
Abstract
In this study, microporous silicon dioxide nanoparticles (SiO2–NPs) were used to improve the physical, chemical, and mechanical properties of bone China ceramics. Microporous SiO2–NPs were prepared economically from sodium metasilicate (SMS) as a precursor with cetyltrimethylammonium bromide (CTAB) as a surfactant at different concentrations. The prepared SiO2–NPs were characterized using scanning electron microscopy, transmission electron microscopy, X-ray diffraction, and Raman spectroscopy to confirm the formation of microporous SiO2–NPs. The optimum concentrations of the precursor and surfactant used in the SiO2–NPs synthesis were set to be 1.5% and 2 g/200 ml, with a size range of 7–96 nm. SiO2–NPs prepared at the optimum concentrations were incorporated into bone China at different concentrations to evaluate their effect on flexural strength and elasticity. The bone China prepared using 1% SiO2–NPs (B1) had the highest flexural strength and Young's modulus values. Sample characteristics, including self-cleaning, differential scanning calorimetry, thermogravimetric analysis, bulk density (BD), apparent porosity (AP), and water absorption (WA), were investigated. The results revealed outstanding characteristic such as self-cleaning ability, remarkable increase in AP and WA, and a decrease in BD.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Bone China is a ceramic body that is traditionally used in the manufacture of translucent white tableware (Yahya et al. 2018; Toludare et al. 2019). Enhancement of the bone China body structure with nanomaterials is proposed to improve its properties and endow it with more functionality. The growing demand for bone China ceramic has led researchers to improve its physical, chemical, and mechanical properties for decorative and household applications as well as advanced applications (Toludare et al. 2019). The composition of bone China includes bone ash, kaolin, and feldspar at mass contents of 50%, 25%, and 25%, while bone China can be sintered at approximately 1250 °C (Nodeh 2017; Zhang et al. 2016). It is a highly crystalline material that contains approximately 70% crystalline components and 30% glass. The crystallinity of bone China is attributed to the presence of large amount of bone ash, which decomposes to CaO (lime), β-tricalcium phosphate (β-TCP), and H2O at high temperatures (up to 1100 °C) (Toludare et al. 2019; Ahsan et al. 2012). The crystalline phases of bone China are formed fundamentally by anorthite (CaAl2Si2O8), β-TCP (Ca3(PO4)2), and calcium aluminosilicate glass (Yahya et al. 2018; Toludare et al. 2019; Bragança and Bergmann 2008). Because of these features, bone China bodies possess high flexural strength, high chipping resistance, and high modulus of rupture; however, the Young's modulus is lower than that of hard porcelain owing to its high porosity (Batista et al. 2001).
Bone China contains recycled materials, such as bone ash (Bragança and Bergmann 2008; Carus and Bragança 2013). It has, therefore, become highly appealing because of the growing interest in recycling materials to maintain the environment. In recent decades, with the advantages of nanotechnology, researchers have made considerable efforts to enhance the properties of ceramics using nanomaterials. Improving the mechanical and physical characteristics of ceramics is challenging. The selection and concentration of nanoparticles (NPs) play a major role in controlling the material properties. There is an increasing interest in the characteristics of nanomaterials that may lead to the development of new ceramics with superior properties. Numerous studies have focused on the use of NPs, such as Al2O3, ZrO2, TiO2, and SiO2–NPs, in ceramic bodies to improve their characteristics (Rambaldi et al. 2010; Ambrosi et al. 2014; Eliyan et al. 2021; la Garza et al. 2020; Aguilar-Elguézabal et al. 2017; Bi et al. 2017; Aly and Abd‐Elhamid, 2018). NPs can be applied to ceramic bodies in various ways, such as airbrush coating on green or fired specimens and the addition of NPs as raw materials (Rambaldi et al. 2010; Ambrosi et al. 2014; Eliyan et al. 2021; la Garza et al. 2020; Aguilar-Elguézabal et al. 2017).
A significant number of experiments have been conducted to prepare SiO2–NPs due to their unique properties. SiO2–NPs have lower toxicity than other NPs, and this increases their applicability, especially in biochemical fields such as drug delivery (Esim et al. 2019). SiO2–NPs can be utilized to produce a strengthening filler for concrete and other construction composites (Jeelani et al. 2020). Furthermore, the nature of microporous SiO2–NPs has a remarkable effect on CO2 adsorption capacity due to their capillary pores as well as their photodegradation ability due to the available active sites found on SiO2–NPs surface that react with dye molecules through redox reactions, especially during radiation exposure (Aguilar-Elguézabal et al. 2017; Rafigh and Heydarinasab 2017; Xu et al 2021).
The effect of SiO2–NPs on porcelain, whose chemical structure is close to that of bone China ceramics, was examined. Desoky et al. added 10% SiO2–NPs during the preparation of porcelain and concluded that the application of these NPs led to a noticeable change in the density, porosity, and water absorption (WA) of the porcelain. The use of SiO2–NPs has also been found to improve dielectric insulating characteristics (Desouky et al. 2020). Anil et al. reported that porcelain samples containing 2% colloidal SiO2–NPs showed an increase in flexural strength and BD (Anil et al. 2009). However, the effect of SiO2–NPs addition on the properties of bone China has not been studied.
In this work, we aim to synthesize microporous SiO2–NPs with a suitable size and pores at the nanoscale and study their applicability at different concentrations in the production of bone China ceramics. Until now, and to the best of our knowledge, no study investigated the effect of the addition of microporous SiO2–NPs to bone china body. In this respect, the physical, chemical, and mechanical properties of bone China were investigated before and after the addition of SiO2–NPs to determine the effect of this addition on bone China characteristics.
Experimental
Materials
Sodium metasilicate (SMS), cetyltrimethylammonium bromide (CTAB), sodium chloride, silver nitrate, methylene blue dye, polyvinyl methacrylate, and 37% hydrochloric acid were purchased from Sigma-Aldrich and used without further purification.
The raw materials used in bone China preparation included sodium feldspar (albite; NaAlSi3O8; Minas Company, Cairo, Egypt), cattle bones from the local market, Cairo, Egypt, and kaolin (Al2Si2O5(OH)4) from Sedlecky Company, Bozicany, Czech Republic.
Methods
Preparation of microporous SiO2 –NPs
A modified method was used to prepare the microporous SiO2–NPs (Abou Rida and Harb 2014). The precursor (silica wet gel) was prepared using SMS at concentrations of 0.15% and 1.5%. The cationic surfactant, CTAB, with different weights (1 and 2 g) was dissolved in 200 ml SMS solution. The temperature was adjusted to 55 °C under constant stirring at 300 rpm, and the mixture was neutralized using 2.5% HCl in two steps. First, HCl was added dropwise until the semi-gelatinous point at pH 9–9.5, and the mixture was stirred without HCl addition for 10 min to prevent the formation of a strong silica gel. HCl was then added to the mixture until it reached a pH of 3–3.5. NaCl (10 mL, 10%) was added to the mixture under constant stirring for 20 min. After this final addition, the silica wet gel was incubated at 50 °C for 24 h without stirring. The prepared silica wet gel was washed well with distilled water using a centrifuge at 5000 rpm until complete remove of the chloride ions, followed by drying in a microwave oven (800 W) for 10 min. This drying process was conducted seven times at 10 min intervals between microwave exposures. The dried samples were calcined at 650 °C for 3 h at a heating rate of 10 °C min−1 and then milled to form a fine powder using a mortar.
SiO2–NPs characterization
The surface morphology of the prepared SiO2–NPs was observed using a scanning electron microscope (JEOL JSM-6510LB, Tokyo, Japan). Gold layer was used as a coating material for high-resolution imaging. Energy-dispersive X-ray spectroscopy (EDX) analysis was performed to determine the elemental composition.
The sizes of the pores inside the prepared SiO2–NPs were determined using a 200 kV transmission electron microscope (JEOL JEM-2100F, Tokyo, Japan) to confirm the formation of microporous SiO2–NPs. The SiO2–NPs powder was sonicated in distilled water, and the specimen was prepared for transmission electron microscopy (TEM) analysis using a drop of colloidal solution loaded onto a 400-mesh copper grid covered by an amorphous carbon film. The solvent was then evaporated at room temperature (Abou Elmaaty et al. 2018).
The crystallinity of the synthesized SiO2–NPs was determined using an X-ray diffractometer (Bruker D8 ADVANCE, Karlsruhe, Germany). The crystallite size was determined using Scherrer's Equation (Rauf et al. 2016) as follows:
where, B = FWHM (Full Width at Half Maximum); θ = Bragg angle; λ = wavelength radiation; L = Crystalline size and k = 0.9 (Scherrer constant).
The types of bonds present in the particles were determined using a confocal Raman microscope covering a range of 200–1000 cm−1 at a laser wavelength and power of 531.95 nm and 7.2 W (Jasco NRS-4500, Tokyo, Japan). The Raman data were acquired and processed using the Jasco spectroscopy software suite.
Preparation of bone China
The cattle bones under study were boiled in water to remove adhering flesh and cartilage bits and then dried in sunlight for several days. This step was repeated thrice to completely clean the bones. The dried bones were calcined at 900 °C and then milled using mortar to obtain the bone ash. The bone ash, sodium feldspar, and kaolin were sieved using nylon sieve (45 μm). The mixture was grounded for 20 min and then mixed in powder mixing machine (Mxbaoheng, USA) for 30 min at speed of 25 rpm/min.
A blank sample of bone China was prepared by mixing bone ash, feldspar, and kaolin in a weight mixing ratio of 50:25:25. These components were mixed with an appropriate amount of water to create a moldable mixture, which was then pressed into a rectangular acrylic mold (75 × 12 × 4 mm). The specimens were sintered inside an electric muffle furnace kiln at 1320 °C and then polished. These steps were repeated for the specimens prepared using microporous SiO2–NPs, as described in Table 1.
Chemical composition analysis of the raw materials in bone China
The chemical composition of the starting raw materials used in bone China preparation was determined using X-ray fluorescence (XRF). Samples of the raw materials were ground through a 200-mesh sieve, and polyvinyl methacrylate was used as the binding agent. The samples were pressed manually for up to 5 t using a Herzog pressing machine (TP 60/2D, Hamburg, Germany).
Quantitative determination of the oxides, including SiO2, Al2O3, CaO, MgO, TiO2, Fe2O3, Na2O, K2O, P2O5, Cr2O3, MnO, SrO, and SO3, was accomplished via computerized XRF (Philips PW-1400, Amsterdam, Netherlands).
DSC and TGA analysis
The thermal behavior of the prepared bone China samples was investigated by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). DSC analysis was performed to identify the difference in the amount of heat required to increase the temperature of the prepared SiO2–NPs and the bone China samples. TGA was conducted to identify weight loss during the sintering process. A differential scanning calorimeter (Setaram S60/58986, Lyon, France) was used for both DSC and TGA analyses. Then, 20 mg of each sample was analyzed in a N2 atmosphere. Alumina powder was used as a reference material, and the heat rate was maintained at 10 °C min−1.
Determination of AP, BD, and WA
The apparent porosity (AP), bulk density (BD), and water absorption (WA) were determined based on the Archimedes method according to ASTM C373 (2006).
The test specimens were dried in an oven at 150 °C until constant mass and then cooled in a desiccator to then obtain the dry weight (WD). The specimens were placed in a pan and boiled in distilled water for 5 h. They were submerged in water while boiling and then soaked in water completely for an additional 24 h without touching the sides of the vessel. After impregnation of the test specimens, the suspended weight (WS) of each specimen suspended in water was determined. Then, all excess water was removed lightly from the surfaces of the specimens using a moistened, lint-free linen or cotton cloth, and the saturated weight (WM) was obtained. The exterior volume (V), in cubic centimeters, was calculated using Eq. (1).
AP, WA, and BD were calculated using Eqs. (2), (3), and (4):
Three-point bending strength test
A three-point bending test was conducted using a Lloyd LRXPlus material testing machine based on ASTM C1341 (2018) at room temperature and preload speed of 21 mm/min to evaluate the flexural strength of the bone China ceramic samples. Three replicates were used for each sample to carry out bending test through the compression direction.
Self-cleaning test
The photodegradation ability of the prepared bone China was also evaluated. The samples were stained by immersing in a methylene blue dye (0.2 g L−1) for 5 min, and then dried and exposed to sunlight for 24 h. The change in color strength was determined as K/S value in the stained samples using a spectrophotometer (CM3600A; Konica Minolta, Japan). The Kubelka–Munk equation was used for the K/S value calculation in Eq (5) as follows:
where R represents the reflectance of the stained samples, K is the absorption coefficient, and S is the scattering coefficient.
A decrease in the K/S value at 600 nm indicates dye degradation, which is expressed as the self-cleaning capacity (SCC) according to Eq. (6) as follows:
where (K/S)a and (K/S)b represent the color strengths before and after exposure to sunlight, respectively (Ibrahim et al. 2017).
Statistical analysis
The obtained data were analyzed based on randomized design using three replicates. The standard deviation (SD) was obtained using SPSS statistical program to evaluate the variability from the measurement values to the mean.
Results and discussion
SiO2–NPs characterization
SEM and EDX
The size and morphology of the prepared SiO2–NPs were characterized using scanning electron microscopy (SEM) after the calcination process. The results showed that the SMS and CTAB concentrations used in the SiO2–NPs preparation evidently affected the SiO2–NPs size, as displayed in Table 2.
The SiO2–NPs prepared using 1.5% SMS and 1 g CTAB ranged from 34 to 385 nm in size, while the change in SMS concentration to 0.15% caused a decrease in their diameter to 15–370 nm. CTAB at different concentrations (1 and 2 g/200 ml SMS) was used as a nonionic surfactant to control the SiO2 particle size at the nanoscale. The SiO2–NPs diameter decreased noticeably with increasing CTAB concentration, as shown in Table 2 and Figs. 1 and 2. The diameter of the SiO2–NPs synthesized using 2 g CTAB/200 ml SMS (1.5%) ranged from 7 to 96 nm. The formation of SiO2–NPs was also confirmed by EDX analysis, which revealed the presence of both silicon and oxygen, as illustrated in Fig. 2. The SEM micrographs showed a clear variation between the traditional silica and the prepared SiO2–NPs. The prepared SiO2–NPs diameter ranged from 7 to 479 nm, while the size of traditional silica particles was in the range of 1883–28,872 nm, as shown in Fig. 1.

SEM images of a SiO2–NPs prepared using SMS (1.5%) and 1 g CTAB, b SiO2–NPs prepared using SMS (1.5%) and 2 g CTAB at different scales, c SiO2–NPs prepared using SMS (0.15%) and 1 g CTAB, d traditional silica used in bone China, e, f TEM images of SiO2–NPs prepared using SMS (1.5%) and 2 g CTAB at different scales
The histogram of the SiO2–NPs revealed that the NPs prepared using 2 g CTAB/200 ml SMS (1.5%) showed the narrowest size range (7–96 nm) in comparison with the other prepared SiO2–NPs. In contrast, the SiO2–NPs synthesized using 1 g CTAB/200 ml SMS (1.5%) had the largest diameter, ranging from 34 to 385 nm, as shown in Fig. 2. In addition, the histogram showed a variation in NPs diameters between the NPs prepared using CTAB at the concentration of 1 g/200 ml, and SMS at different concentrations of 0.15% and 1.5%. The most of SiO2–NPs prepared with 0.15% SMS were around 50 nm, while the most of the NPs synthesized using 1.5% SMS and CTAB (1 g/200 ml) were around 150 nm. The histogram bins were 100 nm wide and centered at 50, 150, 250, and 350 nm, as shown in Fig. 2a. Furthermore, the histogram bins of the SiO2–NPs prepared using 2 g CTAB/200 ml SMS (1.5%) were 20 nm wide and centered at 10, 30, 50, 70, and 90 nm. SiO2–NPs with diameters of 20–40 nm were considered altogether as particles with a diameter of 30 nm, as illustrated in Fig. 2b.

a Histogram of SiO2–NPs prepared using different amounts of SMS and CTAB, b, c histogram and EDX spectrum of SiO2–NPs prepared using SMS (1.5%) and 2 g CTAB, respectively
TEM analysis
SiO2–NPs can be classified into three types depending on the pore diameter: microporous (less than 2 nm), mesoporous (2–50 nm), and macroporous (more than 50 nm) [26]. TEM analysis was conducted to confirm the microporous nature of the prepared SiO2–NPs. The TEM micrographs showed the formation of microporous SiO2–NPs, including pores with diameters ranging from 1 to 2 nm, as shown in Fig. 1. The porous nature of the prepared SiO2–NPs increases the specific area and porosity because of the nanopores that impeded inside, enabling SiO2–NPs capillary action and higher water absorption capacity than traditional SiO2–NPs (Jafari et al. 2019; Gu et al. 2016).
Raman spectrum analysis
The Raman spectrum of the SiO2–NPs showed peaks at approximately 233 and 577 cm−1, indicating the presence of SiO2. The broad peak at 577 cm−1 refers to the formation of amorphous SiO2–NPs (Biswas et al. 2018; Shabir et al. 2011), as shown in Fig. 3. The observed peak in the region 380–445 cm−1 may be corresponding to O–Si–O signal in SiO2. Another peak appeared around 470 cm−1 may be attributed to Si–Si bond (Melinon et al. 1998).

Raman spectrum of microporous SiO2–NPs
Chemical composition of raw materials
The chemical compositions of the raw materials (bone ash, feldspar, and kaolin) were determined using XRF and are listed in Table 3. Feldspar gave the highest SiO2 percentage (75.10%), followed by kaolin (48.16%) and bone ash (0.98%). Kaolin and bone ash had the highest concentrations of Al2O3 (37.04%) and CaO (40.46%). Moreover, bone China raw materials contained other components in small quantities, as tabulated in Table 3. The obtained results revealed that a higher loss of ignition (LOI) was observed in kaolin (12.98%), followed by bone ash and feldspar. Tironi et al. (2012) reported that the LOI of kaolin was 13.3%, which agrees with the results obtained in this study.
Examination of bone China surface
SEM analysis
The surfaces of samples (B0 and B1) were examined using a scanning electron microscope as shown in Fig. 4. Sample B0 did not contain SiO2–NPs, while B1 contained SiO2–NPs, which revealed the stability of the SiO2–NPs in the bone China ceramic after thermal treatment at 1320 °C. The SiO2–NPs found on the surface of the B1 sample were distributed well and were spherical in shape.

SEM micrographs of a, b surface of bone China without SiO2–NPs (B0), c, d surface of bone China with SiO2–NPs (B1), e, f surfaces of fractured bone China without SiO2–NPs (B0), g, h surfaces of fractured bone China with SiO2–NPs (B1) at different scales
Furthermore, the surfaces of fractured bone china samples were examined using SEM analysis to confirm the presence of SiO2–NPs inside the bone china structure and to compare the microstructure of the B0 and B1 samples. The obtained SEM micrographs showed that microporous SiO2–NPs are found on the B1 sample either on the surface or inside its body, comparing to that of B0, which was clear from the prepared SiO2–NPs.
XRD analysis
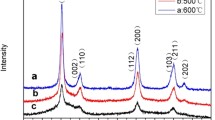
X-ray diffraction (XRD) analysis was conducted for the bone China samples sintered at 1320 °C to identify the different phases formed during the sintering process. B0 showed peaks related to β-TCP, anorthite, and quartz, as shown in Fig. 5 (Biswas et al. 2018; Kara and Stevens 2002; Bimson 1969). The XRD spectra of the SiO2–NPs showed a broad peak in the range of 19.68°–27.32°, indicating the formation of amorphous SiO2–NPs (Rafigh and Heydarinasab 2017; Ghani et al. 2017). The weak and single peak appeared around 27.88° after the broad peak may be attributed to small amounts of crystalline SiO2–NPs. Tsukimura et al. reported that the amorphous nanoparticles are slowly transformed to a crystalline phase because the crystalline phase is more stable than amorphous nanoparticles and found that crystalline SiO2 particles show a strong and sharp peak around 27° (Tsukimura et al. 2021).

XRD spectra of biscuit-fired bone China (B0), bone China with SiO2–NPs (B1), and SiO2–NPs
The fired and sintered sample B1 showed a weak and broad peak at approximately 23.56°, confirming the presence of SiO2–NPs in B1 after thermal treatment at 1320 °C. The obtained peak is weak may be due to the low concentration (1%) of microporous SiO2–NPs. XRD spectrum of B1 showed peaks with lower intensities than B0. In addition, B1 exhibited lower crystallite size (46.83 nm) than B0 (47.43 nm), indicating the decrease in bone china crystallinity nature as a result of SiO2–NPs addition. The low decrease in crystallite size and crystallinity phases may be also due to the low concentration of SiO2–NPs present in B1 sample. (El-Didamony et al. 2020) reported similar results and found that the intensity of the quartz phase in ceramic engobes decreased due to the addition of SiO2–NPs.
Characterization of bone China under study
Bending test
The mechanical characteristics of the bone China samples with SiO2–NPs at different concentrations (1, 5, 10, and 15%) were identified via a bending test in comparison with those of bone China free of SiO2–NPs. Figure 6 shows the effect of the SiO2–Nps concentration on the flexural strength, which is defined as the maximum bending stress that can be applied to a material before it yields in a flexure test. B1 showed a remarkable improvement in flexural strength, which increased to 38.38 MPa compared with B0 (35.46 MPa). On the other hand, the addition of SiO2–NPs at concentrations of 5, 10, and 15% led to a notable decrease in the flexural strength, as shown in Fig. 6. SiO2–NPs at a concentration of 1% may recover the macropores found inside the bone China body, which enhances the flexural strength (Alrumaih 2019). SEM micrographs of the surface of the fractured B1 sample confirmed the presence of microporous SiO2–NPs in the macropores of bone china body as shown in Fig. 4. Therefore, the addition of SiO2–NPs at low concentration (1%) enhances the bone china strength. However, the increase in SiO2–NPs concentrations of 1% maximizes the porosity, which decreases the bone China flexural strength because of the porous nature of SiO2–NPs (Gu et al. 2016).

a Flexural strength, b Young's modulus of pure and nanostructured bone China ceramic samples with SiO2–NPs concentrations of 0%, 1%, 5%, 10%, and 15%
Figure 6 also shows the effect of the SiO2–NPs on Young's modulus of the bone China ceramics. Young's modulus describes the elastic properties of a solid material undergoing compression or tension in only one direction. The Young's modulus of B1 exhibited an evident increase (to 10%) compared with that of B0. Moreover, the bone China samples containing SiO2–NPs at concentrations of 5, 10, and 15% (B5, B10, and B15, respectively) showed a noticeable decrease ranging from 11 to 60% in comparison with the bone China samples that did not have the SiO2 NPs, as shown in Fig. 6. The decrease in Young's modulus may be due to the reduction in the anorthite (CaO·Al2O3·2SiO2) content caused by the addition of SiO2–NPs. Pabst et al. reported that the elastic constant (Young's modulus) of dense, single-phase, isotropic polycrystalline plagioclase depends on the anorthite content (mol.%) and found that the elastic constant increases with the anorthite content (Pabst et al. 2015). In the current study, the addition of SiO2–NPs at a low concentration (1%) led to an increase in elasticity, which reveals that SiO2–NPs could be added at low concentrations to improve the mechanical properties of bone China in a cost-effective process. Based on bending test, the optimum concentration of SiO2–NPs in bone China preparation was 1%, so B1 specimens were selected to be subjected to the following tests.
Physical characteristics
The effect of the addition of SiO2–NPs at the optimum concentration (1%) on the physical characteristics of B0 and B1, namely, BD, AP, and WA, was investigated. The AP and WA percentages of B1 were higher than those of B0, as shown in Table 4. The addition of SiO2–NPs caused a decrease in the BD value compared to the original bone China (prepared without SiO2–NPs addition). El-Didamony et al. studied the physical effect of nano-silica on ceramic engobes and found that BD decreases with an increase in nano-silica concentration, which agrees with the results of the present work (El-Didamony et al. 2020). A slight decrease in BD may be due to the low concentration (1%) of SiO2–NPs used in B1 preparation, which did not significantly affect the BD of bone china. Furthermore, the capillary forces existing in the nanopores filled them with water (Kumar et al. 2015) and led to the increases in WA and AP due to the use of microporous SiO2–NPs, as seen in the TEM micrographs and displayed in Fig. 1.
Self-cleaning
The photodegradation ability of B0 and B1 samples was evaluated against methylene blue dye by direct exposure to ultraviolet radiation from sunlight. Photocatalytic activity is affected by the particle size, crystalline structure, porosity, and specific surface area of the material (Zuo et al. 2014). In photocatalytic activity, UV and visible rays were used to motivate redox reactions via activating the photocatalyst (Zhou et al. 2021). B1 showed an obvious decrease in the blue color and a degradation percentage value of 88.23%, indicating the applicability of the prepared bone China with SiO2–NPs as a photodegradable material. The photocatalytic activity of SiO2–NPs may be due to their porosity and particle size (Abou Elmaaty et al. 2021). The B0 sample showed a lower degradation percentage value of 79.83% than the B1 specimen, indicating that the presence of SiO2–NPs in the microstructure of bone china improved its photodegradation ability.
DSC and TGA
TGA/DSC analysis was performed on B0 and B1, as shown in Fig. 7. The bone China samples were subjected to a temperature range of 20–1550 °C at a heating rate of 10 °C min−1 to evaluate their thermal performance. The TGA/DSC curves of B0 showed four endothermic peaks at 272.5, 370.6, 575.3, and 1435 °C. The first and second peaks (272.5 °C and 370.6 °C, respectively) were due to the removal of water. The endothermic peak at 575.3 °C was a major peak related to the removal of the crystalline water of bone China porcelain minerals, which is the first step in the composition of bone China porcelain structures (Chakraborty 2014; Guo et al. 2020). The endothermic effect at 1435 °C is related to the crystallization of the cristobalite phase (Colombo et al. 2018; Ajanaku et al. 2016; Gualtieri 2007).

TGA/DSC curves of bone China samples a B0, b B1, c SiO2–NPs SiO2–NPs
The TGA/DSC curves of B1 showed nine endothermic peaks at 66.8, 265.1, 576.4, 776.6, 942.9, 1042.3, 1260, 1369.9, and 1427.3 °C, as illustrated in Fig. 7. The first peak (66.8 °C) may correspond to moisture loss (Belhouchet et al. 2019). The second peak (265.1 °C) was due to the removal of water. The peaks at 576.4 °C and 776.6 °C were related to the removal of any chemically bonded water with bone China minerals and to the formation of meta-kaolinite from kaolinite (Chakraborty 2014; Ajanaku et al. 2016). The peaks observed at 942.9 °C and 1042.3 °C may be due to the crystallization of apatite (Riaz et al. 2014). The endothermic peak at 1260 °C signaled the transformation of the spinel phase to the mullite phase (Colombo et al. 2018; Gualtieri 2007). Finally, the endothermic peaks at 1369.9 °C and 1427.3 °C were related to the appearance of the cristobalite phase and the emergence of SiO2–NPs in the sample (Colombo et al. 2018; Ajanaku et al. 2016; Aras and Kristaly 2019; Yang et al. 2012).
Conclusion
In this study, microporous SiO2–NPs were utilized to improve the physical, chemical, and mechanical properties of bone China ceramics. They were prepared using a cost-effective method utilizing SMS as a precursor in the presence of CTAB as a surfactant. The optimum concentrations of SMS and CTAB required for SiO2–NPs preparation at the nanoscale were investigated. The SEM and TEM micrographs, XRD patterns, and Raman spectrum confirmed the formation of microporous SiO2–NPs. These SiO2–NPs were added to the raw materials of bone China at various concentrations to study their effect on bone China ceramic properties. The obtained results showed that B1 had the highest flexural strength and Young's modulus values among all the bone China samples and was selected for further tests. B1 showed a remarkable increase in AP and WA, whereas BD decreased. In addition, the photodegradation ability of B1 specimens was evaluated against methylene blue dye, which decomposed clearly on the B1 surface after exposure to sunlight. Moreover, the thermal behavior of the B0 and B1 samples was studied using TGA/DSC analyses.
The obtained results revealed that SiO2–NPs addition may enable bone China ceramics to be used in new fields, such as filters, bio-scaffolding, and drug delivery. Bone China contains recycled materials; therefore, it is an eco-friendly and sustainable material that can be used in advanced applications.
Data availability
The data presented in this study are available upon request from the corresponding author.
References
Abou Elmaaty T, El-Nagare K, Raouf S, Abdelfattah K, El-Kadi S, Abdelaziz E (2018) One-step green approach for functional printing and finishing of textiles using silver and gold NPs. RSC Adv 8:25546–25557. https://doi.org/10.1039/C8RA02573H
Abou Elmaaty T, Elsisi HG, Elsayad GM, Elhadad HH, Sayed-Ahmed K, Plutino MR (2021) Fabrication of new multifunctional cotton/lycra composites protective textiles through deposition of nano silica coating. Polymers 13:1–12. https://doi.org/10.3390/polym13172888
Abou Rida M, Harb F (2014) Synthesis and characterization of amorphous silica nanoparitcles from aqueous silicates uisng cationic surfactants. J Met Mater Miner 24:37–42. https://doi.org/10.14456/jmmm.2014.7
Aguilar-Elguézabal A, Román-Aguirre M, la Torre-Sáenz L, Pizá-Ruiz P, Bocanegra-Bernal M (2017) Synthesis of CoAl2O4/Al2O3 nanoparticles for ceramic blue pigments. Ceram Int 43:15254–15257. https://doi.org/10.1016/j.ceramint.2017.08.062
Ahsan M, Dewan AH, Mustafi S, Ahmed S (2012) Characterization of crystalline phases of bone ash. Bangladesh J Sci Ind Res 47:265–268. https://doi.org/10.3329/bjsir.v47i3.13057
Ajanaku KO, Aladesuyi O, Mousumi P, Swapan KD (2016) Evaluation of Nigerian source of kaolin as a raw material for mullite synthesis. Orient J Chem 32:1571–1582. https://doi.org/10.13005/ojc/320333
Alrumaih MA (2019) Investigation of the effect of nano SiO2 on porosity. Doctoral dissertation, University of Dayton
Aly HF, Abd-Elhamid AI (2018) Photocatalytic degradation of methylene blue dye using silica oxide nanoparticles as a catalyst. Water Environ Res 90:807–817. https://doi.org/10.2175/106143017X15131012187953
Ambrosi M, Santoni S, Giorgi R, Fratini E, Toccafondi N, Baglioni P (2014) High-performance and anti-stain coating for porcelain stoneware tiles based on nanostructured zirconium compounds. J Colloid Interface Sci 432:117–127. https://doi.org/10.1016/j.jcis.2014.07.002
Anil A, Mohamed AP, Maiti KN (2009) Evaluation of dry as well as thermo-mechanical properties of porcelain compositions containing colloidal silica as an additive. Trans Indian Ceram Soc 68:99–104. https://doi.org/10.1080/0371750X.2009.11082165
Aras A, Kristaly F (2019) α-Cristobalite formation in ceramic tile and sewage pipe bodies derived from Westerwald ball clay and its effect on elastic-properties. Appl Clay Sci 178:105126. https://doi.org/10.1016/j.clay.2019.105126
ASTM (2006) ASTM C 373-88—standard test method for water absorption, bulk density, apparent porosity, and apparent specific gravity of fired whiteware products
ASTM (2018) ASTM C 1341—standard test method for flexural properties of continuous fiber-reinforced
Batista SAF, Messer PF, Hand RJ (2001) Fracture toughness of bone China and hard porcelain. Br Ceram Trans 100:256–259. https://doi.org/10.1179/bct.2001.100.6.256
Belhouchet K, Bayadi A, Belhouchet H, Romero M (2019) Improvement of mechanical and dielectric properties of porcelain insulators using economic raw materials. Bol De La Soc Esp De Cerám y Vidr 58:28–37. https://doi.org/10.1016/j.bsecv.2018.05.004
Bi Y, Zaikova T, Schoepf J, Herckes P, Hutchison JE, Westerhoff EP (2017) The efficacy and environmental implications of engineered TiO2 nanoparticles in a commercial floor coating. Sci Nano 4:2030–2042. https://doi.org/10.1039/c7en00649g
Bimson M (1969) The examination of ceramics by X-ray powder diffraction. Stud Conserv 14:83–89. https://doi.org/10.1179/sic.1969.008
Biswas RK, Khan P, Mukherjee S, Mukhopadhyay AK, Ghosh J, Muraleedharan K (2018) Study of short range structure of amorphous silica from PDF using Ag radiation in laboratory XRD system, RAMAN and NEXAFS. J Non Cryst Solids 488:1–9. https://doi.org/10.1016/j.jnoncrysol.2018.02.037
Bragança SR, Bergmann CP (2008) A comparative study between bone China and hard porcelain. Ind Ceram 28:189
Carus LA, Bragança SR (2013) Bone China formulated with waste glass. Adv Appl Ceram 112:169–175. https://doi.org/10.1179/1743676112Y.0000000065
Chakraborty AK (2014) Phase transformation of kaolinite clay. Springer. https://doi.org/10.1007/978-81-322-1154-9
Colombo AL, Laraz FB, da Silva JF, Lima RHC, Berti LF (2018) Evaluation of kaolin and bauxite from the amazonian region addition on mechanical-, physical-, chemical properties and microstructural characterization of red ceramic industry clay of manaus metropolitan region. Matéria (rio Janeiro) 23:1571–1582. https://doi.org/10.1590/S1517-707620180003.0539
Desouky OA, Belal E, El-Gamal SMA, Abd-Allah MA, Eliyan T (2020) Improving the insulating and physical characteristics of HV porcelain dielectric materials using nano-silica. J Mater Sci Mater Electron 31:12649–12660. https://doi.org/10.1007/s10854-020-03815-8
El-Didamony H, El-Fadaly E, Amer AA, Abazeed IH (2020) Synthesis and characterization of low cost nanosilica from sodium silicate solution and their applications in ceramic engobes. Boletín De La Sociedad Española De Cerámica y Vidrio 59:31–43
Eliyan T, Desouky OA, Belal E, Abd-Allah MA, El-Gamal SM (2021) Utilization of zirconia nanoparticles for improving the electrical and physical characteristics of HV porcelain insulating material. Electr Eng 103:1385–1399. https://doi.org/10.1007/s00202-020-01166-5
Esim O, Kurbanoglu S, Savaser A, Ozkan SA, Ozkan Y (2019) Nanomaterials for drug delivery systems. In: New developments in nanosensors for pharmaceutical analysis. Elsevier. https://doi.org/10.1016/B978-0-12-816144-9.00009-2
Ghani NNA, Saeed MA, Hashim IH (2017) Thermoluminescence (TL) response of silica nanoparticles subjected to 50 Gy gamma irradiation. Malays J Fundam Appl Sci 13:178–180
Gu Y, Li B, Chen M (2016) An experimental study on the cavitation of water with effects of SiO2 nanoparticles. Exp Therm Fluid Sci 79:195–201. https://doi.org/10.1016/j.expthermflusci.2016.07.009
Gualtieri AF (2007) Thermal behavior of the raw materials forming porcelain stoneware mixtures by combined optical and in situ x-ray dilatometry. J Am Ceram Soc 90:1222–1231. https://doi.org/10.1111/j.1551-2916.2007.01614.x
Guo J, Xu S, Qin Y, Li Y, Lin X, He C, Dai S (2020) The temperature influence on the phase behavior of ionic liquid based aqueous two-phase systems and its extraction efficiency of 2-chlorophenol. Fluid Phase Equilib 506:112394. https://doi.org/10.1016/j.fluid.2019.112394
Ibrahim NA, Eid BM, Abd El-Aziz E, Abou Elmaaty TM, Ramadan SM (2017) Loading of chitosan-nano metal oxide hybrids onto cotton/polyester fabrics to impart permanent and effective multifunctions. Int J Biol Macromol 105:769–776. https://doi.org/10.1016/j.ijbiomac.2017.07.099
Jafari S, Derakhshankhah H, Alaei L, Fattahi A, Varnamkhasti BS, Saboury AA (2019) Mesoporous silica nanoparticles for therapeutic/diagnostic applications. Biomed Pharm 109:1100–1111. https://doi.org/10.1016/j.biopha.2018.10.167
Jeelani PG, Mulay P, Venkat R, Ramalingam C (2020) Multifaceted application of silica nanoparticles. A review. SILICON 12:1337–1354. https://doi.org/10.1007/s12633-019-00229-y
Kara A, Stevens R (2002) Characterisation of biscuit fired bone China body microstructure. Part I: XRD and SEM of crystalline phases. J Eur Ceram Soc 22:731–736. https://doi.org/10.1016/S0955-2219(01)00371-5
Kumar S, Kumari M, Mallick MA, Swain BS, Sobral A, Dutta PK (2015) Preparation and characterization of microporous bionanocomposites for biomedical applications. Asian Chitin J 11:23–26
la Garza DA, Rodríguez EA, Contreras JE, López-Perales JF, Díaz-Tato L, Ruiz-Valdés JJ, Vázquez-Rodríguez FJ, Álvarez-Méndez A (2020) Effect of nano-TiO2 content on the mechano-physical properties of electro-technical porcelain. Mater Chem Phys 254:123469. https://doi.org/10.1016/j.matchemphys.2020.123469
Melinon P, Keghelian P, Prével B, Dupuis V, Perez A (1998) Structural, vibrational, and optical properties of silicon cluster assembled films. J Chem Phys 108:4607–4613. https://doi.org/10.1063/1.475871
Nodeh AA (2017) Influence of bone porcelain scraps on the physical characteristics and phase composition of a hard porcelain body. Boletín De La Sociedad Española De Cerámica y Vidrio 56:113–118. https://doi.org/10.1016/j.bsecv.2017.04.003
Pabst W, Gregorova E, Rambaldi E, Bignozzi MC (2015) Effective elastic constants of plagioclase feldspar aggregates in dependence of the anorthite content: a concise review. Ceram-Silikaty 59:326–330
Rafigh SM, Heydarinasab A (2017) Mesoporous chitosan-SiO2 nanoparticles: synthesis, characterization, and CO2 adsorption capacity. ACS Sustain Chem 5:10379–10386. https://doi.org/10.1021/acssuschemeng.7b02388
Rambaldi E, Tucci A, Esposito L, Naldi D, Timellini G (2010) Nano-oxides to improve the surface properties of ceramic tiles. Boletín la Soc. Española Cerámica y Vidr 49:253–258
Rauf N, Tahir D, Arbiansyah M (2016) Structural analysis of bioceramic materials for denture application. AIP Conf Proc. https://doi.org/10.1063/1.4943725
Riaz M, Zia R, Saleemi F, Bashir F, Hossain T, Kayani Z (2014) In vitro evaluation of bioactivity of SiO2–CaO–P2O5–Na2O–CaF2–ZnO glass-ceramics. Mater Sci Pol 32:364–374
Shabir Q, Pokale A, Loni A, Johnson DR, Canham LT, Fenollosa R, Tymczenko M, Rodríguez I, Meseguer F, Cros A, Cantarero A (2011) Medically biodegradable hydrogenated amorphous silicon microspheres. SILICON 3:173–176. https://doi.org/10.1007/s12633-011-9097-4
Tsukimura K, Miyoshi Y, Takagi T, Suzuki M, Wada SI (2021) Amorphous nanoparticles in clays, soils and marine sediments analyzed with a small angle X-ray scattering (SAXS) method. Sci Rep 11:1–11
Tironi A, Trezza MA, Irassar EF, Scian AN (2012) Thermal treatment of kaolin: effect on the pozzolanic activity. Procedia Mater Sci 1:343–350. https://doi.org/10.1016/j.mspro.2012.06.046
Toludare TS, Owoeye SS, Kenneth-Emehige A, Isinkaye OE (2019) Microstructure evolution and physico-mechanical properties of bone China porcelain compositions using two selected kaolinite clays from Nigeria. Sci Afr 3:e00066
Xu S, Zhu Q, Xu S, Yuan M, Lin X, Lin W, Lin W, Qin Y, Li Y (2021) The phase behavior of n-ethylpyridinium tetrafluoroborate and sodium-based salts ATPS and its application in 2-chlorophenol extraction. Chin J Chem Eng 33:76–82. https://doi.org/10.1016/j.cjche.2020.07.024
Yahya H, Mois ARA, Ahmad A (2018) Fabrication and characterization of anorthite-based porcelain using malaysian mineral resources. Malays 1:106–111. https://doi.org/10.30967/ijcrset.1.S1.2018.106-111
Yang S, Yuan P, He H, Qin Z, Zhou Q, Zhu J, Liu D (2012) Effect of reaction temperature on grafting of γ-aminopropyl triethoxysilane (APTES) onto kaolinite. Appl Clay Sci 62:8–14. https://doi.org/10.1016/j.clay.2012.04.006
Zhang Y, Zhou N, Li W, Li J, Nian S, Li X, Sui J (2016) Fabrication and characterization of bone China using synthetic bone powder as raw materials. Ceram Int 42:14910–14917. https://doi.org/10.1016/j.ceramint.2016.06.131
Zhou L, Dai S, Xu S, She Y, Li Y, Leveneur S, Qin Y (2021) Piezoelectric effect synergistically enhances the performance of Ti32-oxo-cluster/BaTiO3/CuS pn heterojunction photocatalytic degradation of pollutants. Appl Catal B 291:120019. https://doi.org/10.1016/j.apcatb.2021.120019
Zuo R, Du G, Zhang W, Liu L, Liu Y, Mei L, Li Z (2014) Photocatalytic degradation of methylene blue using TiO2 impregnated diatomite. Adv Mater Sci Eng. https://doi.org/10.1155/2014/170148
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). The authors received no financial support for the research, authorship, and/or publication of this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abou Elmaaty, T.M., Sayed-Ahmed, K., El Gohari, M.M. et al. Enhancing the properties of bone China ceramics by treatment with microporous SiO2 nanoparticles. Chem. Pap. 76, 5879–5891 (2022). https://doi.org/10.1007/s11696-022-02296-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-022-02296-9