
Abstract
Tuberculosis continues to wreak havoc worldwide and caused around 1.4 million deaths in 2019. Hence, in our pursuit of developing novel antitubercular compounds, we are reporting the e-Pharmacophore-based design of DprE1 (decaprenylphosphoryl-ribose 2′-oxidase) inhibitors. In the present work, we have developed a four-feature e-Pharmacophore model based on the receptor–ligand cavity of DprE1 protein (PDB ID 4P8C) and mapped our previous reported library of compounds against it. The compounds were ranked on phase screen score, and the insights obtained from their alignment were used to design some novel compounds. The designed compounds were docked with DprE1 protein in extra-precision mode using Glide module of Maestro, Schrodinger. Some derivatives like B1, B2, B4, B5 and B12 showed comparable docking score (docking score > − 6.0) with respect to the co-crystallized ligand. The designed compounds were synthesized and characterized. In vitro antitubercular activity was carried out on Mycobacterium tuberculosis H37Rv (ATCC27294) strain using the agar dilution method, and minimum inhibitory concentration (MIC) was determined. The compound B12 showed a MIC value of 1.56 μg/ml which was better than the standard drug ethambutol (3.125 μg/ml). Compounds B7 and B11 were found to be equipotent with ethambutol. Cytotoxicity studies against Vero cell lines proved that these compounds were non-cytotoxic. Molecular dynamic simulation study also suggests that compound B12 will form a stable complex with DprE1 protein and will show the crucial H-bond interaction with LYS418 residue. Further in vitro enzyme inhibition studies are required to validate these findings.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
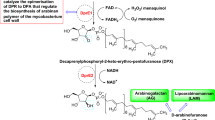
More than 40 years have passed since the introduction of the quadruple drug therapy regimen for tuberculosis (TB), which has proven to be both effective and cost-effective. But despite this, TB continues to wreak havoc worldwide which is evident from the fact that worldwide around 1.4 million died of TB in 2019 (Raviglione et al. 2012; Global tuberculosis report 2020). This state of high mortality rate feeds on various factors like prolonged duration of antitubercular therapy, toxicity induced by the antitubercular drugs, lower patient compliance and the rise of MDR-TB (multidrug-resistant). Bedaquiline, pretomanid and delamanid are the only drugs approved by the FDA (Food and Drug Administration) in the last four decades to treat MDR-TB which highlights the associated challenges in the field of antitubercular drug discovery (Cohen 2013). Hence, it is the urgent need of the hour to develop novel small molecules as antitubercular compounds acting on newer targets of the mycobacterium. Among many of the targets explored so far, DprE1 (Decaprenylphosphoryl-ribose 2′-oxidase) has emerged as a promising target for the development of antitubercular drugs. DprE1 is necessary for the synthesis of the mycobacterial cell wall as it facilitates the conversion of DPR (decaprenylphosphoryl-D-keto-erythro-pentofuranose) to DPX (decaprenylphosphoryl-2-ketoribose). DPX is further reduced to DPA (decaprenylphosporyl arabinose) by DprE2 as shown in Fig. 1 (Mikušová et al. 2005; Brecik et al. 2015).

Epimerization of 2′-hydroxyl group of DPR by DprE1 and DprE2 and rationale behind selecting DprE1 as a potential target for antitubercular drug design
The arabinosyltransferases use DPA as a substrate to synthesize arabinogalactan and lipoarabinomannan, which are major components of the mycobacterial cell wall. DprE1 is an oxidoreductase enzyme-containing FAD (flavin adenine dinucleotide) (Neres et al. 2012; Batt et al. 2012). DprE1 as a potential antitubercular drug target was validated by Makarov et al., who reported BTZ043 (benzothiazinone derivative) as the first covalent inhibitor of DprE1 (Makarov et al. 2009). After this, several authors have reported various heterocyclic derivatives like benzothiazole, azaindoles, triazoles, aminoquinolones, dinitrobenzamides, quinoxalines, pyrazolopyridines, etc. showing antitubercular activity through the inhibition of DprE1 (Landge et al. 2015; Mir et al. 2014; Magnet et al. 2010; Batt et al. 2016; Panda et al. 2014). Readers can refer to some excellent reviews by Chikhale et al. and Gawad et al. for more insights into the development of DprE1 inhibitors (Chikhale et al. 2018; Gawad and Bonde 2018).
This work is in continuation of our previous reported piperidone derivatives as potential anti-HIV/TB agents (Kumar et al. 2019). In the present work, we have designed non-covalent DprE1 inhibitors by developing an e-Pharmacophore model by using the receptor–ligand interactions of DprE1 with 2-carboxyquinoxalines as reported in its X-ray crystal structure (Neres et al. 2015). Several authors have reported e-Pharmacophore modelling combined with molecular docking and molecular dynamic simulations studies to design and identify potent inhibitors of several proteins (Arun et al. 2020; Kumar et al. 2020, 2021; Salam et al. 2009). We have used PDB ID 4P8C to develop a four-feature e-Pharmacophore model and mapped it against our previous reported antitubercular compounds (Kumar et al. 2019). We found that only one aromatic feature matched, and our compounds had very low phase screen score. Based on the insights from the developed e-pharmacophore model, we added a benzimidazole group to the nitrogen of piperidone ring and the resultant derivatives had three out of four features matches with an improved phase screen score. Benzimidazole ring system was selected because historically its derivatives have been reported as antitubercular agents, and recently, Manjunath et al. have found a series of benzimidazole derivatives showing DprE1 inhibition (Manjunatha et al. 2019; Keri et al. 2016; Yeong et al. 2017; Surineni et al. 2019). Detailed design strategy is depicted in Fig. 2 and described under the materials and methods section. Herein, we have reported some potent antitubercular compounds with activity in the lower micromolar range and this design strategy can be used for targeting other proteins as well.

Inter-site angle (a) and inter-site distance (b) between different features of the developed e-Pharmacophore model. Pink spheres with arrow, hydrogen bond acceptor (A); orange open circle, aromatic ring (R); solid red spheres, negatively ionizable (N)
Materials and methods
The chemicals, reagents and solvents for the present work were sourced from Aldrich (Bengaluru, India), Spectrochem P. Ltd. (Mumbai, India), E. Merck (Mumbai, India), S. D. Fine Chem. P. Ltd. (Mumbai, India). TLC (thin-layer chromatography) was done on 0.25 mm silica gel plates purchased from E. Merck (silica gel 60 F254). The melting point was measured by using a laboratory melting point apparatus from Toshniwal P. Ltd., Mumbai, India. IR spectra were recorded on FT‐IR Affinity‐1 IR spectrometer from Shimadzu, Bengaluru, India. Mass spectra of the synthesized compounds were recorded on GC‐MS‐QP5050A from Shimadzu. NMR spectra were recorded on a 400 MHz spectrometer from Bruker, and dimethyl sulphoxide (DMSO)‐d6 was used as the solvent. All in silico experiments were done on an HP computer with Linux (Ubuntu, version 18.04.1) as the operating system and Maestro 11.6 which is a small molecule drug discovery suite from Schrödinger LLC, 2018‐3 (Schrödinger LLC, Bengaluru, India).
Design of non-covalent DprE1 inhibitors
For the present work, we have employed e-Pharmacophore modelling based on the receptor–ligand complex of the non-covalent inhibitor of DprE1 protein as reported by Neres et al.(Neres et al. 2015). The e-Pharmacophore method generates energetically optimized structure-based pharmacophores and has the advantages of both ligand- and structure-based pharmacophore modelling (Salam et al. 2009; Loving et al. 2009). The e-Pharmacophore model was developed employing PHASE module of Maestro suite (Dixon et al. 2006). PHASE has a built-in set of six pharmacophore features like hydrogen bond donor (D), hydrogen bond acceptor (A), hydrophobic group (H), aromatic ring (R), positively ionizable (P) and negatively ionizable (N). The screened compounds were ranked based on the phase screen score. DprE1 protein (PDB ID 4P8C) was imported from the protein data bank followed by pre-processing, removal of water and energy minimization using the protein preparation wizard of Maestro (Madhavi Sastry et al. 2013). For all further studies, this energetically minimized protein structure of DprE1 was used. To further understand the binding of the designed compounds with DprE1 protein, molecular docking simulations were carried out.
Molecular docking and free binding energy calculations
Molecular docking studies are useful in understanding the binding between a protein and ligand. For our study, we carried out extra-precision (XP) docking employing Glide module of Maestro 11.6 interface (Friesner et al. 2006). In the extra-precision mode, false positives are eliminated by extensive sampling and more advanced scoring functions. XP-docking was accomplished in three steps, viz. receptor grid generation, ligand minimization and docking. On the minimized protein structure of DprE1, a receptor grid was generated around the co-crystallized ligand as the site of binding of the bound ligand was the active site where our designed ligands would bind with DprE1. Then the designed compounds were energetically minimized using LigPrep tool of Maestro. In the final step, the structures generated from the LigPrep exercise were docked using the receptor grid file generated in the first step. The docked compounds were ranked based on the docking score. The docking protocol was validated by redocking the co-crystallized ligand and then computing the RMSD (root mean square deviation) value.
Free binding energy calculations were also carried out employing MM-GBSA (molecular mechanics, the generalized Born model and solvent accessibility) under the Prime module (Jacobson et al. 2004). Prime MM-GBSA generates many energy properties which report energies for the receptor, ligand and other complex structures. It can also compute the energy differences produced due to strain and binding. It employs VSGB 2.0 as an optimized implicit solvent model. Five fundamental energy are calculated in Prime, and from these energies, the strain and binding energies are computed (Li et al. 2011). For simplifying our work, we have considered MM-GBSA dG bind expressed in the unit of kcal/mol to predict the binding energy of our designed compounds with the DprE1 protein. For this study, the resultant docked complex files from Glide were directly put for MM-GBSA calculations. Based on the docking score and MM-GBSA dG bind score, twelve compounds were selected for synthesis. The docking score of the selected compounds ranged from high to low in comparison with the co-crystallized ligand.
Synthesis of 1-(1H-benzimidazol-2-ylmethyl)piperidin-4-one (3)
As shown in Scheme 1, 4-piperidone hydrochloride (0.7 gm, 0.005 mol) and 2-chloromethylbenzimidazole (0.81 gm, 0.005 mol) were taken in a round-bottomed flask containing 10 ml of DMF (dimethyl formamide). To this reaction mixture, 0.7 ml (0.005 mol) of TEA (triethylamine) was added and was refluxed for 7 h. The reaction progress was measured by taking TLC at periodic intervals. After the completion of the reaction, the contents were transferred into crushed ice. This mixture was further refrigerated for another 12 h. Finally, the precipitate was filtered under vacuum and recrystallized using methanol to obtain the pure product.

Synthesis of (3E,5E)-1-((1H-benzo[d]imidazol-2-yl)methyl)-3,5-bis(furan-2-ylmethylene)piperidin-4-one (B1) and other derivatives (B2-B12)
Synthesis of (3E,5E)-1-(1H-benzimidazol-2-ylmethyl)-3,5-bis(furan-2-ylmethylidene)piperidin-4-one (B1)
The method reported by Kalai et al. and Katsori et al.was employed to synthesize compound B1 as depicted in Scheme 1 (Kálai et al. 2011; Katsori et al. 2011). Furfural (1.82 ml, 0.022 mol) and 1-(1H-benzimidazol-2-ylmethyl)piperidin-4-one (2.3 g, 0.01 mol) prepared in the first step were taken in an Erlenmeyer flask. A solution of 12 ml ethanol and 20 ml of 10% NaOH was prepared and cooled to 20 °C. At room temperature, it was added to the mixture of furfural and compound 3 with stirring. The reaction was monitored using TLC, and after 2 h, the product was precipitated by cooling the reaction mixture in an ice bath. Finally, the precipitate was filtered under vacuum and recrystallized using methanol and acetonitrile (1:1) to obtain the pure product.
The general method of procedure for the synthesis of B2-12
The synthesis of compounds B2–B12 (Scheme 1) was carried out by using the method reported by Banerjee et al. (Banerjee et al. 2011). Different amines (0.01 mol) and sodium acetate (0.8 gm, 0.01 mol) were added to the ethanolic mixture of compound B1 (3.85 gm, 0.01 mol). The reaction mixture was kept for reflux with continuous stirring using a magnetic stirrer. The reaction was monitored using TLC, and after the completion (after 3.5 h), it was brought to room temperature and then transferred into ice-cold water. Finally, the precipitate was filtered under vacuum and recrystallized using methanol DMF (2:8) to obtain the pure product.
Characterization of the synthesized compounds (B1-B12)
(3E,5E)-1-((1H-benzo[d]imidazol-2-yl)methyl)-3,5-bis(furan-2-ylmethylene)piperidin-4-one (B1)
Yield: 82%, m.p.: 254–256 °C. IR (KBr) cm−1: 3267 (NH); 3101, 3056, 2973 (C–H); 1710 (C=O). 1H NMR (500 MHz, Chloroform-d) δ 9.82 (s, 1H), 7.62 (dd, J = 7.5, 1.5 Hz, 2H), 7.59–7.54 (m, 1H), 7.49–7.43 (m, 5H), 7.15 (t, J = 4.5 Hz, 2H), 6.92 (t, J = 7.5 Hz, 2H), 6.78 (dd, J = 7.5, 1.4 Hz, 2H), 4.06 (s, 2H), 3.62 (d, J = 1.0 Hz, 4H). 13C NMR (125 MHz, Chloroform-d) δ 191.94, 152.72, 149.47, 145.45, 140.31, 138.40, 136.08, 130.68, 123.42, 122.24, 116.90, 116.23, 114.42, 111.38, 55.13, 52.44. MS m/z: 385 (M) + , 369, 343, 301, 258, 212, 173, 131. Elemental analysis (%): Calculated: C, 71.67; H, 4.97; N, 10.90. Found: C, 71.93; H, 5.25; N, 10.77.
(3E,5E)-1-((1H-benzo[d]imidazol-2-yl)methyl)-3,5-bis(furan-2-ylmethylene)piperidin-4-one oxime (B2)
Yield: 76%, m.p.: 275–278 °C. IR (KBr) cm−1: 3364 (OH); 3287 (NH); 3047, 2942 (CH), 1565 (C=N). 1H NMR (500 MHz, Chloroform-d) δ 9.82 (s, 1H), 9.45 (s, 1H), 7.59–7.54 (m, 1H), 7.49–7.43 (m, 1H), 7.25 (dd, J = 7.5, 1.5 Hz, 2H), 7.15 (t, J = 4.5 Hz, 2H), 6.87 (t, J = 7.5 Hz, 2H), 6.59 (s, 3H), 6.26 (dd, J = 7.5, 1.4 Hz, 2H), 4.06 (s, 2H), 3.83 (d, J = 1.0 Hz, 4H). 13C NMR (125 MHz, Chloroform-d) δ 152.72, 149.62, 146.83, 145.45, 140.31, 136.55, 136.08, 135.99, 133.16, 123.42, 122.24, 116.90, 114.71, 114.42, 111.38, 53.39, 52.44. MS m/z: 400(M) + Elemental analysis (%): Calculated: C, 68.99; H, 5.03; N, 13.99. Found: C, 69.20; H, 4.73; N, 14.30.
2-(((3E,5E)-3,5-bis(furan-2-ylmethylene)-4-hydrazonopiperidin-1-yl)methyl)-1H-benzo[d]imidazole (B3)
Yield: 73%, m.p.: 264–265 °C. IR (KBr) cm−1: 3359, 3264 (NH2, NH); 3114, 3024, 2948, (CH), 1657 (C=N). 1H NMR (500 MHz, Chloroform-d) δ 9.82 (s, 1H), 7.59–7.54 (m, 2H), 7.49–7.43 (m, 2H), 7.25 (dd, J = 7.5, 1.5 Hz, 3H), 7.15 (t, J = 4.5 Hz, 3H), 6.87 (t, J = 7.5 Hz, 3H), 6.55 (s, 4H), 6.22 (dd, J = 7.5, 1.4 Hz, 4H), 4.30 (s, 3H), 4.06 (s, 3H), 3.75 (d, J = 1.0 Hz, 6H). 13C NMR (125 MHz, Chloroform-d) δ 152.72, 146.58, 145.45, 144.80, 140.31, 138.94, 138.22, 137.13, 136.08, 123.42, 122.24, 116.90, 114.71, 114.42, 111.38, 53.23, 52.44. MS m/z: 399 (M) + Elemental analysis (%): Calculated: C, 69.16; H, 5.30; N, 17.53. Found: C, 69.95.93; H, 5.26; N, 18.48.
2-((3E,5E)-1-((1H-benzo[d]imidazol-2-yl)methyl)-3,5-bis(furan-2-ylmethylene)piperidin-4-ylidene)hydrazine-1-carbothioamide (B4)
Yield: 77%, m.p.: 302–305 °C. IR (KBr) cm−1: 3314, 3284 (NH2, NH), 3086, 2957 (CH), 1565 (C=N) 1208 (C=S). 1H NMR (500 MHz, Chloroform-d) δ 10.37 (s, 1H), 9.82 (s, 1H), 8.60 (s, 2H), 7.59–7.54 (m, 1H), 7.49–7.43 (m, 1H), 7.25 (dd, J = 7.5, 1.5 Hz, 2H), 7.15 (t, J = 4.5 Hz, 2H), 6.87 (t, J = 7.5 Hz, 2H), 6.59 (s, 3H), 6.26 (dd, J = 7.5, 1.4 Hz, 2H), 4.06 (s, 2H), 3.83 (d, J = 1.0 Hz, 4H). 13C NMR (125 MHz, Chloroform-d) δ 180.13, 152.72, 147.17, 146.60, 145.45, 140.31, 137.51, 137.22, 136.08, 129.17, 123.42, 122.24, 116.90, 114.71, 114.42, 111.38, 53.16, 52.44. MS m/z: 458 (M) + . Elemental analysis (%): Calculated: C, 62.87; H, 4.84; N, 18.33. Found: C, 62.77; H, 4.96; N, 18.91.
2-((3E,5E)-1-((1H-benzo[d]imidazol-2-yl)methyl)-3,5-bis(furan-2-ylmethylene)piperidin-4-ylidene)-N-hydroxyhydrazine-1-carbothioamide (B5)
Yield: 79%, m.p.: 315–318 °C. IR (KBr) cm−1: 3367 (OH); 3251 (NH), 3108, 3049, 2907 (CH), 1636 (C=N). 1H NMR (500 MHz, Chloroform-d) δ 10.52 (s, 1H), 10.05 (d, J = 2.2 Hz, 1H), 9.82 (s, 1H), 9.06 (d, J = 2.2 Hz, 1H), 7.59–7.54 (m, 1H), 7.49–7.43 (m, 1H), 7.25 (dd, J = 7.5, 1.5 Hz, 2H), 7.15 (t, J = 4.5 Hz, 2H), 6.87 (t, J = 7.5 Hz, 2H), 6.59 (s, 3H), 6.26 (dd, J = 7.5, 1.4 Hz, 2H), 4.06 (s, 2H), 3.83 (d, J = 1.0 Hz, 4H). 13C NMR (125 MHz, Chloroform-d) δ 180.51, 152.72, 146.60, 146.35, 145.45, 140.31, 137.51, 137.22, 136.08, 129.17, 123.42, 122.24, 116.90, 114.71, 114.42, 111.38, 53.16, 52.44. MS m/z: 474 (M) + , 390, 369, 301, 173, 131. Elemental analysis (%): Calculated: C, 60.75; H; 4.67; N, 17.71. Found: C, 61.04; H, 4.40; N, 17.61.
3E,5E)-1-((1H-benzo[d]imidazol-2-yl)methyl)-N-(4-fluorophenyl)-3,5-bis(furan-2-ylmethylene)piperidin-4-imine (B6)
Yield: 80%, m.p.: 319–320 °C. IR (KBr) cm−1: 3244 (NH), 3074, 2937 (CH), 1651 (C=N). 1H NMR (500 MHz, DMSO-d6) δ 9.82 (s, 1H), 7.59–7.54 (m, 2H), 7.49–7.43 (m, 2H), 7.28–7.22 (m, 7H), 7.25–7.17 (m, 1H), 7.21–7.11 (m, 6H), 6.87 (t, J = 7.5 Hz, 3H), 6.61 (s, 4H), 6.29 (dd, J = 7.5, 1.5 Hz, 4H), 4.06 (s, 3H), 3.86 (d, J = 1.0 Hz, 6H). 13C NMR (125 MHz, Chloroform-d) δ 160.53, 152.72, 147.07, 145.45, 140.31, 136.08, 133.39, 133.27, 128.17, 123.42, 122.24, 121.72, 121.66, 116.90, 116.38, 116.22, 114.71, 114.42, 111.38, 53.48, 52.44. MS m/z: 478 (M) + Elemental analysis (%): Calculated: C, 72.79; H, 4.84; N, 11.71. Found: C, 72.98; H, 5.13; N, 10.92.
2-(((3E,5E)-3,5-bis(furan-2-ylmethylene)-4-(2-phenylhydrazono)piperidin-1-yl)methyl)-1H-benzo[d]imidazole (B7)
Yield: 71%, m.p.: 325–326 °C. IR (KBr) cm−1: 3276 (NH), 3102, 2986, 2885 (CH), 1571 (C=N). 1H NMR (500 MHz, Chloroform-d) δ 10.37 (s, 1H), 9.82 (s, 1H), 7.59–7.54 (m, 1H), 7.49–7.39 (m, 4H), 7.31–7.23 (m, 5H), 7.15 (t, J = 4.5 Hz, 2H), 7.03 (tt, J = 7.6, 1.5 Hz, 1H), 6.87 (t, J = 7.5 Hz, 2H), 6.59 (s, 3H), 6.26 (dd, J = 7.5, 1.4 Hz, 2H), 4.06 (s, 2H), 3.83 (d, J = 1.0 Hz, 4H). 13C NMR (125 MHz, Chloroform-d) δ 152.72, 146.60, 145.45, 144.73, 143.17, 140.31, 137.54, 137.25, 136.08, 129.17, 128.97, 123.42, 122.24, 119.94, 116.90, 116.07, 114.71, 114.42, 111.38, 53.16, 52.44. MS m/z: 475 (M) + Elemental analysis (%): Calculated: C, 73.25; H, 5.30; N, 14.73. Found: C, 72.89; H, 5.27; N, 15.12.
2-(((3E,5E)-4-((2,4-dimethoxyphenyl)imino)-3,5-bis(furan-2-ylmethylene)piperidin-1-yl)methyl)-1H-benzo[d]imidazol-3-ium (B8)
Yield: 74%, m.p.: > 360 °C. IR (KBr) cm−1: 3224 (NH), 3053, 2947, 2883 (CH), 1626 (C=N). 1H NMR (500 MHz, Chloroform-d) δ 10.95 (s, 1H), 9.23 (s, 1H), 7.81–7.76 (m, 2H), 7.53 (dtd, J = 26.6, 7.5, 1.6 Hz, 2H), 7.35–7.30 (m, 2H), 7.25 (dd, J = 7.5, 1.5 Hz, 2H), 7.18 (d, J = 7.5 Hz, 1H), 6.87 (t, J = 7.5 Hz, 2H), 6.72 (dd, J = 7.5, 1.5 Hz, 1H), 6.67 (d, J = 1.5 Hz, 1H), 6.61 (s, 3H), 6.29 (dd, J = 7.5, 1.5 Hz, 3H), 3.97 (s, 3H), 3.86 (d, J = 1.0 Hz, 4H), 3.78 (d, J = 16.0 Hz, 5H). 13C NMR (125 MHz, Chloroform-d) δ 161.38, 155.84, 154.10, 151.56, 145.88, 145.60, 133.41, 133.30, 132.93, 131.23, 128.33, 126.31, 125.41, 122.75, 115.19, 114.55, 113.91, 111.25, 107.32, 97.30, 56.65, 55.64, 53.26, 52.55. MS m/z: 521 (M) + Elemental analysis (%): Calculated: C, 71.38; H, 5.60; N, 10.74. Found: C, 71.86; H, 5.37; N, 11.13.
(3E,5E)-1-((1H-benzo[d]imidazol-2-yl)methyl)-N-(2,3-dimethylphenyl)-3,5-bis(furan-2-ylmethylene)piperidin-4-imine (B9)
Yield: 70%, m.p.: 290–291 °C. IR (KBr) cm−1: 3273 (NH), 3112, 3038, 2922 (CH), 1637 (C=N). 1H NMR (500 MHz, Chloroform-d) δ 9.82 (s, 1H), 7.60–7.53 (m, 2H), 7.49–7.43 (m, 1H), 7.34 (ddd, J = 7.5, 1.5, 0.7 Hz, 2H), 7.28–7.21 (m, 4H), 7.15 (d, J = 2.7 Hz, 1H), 7.17–7.08 (m, 3H), 6.87 (t, J = 7.5 Hz, 2H), 6.61 (s, 3H), 6.29 (dd, J = 7.5, 1.5 Hz, 3H), 4.06 (s, 2H), 3.86 (d, J = 1.0 Hz, 5H), 2.43 (s, 3H), 2.33 (d, J = 0.7 Hz, 4H). 13C NMR (125 MHz, Chloroform-d) δ 161.00, 152.72, 149.97, 147.07, 145.45, 140.31, 138.94, 136.08, 133.39, 133.28, 133.21, 128.85, 128.06, 127.17, 123.42, 122.24, 116.90, 115.86, 114.71, 114.42, 111.38, 53.57, 52.44, 19.90, 14.02. MS m/z: 488 (M) + Elemental analysis (%): Calculated: C, 76.21; H, 5.78; N, 11.47. Found: C, 75.98; H, 5.65; N, 11.88.
(3E,5E)-1-((1H-benzo[d]imidazol-2-yl)methyl)-N-(2-fluorophenyl)-3,5-bis(furan-2-ylmethylene)piperidin-4-imine (B10)
Yield: 74%, m.p.: 279–280 °C. IR (KBr) cm−1: 3250 (NH), 3085, 3014, 2857 (CH), 1638 (C=N). 1H NMR (500 MHz, Chloroform-d) δ 9.82 (s, 1H), 7.63–7.53 (m, 5H), 7.49–7.43 (m, 1H), 7.41–7.29 (m, 3H), 7.25 (dd, J = 7.5, 1.5 Hz, 2H), 7.19–7.11 (m, 3H), 6.87 (t, J = 7.5 Hz, 2H), 6.61 (s, 3H), 6.29 (dd, J = 7.5, 1.5 Hz, 3H), 4.06 (s, 2H), 3.86 (d, J = 1.0 Hz, 5H). 13C NMR (125 MHz, Chloroform-d) δ 152.72, 147.07, 145.45, 140.31, 136.08, 133.22, 133.10, 128.06, 126.64, 126.57, 126.08, 126.06, 123.42, 122.24, 122.15, 122.09, 116.90, 115.86, 115.70, 114.71, 114.42, 111.38, 53.57, 52.44. MS m/z: 478 (M) + Elemental analysis (%): Calculated: C, 72.79; H, 4.84; N, 11.71. Found: C, 71.81; H, 5.17; N, 12.19.
2-(((3E,5E)-4-((2,4-dichlorophenyl)imino)-3,5-bis(furan-2-ylmethylene)piperidin-1-yl)methyl)-1H-benzo[d]imidazol-3-ium (B11)
Yield: 78%, m.p.: 252–253 °C. IR (KBr) cm−1: 3240 (NH), 3077, 2958 (CH), 1604 (C=N). 1H NMR (500 MHz, Chloroform-d) δ 10.95 (s, 1H), 9.23 (s, 1H), 7.81–7.76 (m, 2H), 7.53 (dtd, J = 26.6, 7.5, 1.6 Hz, 2H), 7.39 (d, J = 1.5 Hz, 1H), 7.35–7.30 (m, 2H), 7.25 (dd, J = 7.5, 1.5 Hz, 2H), 7.18 (dd, J = 7.5, 1.5 Hz, 1H), 6.87 (t, J = 7.5 Hz, 2H), 6.79 (d, J = 7.5 Hz, 1H), 6.61 (s, 3H), 6.29 (dd, J = 7.5, 1.5 Hz, 3H), 3.86 (d, J = 1.0 Hz, 4H), 3.77 (s, 2H). 13C NMR (125 MHz, Chloroform-d) δ 159.92, 154.10, 146.65, 145.88, 145.60, 133.34, 133.13, 133.01, 132.93, 131.23, 130.56, 128.33, 128.20, 127.46, 126.31, 125.41, 121.85, 115.19, 114.55, 113.91, 111.25, 53.26, 52.55. MS m/z: 529 (M) + Elemental analysis (%): Calculated: C, 65.67; H, 4.37; N, 10.56. Found: C, 66.32; H, 4.91; N, 11.27.
(3E,5E)-1-((1H-benzo[d]imidazol-2-yl)methyl)-N-(2,4-difluorophenyl)-3,5-bis(furan-2-ylmethylene)piperidin-4-imine (B12)
Yield: 81%, m.p.: 333–335 °C. IR (KBr) cm−1: 3267 (NH), 3072, 3005, 2944 (CH), 1623 (C=N). 1H NMR (500 MHz, Chloroform-d) δ 9.82 (s, 1H), 7.60–7.53 (m, 2H), 7.50–7.42 (m, 2H), 7.25 (dd, J = 7.5, 1.5 Hz, 3H), 7.21–7.06 (m, 7H), 7.02 (td, J = 8.0, 1.5 Hz, 2H), 6.87 (t, J = 7.5 Hz, 3H), 6.61 (s, 4H), 6.29 (dd, J = 7.5, 1.5 Hz, 4H), 4.06 (s, 3H), 3.86 (d, J = 1.0 Hz, 6H). 13C NMR (125 MHz, Chloroform-d) δ 161.96, 161.90, 160.62, 160.57, 156.97, 154.95, 152.72, 147.07, 145.45, 140.31, 136.08, 133.22, 133.10, 132.43, 132.27, 128.06, 123.60, 123.53, 123.42, 122.24, 116.90, 114.71, 114.42, 113.38, 113.36, 111.38, 104.54, 104.38, 53.57, 52.44. MS m/z: 496 (M) + Elemental analysis (%): Calculated: C, 70.15; H, 4.47; N, 11.28. Found: C, 70.83; H, 5.16; N, 10.96.
In vitro antitubercular evaluation
Antitubercular evaluation of the synthesized derivatives was carried out according the procedure reported by Addla et al. (Addla et al. 2014). In brief, serial dilution of the synthesized compounds and ethambutol (the standard drug used in this study) was carried out followed by inoculation into an agar medium (Middlebrook 7H11) supplemented with OADC (oleic acid, albumin, dextrose and catalase) procured from Difco (Kolkata, India). Inoculum; M. tuberculosis H37Rv ATCC27294 was made from freshly prepared Middlebrook 7H11 agar slants supplemented with OADC. An approximate concentration of 107 cfu/ml of mycobacterial suspension was obtained and 5 µl of this suspension was spotted into 7H11 agar tubes containing graded concentrations of the synthesized derivatives and ethambutol. It was incubated at 37 °C, and readings were taken after 28 days (Hall et al. 2012).
Cytotoxicity assay
Cell toxicity studies for these compounds were carried out on Vero cell line (African green monkey kidney cells) obtained from NCCS (National Centre for Cell Sciences), Pune, India. The assay was carried out using the well stablished protocol reported by Mosmann et al. and Denizot et al. (Mosmann 1983; Denizot and Lang 1986). The growth inhibition percentage was computed by the following formula:
Further, SI (selectivity index) was computed using the formula (\({\text{SI}} = {\text{CC}}50/{\text{MIC}}\)) (Protopopova et al. 2005).
Molecular dynamic simulations study
To better understand the interaction of the most potent antitubercular compound from this study with DprE1 protein, MD simulations studies were carried out with the help of Desmond module (Schrödinger Release 2018-3) (Bowers et al. 2006). MD simulations were carried out in three steps which included system builder, minimization and simulation. In the system builder step, the DprE1 protein–ligand complex was solvated using SPC as a solvent system in an orthorhombic box shape. This solvated complex was then minimized employing SD method with iterations fixed at 2000. The system was slowly equilibrated at 300 K (temperature) and 1.01325 bar (pressure) with the help of Nose–Hoover thermostat method and Martina–Tobias–Klein barostat method. In the last step, the solvated and minimized complex was simulated for 20,000 ps.
Results and discussion
Design of non-covalent DprE1 inhibitors
Based on the non-covalent interactions between the amino acid residues in the active pocket of DprE1 protein (PDB ID 4P8C) and the co-crystallized ligand with the protein, i.e. 6-(trifluoromethyl)-3-{[4-(trifluoromethyl)benzyl]amino}quinoxaline-2-carboxylic acid, we developed a four-feature e-Pharmacophore model (ANRR) as shown in Fig. 2. Recently, Zhang et al. have also reported a three-feature pharmacophore (3-feature) model which had two hydrophobic and one hydrogen bond acceptor feature (Zhang et al. 2018). They had selected trifluoromethyl group substituted at the 6th position of quinoxaline ring as one of the hydrophobic features, while we have selected the fused phenyl ring of the quinoxaline ring system. The distance reported by Zhang et al., between the two hydrophobic groups, was different than ours. We found it to be 6.65 Å while Zhang et al. have reported it to be around 10.5 Å. The inter-site angle and inter-site distance are depicted in Fig. 2a and b. Since the developed model had features of a known non-covalent inhibitor of DprE1, it was assumed that the potential DprE1 inhibitors should have similar features. The developed model had two aromatic ring features (R7 and R9) at 6.65 Å and an acceptor feature (A2) at 6.41 Å from R7 and 2.77 Å from R9. A2 was at 2.97 Å from the negatively charged feature (N6) while N6 was at 5.64 Å from R9 and 6.49 Å from R7. The angle was computed between three features with the second feature at the centre. The angle between A2-R9-R7 was 72.9°, R9-R7-A2 was 24.4°, R9-R7-N6 was 50.8°, R7-N6-A2 was 75.1°, and R9-A2-N6 was 158.3°. Based on this insight, we propose that any ligand with all these four features at this specific distance and angle might be a potential DprE1 non-covalent inhibitor.
The developed e-Pharmacophore model was used to map ligands to assess whether they possessed pharmacophoric features essential for DprE1 inhibition. In our previous work, we had found some piperidone derivatives showing good antitubercular activity. We mapped all those 25 compounds on the developed e-Pharmacophore model and found none of the compounds had even two matching features. The best compound was R7 ((3E,5E)‐3,5‐Bis(furan‐2‐ylmethylidene)‐4‐(2‐phenylhydrazinylidene)piperidine) with one match and a phase screen score of 0.245. The detailed design strategy is depicted in Fig. 3, where we found that if -NH group of piperidone ring could be substituted, it could increase the matches. As discussed in “Introduction”, benzimidazole derivatives have been reported as DprE1 inhibitors with potent antitubercular activity; hence, we added benzimidazole ring system and this indeed led to increasing in the matches of features as well as phase screen score. All the 25 derivatives were substituted with 1H-benzo[d]imidazol-2-yl)methyl group. This exercise led to an increase in the number of matches to three out of four features. N6 pharmacophoric did not match, but there was a substantial increase in the phase screen score of the compounds. We decided to synthesize all those compounds which had at least three matches and a phase screen score more than 1.60 and twelve compounds passed this test. Derivative B1, i.e. (3E,5E)-1-((1H-benzo[d]imidazol-2-yl)methyl)-3,5-bis(furan-2-ylmethylene)piperidin-4-one, showed the highest screen score of 1.84 while compound B4, i.e. 2-((3E,5E)-1-((1H-benzo[d]imidazol-2-yl)methyl)-3,5-bis(furan-2-ylmethylene)piperidin-4-ylidene)hydrazine-1-carbothioamide, showed the least screen score of 1.60. The rest of the compounds had a phase screen score between 1.84 and 1.60 as shown in Table 1.

e-Pharmacophore-based design strategy of piperidone derivatives as potential DprE1 inhibitors
Molecular docking and free binding energy calculations
Molecular docking study (XP mode) was done to understand the binding of the designed compounds with DprE1 protein. The docking protocol was validated by redocking the co-crystallized ligand at its reported binding site and then by calculating the RMS value by superimposing both the poses. As shown in Fig. 4, both poses of the co-crystallized ligand were similar to an RMS value of 0.5149.

Validation of the docking protocol by the superimposition of two poses of the co-crystallized ligand
The compounds were ranked by their docking score, and non-bonding amino acid interactions were compared with the co-crystallized ligand. As shown in Fig. 5f, the co-crystallized ligand showed H-bond interaction with LYS418 amino acid residue. It also showed hydrophobic interactions with TYR60, TRP230, PRO316, LEU317, LUE363, VAL365, PHE369 and CYS387 residues. It showed a docking score of − 6.728 and free binding energy of − 47.92 kcal/mol as computed by prime MM-GBSA and shown by MM-GBSA dG bind. Compound B1 which had the highest phase screen score also showed the highest docking score of − 7.644, even better than the co-crystallized ligand. The dG bind value of B1 was also more than − 49.64 kcal/mol. It also showed possible H-bond interactions with LYS418 residue and its other interactions were also like the co-crystallized ligand. The docking score for other five compounds like B2, B3, B4, B5 and B12 was more than − 6.0 and was comparable to the co-crystallized ligand. Compound B3 showed potential H-bond interaction with GLN336 residue and not with LYS418 residue. The rest of the compounds had docking scores below − 6.0 and more than − 2.50. Docking study and MM-GBSA free binding studies suggest that few of the designed compounds like B1, B2, B3, B4, B5 and B12 might form a stable complex with DprE1 protein. Ligand interaction diagram (2D) is depicted for the top five ranked compounds in Fig. 5a–e.

Ligand interaction diagram (2D) of the top five ranked compounds based on docking score and the co-crystallized compound. a B1, b B2, c B3, d B4, e B12, f co-crystallized ligand
Chemistry
The reaction between piperidin-4-one (1) and 2-chloromethylbenzimidazole (2) resulted in the synthesis of 1-(1H-benzimidazol-2-ylmethyl)piperidin-4-one (3). In this reaction, chloro group of the 2-chloromethylbenzimidazole acts as a good leaving group. This leads to a nucleophilic attack on the methylene carbon of compound 2 by the secondary amine of 4-piperidone. Hydrochloric acid formed as a by-product of this reaction is neutralized by TEA thus driving the reaction forward. In the next step, intermediate 3 undergoes aldol condensation when treated with 2-furfural and NaOH to form an aldol product which upon loss of water molecule forms the first derivative B1. In the last step, B1 was treated with different substituted amines to yield compounds B2-B12 through sodium acetate catalysed Schiff’s reaction.
In vitro antitubercular evaluation
Agar dilution method was employed for the in vitro antitubercular assessment of the synthesized derivatives against M. tuberculosis H37Rv (ATCC27294) strain, and minimum inhibitory concentration (MIC) was determined in triplicates. The MIC values (μg/ml) are given in Table 1. The compound B12 showed the most potent antitubercular activity with a MIC of 1.56 μg/ml which was better than the standard drug ethambutol (3.125 μg/ml). Compounds B7 and B11 were found to be equipotent with ethambutol and had a MIC value of 3.125 μg/ml. Compound B1 had a MIC value of 50 μg/ml which was the least potent among all the tested compounds. Compounds B2, B4, B5, B6 and B10 showed a MIC value of 6.25 μg/ml which was comparable with ethambutol activity. It was observed that non-substitution at the 4th position of piperidone ring like in B1 resulted in the least potent compound of the series. Hence, substitution at 4th position was favourable for the activity. The most active compound B12 had o,p-difluoro substitution at the phenyl ring attached to the 4th position of the piperidine ring. Similarly, B11 with o,p-dichloro substitution was equipotent with ethambutol. From this, we can conclude that electron-withdrawing groups at the phenyl ring on o,p-position led to an increase in the activity. Single fluoro substitution at para position (B6) was less potent than o,p-difluoro substitution (B12). Hence, di-substitution at o,p-position was more favourable. Electron-donating groups like methyl (B9) and methoxy (B8) at the phenyl ring led to a decrease in the activity as both compounds showed a MIC value of 25 μg/ml. From this study, we can conclude that the piperidine ring substituted with furylidene rings and methyl benzimidazole ring showed potent antitubercular activity. But if we compare the antitubercular activity of parent compounds without substitution at the 4th position of piperidine ring like R2, R3, R8, R9 and R11 for which the corresponding compounds are B2, B3, B8, B9 and B11, we found that the activity was same in both the cases (Kumar et al. 2019). The compounds like B4, B5, B6 and B12 were twice less potent than their unsubstituted derivatives. Hence, we can conclude that the substitution at the 4th position of piperidine ring was well tolerated.
Cytotoxicity studies
As antitubercular drugs should be non-toxic to the normal cells; hence, all the synthesized derivatives were tested for their cytotoxicity potential using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay against Vero cell lines. MTT is a colorimetric assay where yellow-coloured MTT dye is reduced to insoluble purple-coloured formazan in living cells. We selected Vero cell lines as several authors have reported such cell safety study using Vero cell lines (Sukheja et al. 2017; Logu et al. 2002). The compounds were tested at different concentrations of 62.5. 125, 250 and 500 μg/ml, and CC50 (cytotoxic concentration) value was calculated. The results are tabulated in Table 1, and we found that all tested compounds showed CC50 value more than 200 μg/ml. The most active compound B12 showed a CC50 value of 330 μg/ml which indicates its cell safety profile. Further SI (selectivity index) values were computed using CC50 and MIC values. All the compounds except B1 exhibited excellent SI values. SI value for B1 was 8.24, and ideally, it should have been more than 10. The higher the SI value, the better is the cell safety profile of the drugs. For compound B12, SI value was 211.53. Hence, from the above in vitro studies, we can conclude that compound B12 is a potent antitubercular compound and can be considered non-toxic to cells.
MD simulations studies
Molecular docking studies and MM-GBSA analysis also showed that compound B12 might form a stable complex with DprE1 enzyme, but crucial H-bond interaction with LYS418 residue was missing. Hence, to better understand the interaction of compound B12 with DprE1 protein, MD simulations were carried out. The trajectory file obtained after 20,000 ps simulation was analysed, the simulation interaction diagram report was generated, and the detailed result is shown in Fig. 6. One frame was captured every 20 ps, and thus, a total of 1000 frames were generated for the study which was superimposed on the first frame, and RMSD value was calculated as shown in Fig. 6a. DprE1-B12 RMSD plot suggests that the complex was stable throughout the simulation period as the deviation was less than 2 Å at any given point of time. However, a slight drift was observed for the first half of simulation, i.e. first 10 ns; after that, the complex stabilized and drifted together. The most interesting finding was observed in the 2D protein–ligand contact plot as shown in Fig. 6b. We observed that B12 showed crucial H-bond interaction with LYS418 residue for 75% of the simulations time. This interaction was not observed in the docking studies. Also, it was evident from the histogram plot (Fig. 6c) that B12 showed similar non-bonding interactions with DprE1 enzyme as shown by its co-crystallized ligand. It showed hydrophobic interactions with LYS60, TRP230, LUE283, TYR314, PRO316, LEU317, PHE362, LEU363, VAL385 and CYS387 residues. A water bridge interaction was also observed with TYR314 residue. Thus, MD simulation analysis supports our hypothesis that B12 will inhibit DprE1 protein.

MD simulation analysis of compound B12 with DprE1 protein. a B12-DprE1 RMSD plot, b B12-DprE1 protein–ligand 2D interaction, c B12-DprE1 histogram plot of protein–ligand contact
Conclusion
In the present work, we have developed a four-feature e-Pharmacophore model based on the receptor–ligand cavity of DprE1 protein and mapped our previous reported library of compounds against it. The compounds were ranked on phase screen score, and the insights obtained from their alignment was used to design some novel benzimidazole-substituted compounds which might act as DprE1 inhibitors. These designed compounds had three matches out of four pharmacophoric features and favourable phase screen score up to 1.84. The designed compounds were also put for docking studies in XP mode, and some derivatives like B1, B2, B4, B5 and B12 showed comparable docking score with respect to the co-crystallized ligand. The designed compounds were synthesized in good yield, purified and characterized using various techniques like IR, NMR, LC-MS and elemental analysis. In vitro antitubercular activity was carried out on M. tuberculosis H37Rv (ATCC27294) strain using agar dilution method, and minimum inhibitory concentration (MIC) was determined in triplicates. All the compounds showed potent antitubercular activity in comparison with ethambutol. The compound B12 showed the most potent antitubercular activity with a MIC of 1.56 μg/ml which was better than the standard drug ethambutol (3.125 μg/ml). Compounds B7 and B11 were found to be equipotent with ethambutol and had a MIC value of 3.125 μg/ml. From this study, we can conclude that the piperidine ring substituted with furylidene rings and methyl benzimidazole ring showed potent antitubercular activity. Further substitution at the 4th position of piperidine ring was well tolerated with electron-withdrawing groups at the phenyl ring on o,p-position that led to an increase in the activity. Cytotoxicity studies against Vero cell lines proved that these compounds can be considered to be safe which is reflected in their high SI values. MD simulation study was done for the most potent compound B12 in complex with DprE1 protein and it also suggests that B12 will form a stable complex with DprE1 protein and showed the crucial H-bond interaction with LYS418 residue. To the best of our knowledge, this is the first work reported where e-Pharmacophore modelling has resulted in potent antitubercular compounds with the potential to inhibit DprE1 protein. Further in vitro enzyme inhibition studies are required to validate these findings.
Availability of data and material
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Change history
05 July 2021
A Correction to this paper has been published: https://doi.org/10.1007/s11696-021-01762-0
References
Addla D, Jallapally A, Gurram D et al (2014) Rational design, synthesis and antitubercular evaluation of novel 2-(trifluoromethyl)phenothiazine-[1,2,3]triazole hybrids. Bioorganic Med Chem Lett 24:233–236. https://doi.org/10.1016/j.bmcl.2013.11.031
Arun KG, Sharanya CS, Abhithaj J et al (2020) Drug repurposing against SARS-CoV-2 using E-pharmacophore based virtual screening, molecular docking and molecular dynamics with main protease as the target. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1779819
Banerjee D, Yogeeswari P, Bhat P et al (2011) Novel isatinyl thiosemicarbazones derivatives as potential molecule to combat HIV-TB co-infection. Eur J Med Chem 46:106–121. https://doi.org/10.1016/j.ejmech.2010.10.020
Batt SM, Jabeen T, Bhowruth V et al (2012) Structural basis of inhibition of Mycobacterium tuberculosis DprE1 by benzothiazinone inhibitors. Proc Natl Acad Sci U S A 109:11354–11359. https://doi.org/10.1073/pnas.1205735109
Batt SM, Cacho Izquierdo M, Castro Pichel J et al (2016) Whole cell target engagement identifies novel inhibitors of mycobacterium tuberculosis decaprenylphosphoryl-β- d -ribose oxidase. ACS Infect Dis 1:615–626. https://doi.org/10.1021/acsinfecdis.5b00065
Bowers KJ, Chow E, Xu H et al (2006) Scalable algorithms for molecular dynamics simulations on commodity clusters. In: Proceedings of the 2006 ACM/IEEE conference on supercomputing, SC’06, ACM Press, New York, New York, USA, p 84
Brecik M, Centárová I, Mukherjee R et al (2015) DprE1 is a vulnerable tuberculosis drug target due to its cell wall localization. ACS Chem Biol 10:1631–1636. https://doi.org/10.1021/acschembio.5b00237
Chikhale RV, Barmade MA, Murumkar PR, Yadav MR (2018) Overview of the development of DprE1 inhibitors for combating the menace of tuberculosis. J Med Chem 61:8563–8593. https://doi.org/10.1021/acs.jmedchem.8b00281
Cohen J (2013) Approval of novel TB drug celebrated—With restraint. Science 339:130
De Logu A, Onnis V, Saddi B et al (2002) Activity of a new class of isonicotinoylhydrazones used alone and in combination with isoniazid, rifampicin, ethambutol, para-aminosalicylic acid and clofazimine against Mycobacterium tuberculosis. J Antimicrob Chemother 49:275–282. https://doi.org/10.1093/jac/49.2.275
Denizot F, Lang R (1986) Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 89:271–277. https://doi.org/10.1016/0022-1759(86)90368-6
Dixon SL, Smondyrev AM, Knoll EH et al (2006) PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J Comput Aided Mol Des 20:647–671. https://doi.org/10.1007/s10822-006-9087-6
Friesner RA, Murphy RB, Repasky MP et al (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49:6177–6196. https://doi.org/10.1021/jm051256o
Gawad J, Bonde C (2018) Decaprenyl-phosphoryl-ribose 2′-epimerase (DprE1): challenging target for antitubercular drug discovery. Chem Cent J. https://doi.org/10.1186/s13065-018-0441-2
Global tuberculosis report (2020) https://www.who.int/publications/i/item/9789240013131. Accessed 1 Mar 2021
Hall L, Jude KP, Clark SL et al (2012) Evaluation of the sensititre MycoTB plate for susceptibility testing of the Mycobacterium tuberculosis complex against first- and second-line agents. J Clin Microbiol 50:3732–3734. https://doi.org/10.1128/JCM.02048-12
Jacobson MP, Pincus DL, Rapp CS et al (2004) A hierarchical approach to all-atom protein loop prediction. Proteins Struct Funct Genet 55:351–367. https://doi.org/10.1002/prot.10613
Kálai T, Kuppusamy ML, Balog M et al (2011) Synthesis of N-substituted 3,5-bis(arylidene)-4-piperidones with high antitumor and antioxidant activity. J Med Chem 54:5414–5421. https://doi.org/10.1021/jm200353f
Katsori AM, Chatzopoulou M, Dimas K et al (2011) Curcumin analogues as possible anti-proliferative & anti-inflammatory agents. Eur J Med Chem 46:2722–2735. https://doi.org/10.1016/j.ejmech.2011.03.060
Keri RS, Rajappa CK, Patil SA, Nagaraja BM (2016) Benzimidazole-core as an antimycobacterial agent. Pharmacol Reports 68:1254–1265
Kumar A, Revathi R, Sriram D et al (2019) Targeting HIV-TB coinfection by developing novel piperidin-4-substituted imines: design, synthesis, in vitro and in silico studies. Arch Pharm (weinheim) 352:1800358. https://doi.org/10.1002/ardp.201800358
Kumar A, Rathi E, Kini SG (2020) Drug repurposing approach for the identification and designing of potential E6 inhibitors against cervical cancer: an in silico investigation. Struct Chem 31:141–153. https://doi.org/10.1007/s11224-019-01378-x
Kumar A, Rai S, Rathi E et al (2021) Pharmacophore-guided fragment-based design of novel mammalian target of rapamycin inhibitors: extra precision docking, fingerprint-based 2D and atom-based 3D-QSAR modelling. J Biomol Struct Dyn 39:1155–1173. https://doi.org/10.1080/07391102.2020.1726816
Landge S, Mullick AB, Nagalapur K et al (2015) Discovery of benzothiazoles as antimycobacterial agents: synthesis, structure-activity relationships and binding studies with Mycobacterium tuberculosis decaprenylphosphoryl-β-d-ribose 2′-oxidase. Bioorganic Med Chem 23:7694–7710. https://doi.org/10.1016/j.bmc.2015.11.017
Li J, Abel R, Zhu K et al (2011) The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins Struct Funct Bioinform 79:2794–2812. https://doi.org/10.1002/prot.23106
Loving K, Salam NK, Sherman W (2009) Energetic analysis of fragment docking and application to structure-based pharmacophore hypothesis generation. J Comput Aided Mol Des 23:541–554. https://doi.org/10.1007/s10822-009-9268-1
Madhavi Sastry G, Adzhigirey M, Day T et al (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27:221–234. https://doi.org/10.1007/s10822-013-9644-8
Magnet S, Hartkoorn RC, Székely R et al (2010) Leads for antitubercular compounds from kinase inhibitor library screens. Tuberculosis 90:354–360. https://doi.org/10.1016/j.tube.2010.09.001
Makarov V, Manina G, Mikusova K et al (2009) Benzothiazinones Kill Mycobacterium tuberculosis by blocking Arabinan synthesis. Science 324(5928):801–804. https://doi.org/10.1126/science.1171583
Manjunatha R, Shandil R, Panda M et al (2019) Scaffold morphing to identify Novel DprE1 inhibitors with antimycobacterial activity. ACS Med Chem Lett 10:1480–1485. https://doi.org/10.1021/acsmedchemlett.9b00343
Mikušová K, Huang H, Yagi T et al (2005) Decaprenylphosphoryl arabinofuranose, the donor of the D-arabinofuranosyl residues of mycobacterial arabinan, is formed via a two-step epimerization of decaprenylphosphoryl ribose. J Bacteriol 187:8020–8025. https://doi.org/10.1128/JB.187.23.8020-8025.2005
Mir F, Shafi S, Zaman MS et al (2014) Sulfur rich 2-mercaptobenzothiazole and 1,2,3-triazole conjugates as novel antitubercular agents. Eur J Med Chem 76:274–283. https://doi.org/10.1016/j.ejmech.2014.02.017
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63. https://doi.org/10.1016/0022-1759(83)90303-4
Neres J, Pojer F, Molteni E et al (2012) Structural basis for benzothiazinone-mediated killing of Mycobacterium tuberculosis. Sci Transl Med 4:150ra121-150ra121. https://doi.org/10.1126/scitranslmed.3004395
Neres J, Hartkoorn RC, Chiarelli LR et al (2015) 2-carboxyquinoxalines kill mycobacterium tuberculosis through noncovalent inhibition of DprE1. ACS Chem Biol 10:705–714. https://doi.org/10.1021/cb5007163
Panda M, Ramachandran S, Ramachandran V et al (2014) Discovery of pyrazolopyridones as a novel class of noncovalent DprE1 inhibitor with potent anti-mycobacterial activity. J Med Chem 57:4761–4771. https://doi.org/10.1021/jm5002937
Protopopova M, Hanrahan C, Nikonenko B et al (2005) Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J Antimicrob Chemother 56:968–974. https://doi.org/10.1093/jac/dki319
Raviglione M, Marais B, Floyd K et al (2012) Scaling up interventions to achieve global tuberculosis control: progress and new developments. Lancet 379:1902–1913
Salam NK, Nuti R, Sherman W (2009) Novel method for generating structure-based pharmacophores using energetic analysis. J Chem Inf Model 49:2356–2368. https://doi.org/10.1021/ci900212v
Sukheja P, Kumar P, Mittal N et al (2017) A novel small-molecule inhibitor of the mycobacterium tuberculosis demethylmenaquinone methyltransferase menG is bactericidal to both growing and nutritionally deprived persister cells. mBio https://doi.org/10.1128/mBio.02022-16
Surineni G, Gao Y, Hussain M et al (2019) Design, synthesis, and in vitro biological evaluation of novel benzimidazole tethered allylidenehydrazinylmethylthiazole derivatives as potent inhibitors of Mycobacterium tuberculosis. Medchemcomm 10:49–60. https://doi.org/10.1039/c8md00389k
Yeong KY, Ang CW, Ali MA et al (2017) Antituberculosis agents bearing the 1,2-disubstituted benzimidazole scaffold. Med Chem Res 26:770–778. https://doi.org/10.1007/s00044-017-1784-2
Zhang G, Guo S, Cui H, Qi J (2018) Virtual screening of small molecular inhibitors against DprE1. Molecules 23(3):524. https://doi.org/10.3390/molecules23030524
Funding
Open access funding provided by Manipal Academy of Higher Education, Manipal. No funding was received for conducting this study.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by AK and RR. The first draft of the manuscript was written by AK and ER, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original Online version of this article was revised due to the superscription of incorrect affiliation numbers.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kumar, A., Rajappan, R., Kini, S.G. et al. e-Pharmacophore model-guided design of potential DprE1 inhibitors: synthesis, in vitro antitubercular assay and molecular modelling studies. Chem. Pap. 75, 5571–5585 (2021). https://doi.org/10.1007/s11696-021-01743-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-021-01743-3