
Abstract
COVID-19 is an unprecedented pandemic threatening global health, and variants were discovered rapidly after the pandemic. The two variants, namely the SARS-CoV-2 B.1.1.7 (Alpha) and P.1 (Gamma), were formed by the mutations in the receptor binding domain of spike glycoprotein (SGP). These two variants are known to possess a high binding affinity with the angiotensin-converting enzyme 2. Amidst the rapid spread of these mutant strains, research and development of novel molecules become tedious and labour-intensive. Imidazole and benzimidazole scaffolds were selected in this study based on their unique structural features and electron-rich environment, resulting in increased affinity against a variety of therapeutic targets. In the current study, imidazole- and benzimidazole-based anti-parasitic drugs are repurposed against SARS-CoV-2 Alpha and Gamma variant spike glycoproteins using computational strategies. Out of the screened 15 molecules, flubendazole and mebendazole have exhibited promising binding features to the two receptors (PDB ID: 7NEH and 7NXC), as evidenced by their glide score and binding free energy. The results are compared with that of the two standard drugs, remdesivir and hydroxychloroquine. Flubendazole and mebendazole have become convenient treatment options against mutant lineages of SARS-CoV-2. The edge of the flubendazole was further established by its stability in MD simulation conducted for 100 ns employing GROMACS software. Further, in vitro and in vivo studies are essential to understand, if flubendazole and mebendazole indeed hold the promise to manage SARS-CoV-2 mutant stains.
Graphic abstract

Similar content being viewed by others


Introduction
The rampant dissemination of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) or COVID-19 amongst humans hastened its molecular evolution (Hui et al. 2020). SARS-CoV-2 has been accumulating mutations at a rate of up to two nucleotide modifications per month. Current SARS-CoV-2 isolates have at least 20 nucleotide variations in their genomic sequences than the earliest isolates (Faria et al. 2021). The majority of the heterogeneity observed in SARS-CoV-2 isolates is due to mutations in the receptor binding domain (RBD) of spike glycoprotein (SGP), which facilitates virion adhesion to the angiotensin-converting enzyme 2 (ACE2) on human cell surfaces (Shang et al. 2020, Burki 2021).
The appearance of the SARS-CoV-2, B.1.1.7 lineage (UK strain) (Alpha), discovered in early December 2020 by the UK Consortium of COVID-19 Genomics, is one example of rapid viral (Kupferschmidt 2020) molecular evolution . The Alpha variant is interesting because it has 17 mutations in its genomic sequence, and 8 out of them are found in the gene that encodes the SGP. The Alpha lineage phenotype has also received recognition because of its rapid transmission rates in humans than the other recognized SARS-CoV-2 lineages. N501Y has been recognized as a residue that enhances the binding affinity with human ACE2. As per the studies by Faria et al. 2021, it is clear that the N501Y mutation observed in the Alpha lineage at the Spike gene covers one of the six primary interacting amino acid residues within the SGP (Starr et al. 2020; Sternberg and Naujokat 2020).
One more strain of SARS-CoV-2, P.1 lineage (Brazil strain) (Gamma), or 20 J/401Y.V3 variant, was first identified by the National Institute of Infectious Diseases (NIID) on 6 January 2021 in Japan (Wang et al. 2021; Maggi et al. 1249). This variant also has 17 mutations, 10 of which are in its SGP. However, three mutations were designated to be of particular concern: K417T, E484K, and N501Y. In March 2021, Gamma lineage has been designated as a deadly variant (10–80% lethal), producing nearly 10 times more viral load than in other COVID-19 patients and exhibited 1.4–2.2 times higher transmissibility. Besides, it can evade 25–61% of immunity from previous SARS-CoV-2 diseases, increasing the probability of recurring infection. These two mutant lineages were classified as variants of concern by Centers for Drug Control and Prevention (CDC). In addition, WHO has currently discovered some standard therapies using some drug combinations, investigational drugs, and vaccines (Villoutreix et al. 1695; Ramírez-Salinas et al. 2020). Besides, many available therapeutics to reposition against SARS-CoV-2 infection have been subjected to clinical trials (Hoffmann et al. 2021; Abdool Karim and Oliveira 2021). Nevertheless, the search for other novel molecules cannot pause while waiting for the outcomes because the demand for new therapeutic agents is inflating (Singh et al. 2021).
Since the commencement of the SARS-CoV-2 infection, various strategies have been implemented to tackle the pandemic. One of them consists of drug repositioning using computational techniques (Novac 2013). With this approach, the emergence of commercial medications with therapeutic potential against SARS-CoV-2 infection has been proposed. One such example is the repurposing of imidazole- and benzimidazole-based anti-parasitic drugs. Imidazole and benzimidazole rings are the most important nitrogen-containing heterocycles, which are widely explored and utilized by the pharmaceutical industry for the drug discovery and development. Chemically, imidazole is a five-membered aromatic heterocycle present in the biological building blocks, such as amino acid (histidine), histamine, purines, and biotin (Fei and Zhou 2013), whereas benzimidazole is a six-membered bicyclic heteroaromatic cycle present in vitamin B12 and many other biological compounds (Wright 1951; Barker et al. 1960).
Both the heterocycles contain two nitrogen atoms with amphoteric nature and exist in two equivalent tautomeric forms. In addition, the electron-rich nitrogen heterocycles could readily accept/donate electrons and form diverse weak interactions easily. These unique characteristic features are beneficial for the imidazole and benzimidazole derivatives to readily bind with a variety of therapeutic targets, thereby exhibiting broad range of pharmacological or biological activities (Wright 1951; Bhatnagar et al. 2011; Ingle and Magar 2011; Gaba et al. 2010, 2014; Narasimhan et al. 2011; Yadav and Ganguly 2015). Their ubiquitous properties and key role in management of diseases have attracted special interest in imidazole- and benzimidazole-based medicinal chemistry.
In this paper, we performed docking and binding free energy calculations of available imidazole- and benzimidazole-based anti-parasitic drugs within the catalytic pockets of SARS-CoV-2 Alpha and Gamma variant SGP.
Materials and methods
Preparation of protein
To circumvent the current pandemic of SARS-CoV-2 mutants, X-ray crystallographic protein structures of mutated SGPs, with accession IDs 7NEH (resolution 1.77 Å; R-value work 0.188 Å; R-value free 0.198 Å) (Supasa et al. 2021) and 7NXC (resolution 3.14 Å; R-value work 0.226 Å; R-value free 0.278 Å) (Dejnirattisai et al. 2021), were downloaded from the RCSB protein data bank. Crystal structures were prepared using a protein preparation wizard available in the Maestro interface (Sastry et al. 2013). While preparing, hydrogen atoms were added, and bond orders were assigned using the PROPKA method (Beard et al. 2021). Further, filling of missed side chains and loops was achieved using the module Prime (Iwaloye et al. 2020). Minimization of protein structure was carried out using OPLS3e force field (Roos et al. 2019). A 10 Å grid boxes for the two receptors were generated using the wizard Receptor Grid Generation available in Glide module (Halgren et al. 2004) by selecting the centroid of active amino acid residues Tyr52, Val58, Ser60, Arg61 Gly158, and Asn159 for PDB ID: 7NEH (Supasa et al. xxxx) and Lys26, Asn53, Asn103, Asn343, and Leu368 for PDB ID: 7NXC (Dejnirattisai et al. 2021).
Preparation of ligand
A total of 15 reported imidazole- and benzimidazole-based anti-parasitic drugs were selected for molecular docking studies to discover the potential therapeutics against SARS-CoV-2 mutants. The ligands and standard drugs (hydroxychloroquine and remdesivir) used are given in Table 1. The 3D structures were taken from the PubChem database in .sdf format. Geometry optimization and minimization of the molecules were carried out using the algorithms available in the LigPrep Wizard. Ligands were prepared by adding hydrogens, desalting and ionizing at pH 7 ± 2. OPLS3e force field was used for energy minimization of ligands by keeping root-mean-square deviation (RMSD) cut-off 0.01 Å and standard energy functions of molecular mechanics to produce low-energy conformers (Friesner et al. 2006).
Molecular docking studies
Molecular docking is an in silico technique used to discover the critical amino acid interactions between the chosen receptors and low-energy conformers prepared using LigPrep. Protein–ligand interactions were categorized by the scoring function, which indirectly demonstrates the ligand’s binding affinity with the chosen receptor. Glide extra-precision (XP) docking protocol was employed without applying any constraints. XP docking was carried out to speculate the ligand efficiency and binding affinity as potent inhibitors of SARS-CoV-2 mutants. Maestro interface was used to visualize the poses of docked complexes.
where a and b = coefficient constants of Coulombic and van der Waals energy; Metal = binding with metals; Lipo = lipophilic term; Hbond = hydrogen bonding; RotB = rotatable bonds penalty; BuryP = buried polar groups penalty; Site = catalytic pocket polar interaction.
Binding free energy (BFG) calculations
BFG was calculated to determine the strength of the ligands using molecular mechanics-generalized born surface area (MM-GBSA) technique, Prime module (Jacobson et al. 2004). Energy minimization of protein–ligand docked complex is done using the local optimization feature present in the Prime module. Simulation of the complexes was achieved by input ligand partial charges without smearing any constraints by employing VSGB 2.0 energy model (Li et al. 2011). The energy of the docked complexes was calculated using OPLS3e force field. The BFG (Gbind) was calculated using the following formula:
Gcpx = protein–ligand complex free energy; Glig = ligand energy; Grec = unbound receptor energy.
Molecular dynamic (MD) simulations
GROMACS 2018.1 software package was used to perform a 100 ns of MD simulation for flubendazole/7NEH complex. Gromos54a7 force field and PRODRG web server were utilized for the energy optimization and constructing structural topologies of the complex (Schüttelkopf and Aalten 2004). SPC water model and cubic boundary box were employed for the simulation of a system. Neutralization was attained using inorganic ions such as sodium (Na+) and chloride (Cl−) ions, controlling periodic limit constraints. System equilibration was attained at a pressure of 1 atm and temperature of 300 K (Berendsen et al. 1984). Energy minimization of the system was done using the steepest descent algorithm.
The command lines g_gyrate, g_cluster, g_rmsd, and g_rmsf were used for analysing radius of gyration (ROG), RMSD, mean square displacement, and root-mean-square fluctuation (RMSF), respectively. The docked complex flubendazole/7NEH was employed to observe the migration over the simulation time.
Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) calculation
Binding free energy calculation of flubendazole/7NEH complex was performed using the MM-PBSA approach (Kollman et al. 2000). This technique calculates the binding free energy of the end states directly, by omitting the intermediate states simulation, and integrates the molecular mechanical energies with the continuum solvent strategies. Explicit water molecules were eliminated from the trajectories.
The binding free energy (ΔGbind) was calculated using the following formula:
where TΔSconf is the solute entropies calculated by the module NMODE in GROMACS.
ΔEgas indicates the interaction energy between protein and ligand in the gaseous phase:
where ΔEelec and ΔEvdW denote the protein–ligand electrostatic and van der Waal interactions, respectively.
Further, total solvation free energy (ΔGsolv) is the sum of a polar (ΔGNP) and electrostatic (ΔGPB) solvation free energies (Luo et al. 2002):
Results and discussion
Molecular docking analysis
To determine a possible drug candidate to combat SARS-CoV-2 mutant strains, molecular docking studies were employed using 15 imidazole- and benzimidazole-based anti-parasitic drugs on the catalytic sites of Alpha and Gamma variant SGP. All these compounds were docked against SARS-CoV-2 Alpha and Gamma variant SPGs and ranked based on their glide score (Gscore). The docking results were compared with standards (hydroxychloroquine and remdesivir) used in the study and are summarized in Tables 2 and 3.
Interaction analysis of ligands with SARS-CoV-2 Alpha variant SGP
All the compounds exhibited significant activity in the range of − 2.367 to − 4.418 kcal/mol in the catalytic pocket of SARS-CoV-2 Alpha variant SGP (Table 2 and Supplementary Figure S1a-q). Gratifyingly, all the imidazole- and benzimidazole-based anti-parasitic drugs showed excellent results in computational docking studies. Glide score exhibited a good correlation with glide energy (R2 = 0.8541), glide emodel (R2 = 0.8482), and Hbond (R2 = 0.8398), while the moderate correlation with glide van der Waal (R2 = 0.7601) and glide Coulomb (R2 = 0.7284). Further, the ligands exhibited significant interactions at the same catalytic site compared to the standard drugs hydroxychloroquine and remdesivir. Hydroxychloroquine exhibited significant interactions with the Ser349 and Tyr351 with a Gscore of − 3.471 kcal/mol (Supplementary Figure S1p), whereas remdesivir has exhibited significant interactions with amino acid residues Ser349, Ala352, Asn354, and Asn450 with a Gscore of − 6.266 kcal/mol (Supplementary Figure S1q).
Out of 15 test compounds, flubendazole exhibited more potent inhibitory activity as indicated with its high Gscore of − 4.481 kcal/mol. The other compound, mebendazole, also showed a promising Gscore of − 4.066 kcal/mol within the catalytic pocket of protein structure.
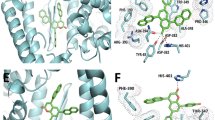
Flubendazole has shown five hydrogen bonding (HB) interactions similar to that of remdesivir with the critical interacting residues of the catalytic pocket of SARS-CoV-2 Alpha lineage SGP (Fig. 1). The oxygen of the terminal ester attached at position two of benzimidazole formed a HB network with the protonated amino group of Asn354 ((> C = O)O⋯HN(H), 2.41 Å), whereas -OH and -NH of Ser349 residue involved in the formation of two HB interactions with the carbonyl group of carbamate linker attached to benzimidazole group at position two (> C = O⋯HN(H), 1.85 Å; > C = O⋯HO, 2.09 Å). The carbonyl group between the two aromatic rings has shown two HB interactions with the -NH moieties of Asn448 (> C = O⋯HN(H), 1.79 Å) and Asn450 (> C = O⋯HN(H), 2.00 Å), respectively. Moreover, flubendazole is stabilized by two π–π stacking interactions, observed between electron clouds of the benzimidazole nucleus and the phenyl ring of Tyr451.

3D interaction diagram of flubendazole with amino acid residues necessary for the inhibition of SARS-CoV-2 Alpha variant
Further, the hydrophilic–hydrophobic map of the flubendazole reveals that the nitrogen moieties present in the benzimidazole ring and HNO = C(O)CH3 fragment are located well in the hydrophilic region. In contrast, phenyl rings are buried in the hydrophobic part of the receptor. On the other hand, the fluorobenzene group at position five of the benzimidazole ring is well buried in the hydrophilic pocket because of the high dielectric constant of fluorinated compounds compared to the simple aromatic rings (Biffinger et al. 2004) (Supplementary Figure S2).
Interaction analysis of ligands with SARS-CoV-2 Gamma variant SGP
Comparison of the docked poses of conformationally flexible ligands along with the standards revealed similar kinds of binding poses within the catalytic pocket of SARS-CoV-2 Gamma variant SGP (Table 3 and Supplementary Figure S3). Glide score of selected analogues in the catalytic pocket of SARS-CoV-2 Gamma variant SGP was observed in the range of − 1.251 to − 4.162 kcal/mol. The critical amino acid interactions were dominated in Phe347-Ser357, Tyr396-Pro426, Asn450-Leu475, and Ser514-Glu516. Glide score exhibited a good correlation with glide emodel (R2 = 0.6077), while the moderate correlation with glide van der Waal (R2 = 0.5853) and glide energy (R2 = 0.5631). The results obtained were compared with that of standards; hydroxychloroquine exhibited significant interactions with the Thr430 and Glu516 with a Gscore of − 2.536 kcal/mol (Supplementary Figure S3p). In comparison, remdesivir has exhibited significant interactions with amino acid residues Arg355, Phe464, Arg466, and Glu516 with a Gscore of −3.115 kcal/mol (Supplementary Figure S3q) (Table 3).
Amongst 15 test ligands, mebendazole exhibited more potent inhibitory activity as indicated with its high Gscore of − 4.162 kcal/mol (Fig. 2). The other compounds showed moderate biological activity within the binding pocket of protein structure. Mebendazole has shown three HB interactions with the critical interacting residues of SARS-CoV-2 Gamma variant SGP. The NH moiety of the benzimidazole nucleus exhibited a HB interaction with the carbonyl group of Phe347 (NH⋯O = C < , 1.92 Å). The Ser349 formed HB two interactions with the carbonyl group of carbamate fragment (> C = O⋯HN, 1.85 and 2.05 Å). Further, the carbamate fragment’s NH moiety showed a HB interaction with the carbonyl group of Asn450 (NH⋯O = C < , 2.57 Å).

3D interaction diagram of mebendazole with amino acid residues necessary for the inhibition of SARS-CoV-2 Gamma variant
In addition, the hydrophobic–hydrophilic map of the mebendazole demonstrated that the imidazole ring of the benzimidazole scaffold is well located in the hydrophilic region of the catalytic pocket, while the aromatic group is present at position five of the benzimidazole ring. At the same time, the carbonyl group present between the two aromatic rings is well buried in the hydrophilic pocket of the receptor (Supplementary Figure S4).
Binding free energy calculation analysis
In the current study, the BFG of selected 15 molecules was calculated using the MM-GBSA strategy to get insight into the relative potencies and binding strengths against SARS-CoV-2 Alpha and Gamma variant SGP.
Binding affinity and free energy analysis of ligands against SARS-CoV-2 Alpha variant SGP
The predicted BFG score of docked poses of ligands was found to be in the range of − 24.31 to − 66.05 kcal/mol (Table 4). Similar results were observed for the standard compounds hydroxychloroquine (ΔGbind = -59.25 kcal/mol) and remdesivir (ΔGbind = -49.95 kcal/mol). It is evident from the study that van der Waal (ΔGvdW = − 10.15 to – 43.03 kcal/mol) energy term is the major contributor to inhibitory activity in the catalytic pocket of 7NEH, while Coulombic energy term (ΔGcou = 36.06 to – 48.46 kcal/mol) moderately favours the ligand binding in most of the active ligands. In all the active compounds, non-polar solvation (ΔGlipo = − 2.6 to – 15.18 kcal/mol) and covalent energy term are considered unfavourable for the ligand’s inhibitory activity within the binding site of 7NEH. Nevertheless, the compound flubendazole showed relatively BFG (ΔGbind = − 66.05 kcal/mol), van der Waal energy (ΔGvdW = − 45.62 kcal/mol), and Coulombic energy (ΔGcou = − 40.26 kcal/mol). It is evident from the result that non-polar van der Waals (ΔGvdW) is considered to be the main contributor to the inhibitory activity in the binding pocket of 7NEH.
Binding affinity and free energy analysis of ligands against SARS-CoV-2 Gamma variant SGP
The results of BFG of selected compounds against SARS-CoV-2 Gamma variant SGP are represented in Table 5. Similar results were observed for the standard compound drugs hydroxychloroquine (ΔGbind = − 66.43 kcal/mol) and remdesivir (ΔGbind = − 75.45 kcal/mol). The predicted BFG was observed to be in the range of − 22.35 to − 71.54 kcal/mol. It is clear from the studies that van der Waals (ΔGvdW = − 10.15 to − 43.03 kcal/mol) and Coulombic energy (ΔGcou = − 10.15 to − 43.03 kcal/mol) terms are found to be highly favourable for the inhibitory activity of the ligands in the active site of 7NXC, whereas non-polar solvation energy terms and covalent energy terms fluctuate between 19.66 to − 5.83 kcal/mol and − 12.08 to − 6.16 kcal/mol, clearly showing the low preference of their components in the ligand binding. Nonetheless, the compound mebendazole showed relatively high BFG (ΔGbind = − 71.54 kcal/mol), van der Waal energy (ΔGvdW = − 29.7 kcal/mol) and Coulombic energy (ΔGcou = − 25.51 kcal/mol). Thus, it is evident that van der Waals term and Coulombic energy terms are the key driving forces for the inhibitory activity of these ligands.
MD simulation analysis
To investigate the docking validity, the docked complex of flubendazole/7NEH was subjected to 100 ns MD simulation. RMSD provides data related to the protein’s backbone, and the value was found to be 3.6 nm; small initial fluctuation was observed, which was stabilized after 10 ns due to the decrease in amplitude of fluctuation with time (Fig. 3).

RMSD plot of backbone atoms of flubendazole/7NEH over a time period of 100 ns MD simulation
In addition, RMSF was computed to determine the dynamic behaviour of atomic positional fluctuations of backbone residues and the value was found to be 0.45 nm at 200–300 residues (Fig. 4).

RMSF plot of backbone atoms of 7NEH over a time period of 100 ns MD simulation
ROG was computed to determine the compactness of the complex and protein structure, and the value was observed between the range of 1.42 to 1.49 nm (Fig. 5).

Radius of gyration (ROG) plot of flubendazole/7NEH complex for 100 ns MD simulation
MM-PBSA
MM-PBSA analysis aids in segregating the total binding free energy into electrostatic, van der Waals, and solute–solvent interactions, to get insight into the binding modes and protein–ligand interaction process. The total binding free energy of the complex was found to be -68.831 kcal/mol. From the results, it is evident that van der Waals interactions (−79.735 kcal/mol) play an essential role in the MD simulation, contributing noticeably more to total interaction energy than electrostatic interactions (-55.919 kcal/mol) indicating dominating effect of polar components in the system. The polar/electrostatic and apolar/hydrophobic contributions to free energy are also recorded in Table 6. Further, SASA was calculated to determine the hydrophobic core in the protein structure and the value lies between 80 and 95 nm2, indicating the stability throughout the MD simulation (Fig. 6). SASA has not exhibited any fluctuations after attaining equilibrium throughout the MD simulation study. In disparity, the ΔEPB and SASA energy display limited contribution to the energy.

Solvent accessible surface area (SASA) of flubendazole/7NEH complex
Conclusion
Several studies are ongoing using small molecule inhibitors to circumvent the life-threatening infections caused by the mutant strains of SARS-CoV-2. Imidazole- and benzimidazole-based anti-parasitic drugs have been suggested as effective treatments in tackling SARS-CoV-2 mutants. In this study, molecular docking studies were performed on 15 imidazole- and benzimidazole-based anti-parasitic drug molecules along with the standard drugs hydroxychloroquine and remdesivir. Amongst these compounds, flubendazole and mebendazole are useful as potential drug candidates against SARS-CoV-2 mutants, as evidenced by the glide score (− 4.418 kcal/mol for 7NEH and − 4.162 kcal/mol for 7NXC kcal/mol) and BFG (− 66.05 kcal/mol for 7NEH and − 71.54 kcal/mol for 7NXC). Further, flubendazole/7NEH complex was subjected to MD simulation and the results obtained satisfied all the parameters of trajectory analysis. We, therefore, propose that the inhibitory potentials of flubendazole and mebendazole against spike glycoprotein variants of SARS-CoV-2 should further be explored in the subsequent wet laboratory experiments. This study facilitates the discovery process of potent inhibitors against SARS-CoV-2 mutants.
References
Abdool Karim SS, de Oliveira T. 2021 New SARS-CoV-2 variants—clinical, public health, and vaccine implications. New England Journal of Medicine
Barker H, Smyth R, Weissbach H, Toohey J, Ladd J, Volcani B (1960) Isolation and properties of crystalline cobamide coenzymes containing benzimidazole or 5, 6-dimethylbenzimidazole. J Biol Chem 235(2):480–488
Beard H, Cholleti A, Pearlman D, Sherman W, Loving KA (2013) Applying physics-based scoring to calculate free energies of binding for single amino acid mutations in protein-protein complexes. PloS one. 8(12):e82849
Berendsen HJ, Postma Jv, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys 1984;81(8):3684–90.
Bhatnagar A, Sharma P, Kumar N (2011) A review on “Imidazoles”: their chemistry and pharmacological potentials. Int J PharmTech Res 3(1):268–282
Biffinger JC, Kim HW, DiMagno SG (2004) The polar hydrophobicity of fluorinated compounds. ChemBioChem 5(5):622–627
Burki T (2021) Understanding variants of SARS-CoV-2. The Lancet 397(10273):462
Dejnirattisai W, Zhou D, Supasa P, Liu C, Mentzer AJ, Ginn HM et al (2021) Antibody evasion by the P 1 strain of SARS-CoV-2. Cell. https://doi.org/10.1016/j.cell.2021.03.055
Faria NR, Claro IM, Candido D, Moyses Franco L, Andrade PS, Coletti TM, et al. 2021 Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: preliminary findings. Virological.
Fei F, Zhou Z (2013) New substituted benzimidazole derivatives: a patent review (2010–2012). Expert Opin Ther Pat 23(9):1157–1179
Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA et al (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein− ligand complexes. J Med Chem 49(21):6177–6196
Gaba M, Singh S, Mohan C (2014) Benzimidazole: an emerging scaffold for analgesic and anti-inflammatory agents. Eur J Med Chem 76:494–505
Gaba M, Singh D, Singh S, Sharma V, Gaba P (2010) Synthesis and pharmacological evaluation of novel 5-substituted-1-(phenylsulfonyl)-2-methylbenzimidazole derivatives as anti-inflammatory and analgesic agents. Eur J Med Chem 45(6):2245–2249
Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT et al (2004) Glide: a new approach for rapid accurate docking and scoring. 2. Enrichment factors in database screening. J med chem 47(7):1750–9
Hoffmann M, Arora P, Groß R, Seidel A, Hörnich BF, Hahn AS, et al. 2021 SARS-CoV-2 variants B. 1.351 and P. 1 escape from neutralizing antibodies. Cell. doi: https://doi.org/10.1016/j.cell.2021.03.036
Hui DS, Azhar EI, Madani TA, Ntoumi F, Kock R, Dar O et al (2020) The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan China. Int J Infect Diseases 91:264–266
Ingle R, Magar D (2011) Heterocyclic chemistry of benzimidazoles and potential activities of derivatives. Int J Drug Res Tech 1(1):26–32
Iwaloye O, Elekofehinti OO, Momoh AI, Babatomiwa K, Ariyo EO (2020) In silico molecular studies of natural compounds as possible anti-Alzheimer’s agents: ligand-based design. Network Modeling Analysis in Health Informatics and Bioinformatics 9(1):1–14
Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE et al (2004) A hierarchical approach to all‐atom protein loop prediction. Proteins: Structure, Function, and Bioinformatics. 55(2): 351–67
Kollman P, Massova I, Reyes C, Kuhn B, Huo S, Chong L, O. Donin i, P. Cieplak, J. Srinivasan, DA Case, and r. Cheatham, TE, et al (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 33(12):889–97
Kupferschmidt K (2020) Mutant coronavirus in the United Kingdom sets off alarms, but its importance remains unclear. Science, December. https://doi.org/10.1126/science.abg2626
Li J, Abel R, Zhu K, Cao Y, Zhao S, Friesner RA (2011) The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins: Structure, Function, and Bioinformatics. 79(10):2794–812
Luo R, David L, Gilson MK (2002) Accelerated Poisson-Boltzmann calculations for static and dynamic systems. J Comput Chem 23(13):1244–1253
Maggi F, Novazzi F, Genoni A, Baj A, Spezia PG, Focosi D et al (2021) Imported SARS-CoV-2 variant P 1 in traveler returning from Brazil to Italy. Emerging infectious diseases. 27(4):1249
Narasimhan B, Sharma D, Kumar P (2011) Biological importance of imidazole nucleus in the new millennium. Med Chem Res 20(8):1119–1140
Novac N (2013) Challenges and opportunities of drug repositioning. Trends Pharmacol Sci 34(5):267–272
Ramírez-Salinas GL, Martínez-Archundia M, Correa-Basurto J, García-Machorro J (2020) Repositioning of ligands that target the spike glycoprotein as potential drugs for SARS-CoV-2 in an In Silico Study. Molecules 25(23):5615
Roos K, Wu C, Damm W, Reboul M, Stevenson JM, Lu C et al (2019) OPLS3e: Extending force field coverage for drug-like small molecules. J Chem Theory Comput 15(3):1863–1874
Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27(3):221–234
Schüttelkopf AW, Van Aalten DM (2004) PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr D Biol Crystallogr 60(8):1355–1363
Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H et al (2020) Structural basis of receptor recognition by SARS-CoV-2. Nature 581(7807):221–224
Singh J, Samal J, Kumar V, Sharma J, Agrawal U, Ehtesham NZ et al (2021) Structure-Function Analyses of New SARS-CoV-2 Variants B. 1.1. 7, B. 1.351 and B. 1.1. 28.1: Clinical, Diagnostic, Therapeutic and Public Health Implications. Viruses. 13(3):439.
Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KH, Dingens AS et al (2020) Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 182(5):1295–310
Sternberg A, Naujokat C (2020) Structural features of coronavirus SARS-CoV-2 spike protein: targets for vaccination. Life sciences. 257:118056
Supasa P, Zhou D, Dejnirattisai W, Liu C, Mentzer AJ, Ginn HM et al (2021) Reduced neutralization of SARS-CoV-2 B. 1.1. 7 variant by convalescent and vaccine sera. Cell. 184(8):2201–11. e7
Villoutreix BO, Calvez V, Marcelin A-G, Khatib A-M (2021) In silico investigation of the new UK (B. 1.1. 7) and South African (501y. v2) SARS-CoV-2 variants with a focus at the ace2–spike rbd interface. International journal of molecular sciences. 22(4):1695.
Wang P, Casner RG, Nair MS, Wang M, Yu J, Cerutti G et al (2020) Increased resistance of sars-cov-2 variant p 1 to antibody neutralization. Cell Host & Microbe. 383:2427
Wright JB (1951) The chemistry of the benzimidazoles. Chem Rev 48(3):397–541
Yadav G, Ganguly S (2015) Structure activity relationship (SAR) study of benzimidazole scaffold for different biological activities: A mini-review. Eur J Med Chem 97:419–443
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
Vidyasrilekha Yele and Bharat Kumar Reddy Sanapalli contributed equally as first authors by synthesizing the literature, drafting the paper, and involving in the critical revision of the manuscript. Vidyasrilekha Yele and Afzal Azam Mohammed were involved in conceptual inputs. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest in this study. The authors alone are responsible for the content and writing of the paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yele, V., Sanapalli, B.K.R. & Mohammed, A.A. Imidazoles and benzimidazoles as putative inhibitors of SARS-CoV-2 B.1.1.7 (Alpha) and P.1 (Gamma) variant spike glycoproteins: A computational approach. Chem. Pap. 76, 1107–1117 (2022). https://doi.org/10.1007/s11696-021-01900-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-021-01900-8