pediatr 87(4):281–286
Sinha S, Wang SM (2020) Classification of VUS and unclassified variants in BRCA1 BRCT repeats by molecular dynamics simulation. Comput Struct Biotechnol J 18:723–736
Sousa SF et al (2013) Protein-ligand docking in the new millennium–a retrospective of 10 years in the field. Curr Med Chem 20(18):2296–2314
Tallei TE et al (2020) Potential of plant bioactive compounds as SARS-CoV-2 main protease (Mpro) and spike (S) glycoprotein inhibitors: a molecular docking study. Scientifica. https://doi.org/10.1155/2020/6307457
Tian W et al (2018) CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acid Res 46(W1):W363–W367
Trott O, Olson AJ (2010) AutoDock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461
Verma M et al (2004) Anticonvulsant activity of schiff bases of isatin derivatives. Acta Pharm 54(1):49–56
Wadood A et al (2013) In-silico drug design: an approach which revolutionarised the drug discovery process. OA Drug Des Deliv 1(1):3
Wang L et al (2017) Switchable access to different spirocyclopentane oxindoles by N-heterocyclic carbene catalyzed reactions of isatin-derived enals and N-sulfonyl ketimines. Angew Chem 129(29):8636–8641
Wang K et al (2020) Clinical and laboratory predictors of in-hospital mortality in patients with coronavirus disease-2019: a cohort study in wuhan. China Clin Infect Dis 71(16):2079–2088
Xu Z et al (2017) Isatin hybrids and their anti-tuberculosis activity. Chin Chem Lett 28(2):159–167
Xu Y et al (2018) CavityPlus: a web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acid Res 46(W1):W374–W379
Zhang L et al (2020) Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368(6489):409–412
Zheng J (2020) SARS-CoV-2: an emerging coronavirus that causes a global threat. Int J Biol Sci 16(10):1678
Zmudzinski M et al (2020) Ebselen derivatives are very potent dual inhibitors of SARS-CoV-2 proteases-PLpro and Mpro in in vitro studies. BioRxiv. https://doi.org/10.1101/2020.08.30.273979
Acknowledgements
The author from King Khalid University extends his appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through Large Groups RGP.1/318/43. For computer time, this research used the resources of the Supercomputing Laboratory at King Abdullah University of Science & Technology (KAUST) in Thuwal, Saudi Arabia.
Funding
King Khalid University,RGP.1/318/43,Shabbir Muhammad
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors disclose the following financial interest/personal ties as possible conflicting interests. All the co-authors are aware of and approver of our current submission
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ilyas, M., Muhammad, S., Iqbal, J. et al. Insighting isatin derivatives as potential antiviral agents against NSP3 of COVID-19. Chem. Pap. 76, 6271–6285 (2022). https://doi.org/10.1007/s11696-022-02298-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-022-02298-7