
Abstract
2,5-Lutidinium bromanilate is a molecular complex that consists of bromanilic acid and 2,5-lutidine in which hydrogen-bonding interactions occur between them, producing a charge-assisted bifurcated N–H…O hydrogen bond. Bond characteristics are determined from the experimental charge density distribution of the molecular complex using the Hansen–Coppens model. The electron density, topological properties, electrostatic potential and atomic charges of the molecule have been investigated to better understand the atomic, molecular and electronic properties in a detailed manner. The electronic nature of the significantly important charge-assisted bifurcated hydrogen bond has been analyzed with the help of topological properties.
Graphic abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In recent years, the occurrence of molecular complexes in molecular crystals has received considerable attention, especially from the viewpoint of drug design and crystal engineering. The significant behavior of hydrogen-bonding interactions plays a key role in the effective production of ionic molecular complexes (Thomas et al. 2013), particularly in the design of pharmaceutical materials. In such a molecular complex that consists of bromanilic acid with the N-heterocyclic compound 2,5-lutidine, intra- and/or intermolecular interaction occurs between the bromanilic acid molecule and the N heteroatom of the pyridine ring of 2,5-lutidine, producing a charge-assisted bifurcated N–H…O hydrogen bond. In a charge transfer process, the electrophile receives charge from the nucleophile, and hence, in many cases electrophile acts as a Lewis acid and nucleophile acts as a Lewis base.
Intra- and intermolecular interactions are drawing special attention due to their crucial role in the determination of chemical structure and reactivity of the molecules in the field of crystallography (Scheiner 1997). Due to the productive nature of hydrogen bonding, it is accountable for the chemical reactions as commencement in crystal engineering (Aakeröy et al. 2016; Braga et al. 1998; Cleland 1997; Desiraju and Steiner 1999; Jeffrey and Jeffrey 1997; Perrin and Nielson 1997; Sobczyk et al. 2005) which allows the controlled growth of supramolecular groups in pharmaceuticals. Moreover, the charge-assisted hydrogen bond helps in the salt formation process with desired properties in drug engineering.
Besides, it is highly crucial to provide accurate electrostatics parameters for drugs, pharmaceutical compounds and molecular complexes in order to understand their mode of action and will be useful in further investigations such as modeling and docking them into the binding sites of enzymes and proteins. Extraction of the precise electron density distribution in crystalline materials is achieved using ultra-high-resolution X-ray diffraction experiments, allowing us to possibly derive one-electron properties such as the electrostatic potential and other atomic or molecular moments.
In this paper, the experimental charge density study of 2,5-LB has been performed successfully from high-resolution single-crystal X-ray diffraction at low temperature (100 K) to understand the bonding and topological properties of the chosen molecule. The topologies of the electron density and the electrostatic potential are carefully analyzed. Special attention has been paid to one of the important aspects of hydrogen bonding: the bifurcated hydrogen bonding present in the compound.
Materials and methods
Data information
A high-resolution dataset for 2,5-lutidinium bromanilate was collected up to (sinθ/λ)max = 0.94 Å−1 with 99.1% completeness and redundancy. The crystallographic data for this study were taken from the article (Aakeröy et al. 2016), and the structure was refined and then used for the charge density refinement.
Refinement details
The experimental charge density refinement was accomplished against diffraction intensities using the software MOPRO (Jelsch et al. 2005). Some of the major parameters such as restraints/constraints on the stereochemistry, thermal displacement, and charge density parameters have been included in the program itself, and multipolar scattering factor formalism has been utilized in the program. For Br atom, the multipole expansion was done up to the hexadecapole level, for O, C and N atoms up to the octupole level and for H atom up to the dipole level. The Uij parameters were fixed to the values taken from the SHADE server (Madsen 2006), and the H–X distances of H atoms were restricted to the values derived from neutron diffraction studies (Allen and Bruno 2010) with a restrained sigma of 0.01 Å. The core and valence spherical scattering factors have been calculated from Su and Coppens (1997) which were obtained from the International Tables for Crystallography (Prince 2004). Using multipolar Hansen–Coppens formalism (Hansen and Coppens 1978), the charge density was refined against structure-factor amplitudes finally for pseudo-atom electron density. Refinement on the parameters such as scale factor, XYZ, Uij, multipole Plm, valence populations Pval and contraction/expansion coefficients κ and κ′ was done as far as convergence is reached. The refinement details are given in Table S1.
The static and deformation maps were computed and plotted using the VMoPro module of the MoPro software (Jelsch et al. 2005,) and the molecular view with atomic displacement parameter ellipsoids was produced with MoProViewer (Guillot et al. 2001).
Results and discussion
Structure description
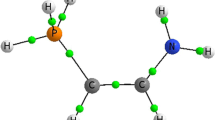
For obtaining information about the geometric arrangement of the atoms in the unit cell, the molecular structure of 2,5-LB has been discussed with the help of bond length, bond angle and dihedral angle. The molecular structure of 2,5-LB with atomic labeling as derived from X-ray experiments is depicted in Fig. 1. Among the various investigated bonds, the C1–C2 bond is found with the shortest bond distance (1.349 Å), which reveals the partial double-bond nature and the possibility of the occurrence of resonance in the bond. The bond distances for all C=O bonds are in the range 1.221–1.328 Å, which agrees with the standard values (Allen et al. 1987).

Molecular structure of 2,5-LB showing the bifurcated H bond (Dolomanov et al. 2009)
It is interesting to take a closer look at the geometries of the two different methyl (CH3) groups of 2,5-LB as the calculated C–C–H bond angles are equal to 109.5°, which shows the tetrahedral shape and the hybridization involved associated with the carbon atoms C12 and C13 are sp3 hybridizations. The C7–N1–C11 bond angle of the pyridine ring of 2,5-lutidine is 123.63° which is greater than the ideal value 120°; this slight variation may be due to the lone-pair electrons of nitrogen involved in sp3 hybridization and the occurrence of delocalization of double bonds.
Intermolecular interactions
The crystal structure of 2,5-LB is mainly stabilized by typical O–H…O, N–H…O and C–H…O types of hydrogen-bonding interactions. The structure shows the formation of bifurcated hydrogen bonding between bromanilate and the 2,5-lutidinium compound as shown in Fig. 1. Generally, the bifurcated hydrogen bond is formed when there is larger flexibility found in moderate and weak hydrogen bonds involving two acceptor sites (Taylor et al. 1984). In the molecule, the type of bifurcated hydrogen bond found is Y type and is termed as a three-center hydrogen bond (Rozas et al. 1998). Interestingly, the sum of the angles in the bifurcated hydrogen bond is exactly equal to 360°, and hence, the bond is found to be planar (Martin 2012; Rozas et al. 1998).
The hydrogen-bonding parameters of the two medium-strength O1–H1…O2 interactions are: H1…O2i; 2.14 Å, O1–H1i; 0.82 Å, and the angles are 119.71° and H1…O2ii; 2.04 Å, O1–H1ii; 0.82 Å, and the angles are 137.93°, respectively. In addition to these interactions, three weak N1–H1A…O3, N1–H1A…O4 and C11–H11…O3 hydrogen bonds are also involved in the packing of the molecule. The hydrogen-bonding interactions are shown in Fig. 2a. The experimental hydrogen-bonding parameters are given in Table S2.

a The intermolecular interactions (c* axis). b View of Hishfeld surface of 2,5-LB
One of the best ways to define the molecular surface and to picture the intermolecular interactions is the Hirshfeld surface analysis (Hirshfeld 1977a, b; McKinnon et al. 2004; Wolff et al. 2012). Figure 2b shows the Hirshfeld surface of 2,5-LB mapped with the function dnorm since this function displays the stronger O–H…O interactions sensitively.
Here, the strong O–H…O interactions are clearly visible and are identified from the light red-colored circular patch. The values of di and de for any Hirshfeld surface point are directly plotted against each other for generating the fingerprint plot (Fig. 3a), which shows typical motifs and reveals the individual interaction types which are present in the complex (Spackman and McKinnon 2002). The two long and sharp spikes at the most top and bottom of the fingerprint plot are due to the O…H/H…O contacts of 2,5-LB.

Fingerprint plots: a full, b resolved into O…H/H…O contacts and c the percentages of contact contributing to the total Hirshfeld surface area of the 2,5-LB molecule in the crystal
The percentage contribution of various intermolecular contacts (C…H, O…H, Br…H and others) is shown in Fig. 3c. Here, 24.2% of the surface area of the molecule is covered by O…H contacts. The Br…H interactions contribute to a larger area (25.0%) of the total Hirshfeld surface. It has small spikes in the right and left most corner on both sides in the fingerprint plot (Fig. 3a). The other type of intermolecular interactions involving π electrons is the π…π interactions that also have to be considered in the case of 2,5-LB.
Topological analysis of 2,5-LB
For obtaining a quantitative description of the electronic structure of 2,5-LB, a full topological analysis was carried out on the basis of the theory of atoms in molecules (AIM) as developed by Laidig and Bader (1990). The topological properties serve as an efficient tool to discover potential correlations between the physical properties and the molecular density distributions.
For highlighting the quality of the data, the featureless residual density map is plotted (Fig. 4a) and the maps contain very little noise, attesting to the quality of the data. The topological properties of the electron density of 2,5-lutidinium bromanilate are listed in Table S3. The static deformation density maps of the bromanilate of 2,5-LB portray the concentration of electron density between connected atoms and are also associated with the lone pairs of oxygen atoms (O1, O2, O3 and O4) as well as nitrogen (N1) atom of the 2,5-LB. The depletion of electron density shown in red-colored contours in Fig. 4b, c found near the H atoms reveals their polar character. The three-dimensional static multipole deformation density map of 2,5-LB is shown in Fig. 4d. The range of electron density of the C–C bonds of the 2,5-LB molecule is 1.513–2.289 e/Å3 and the average is 1.897 e/Å3, and this range is comparable with the C–C bonds of the aromatic ring of DL-histidine (Coppens et al. 1999). Among all the C–C bonds in the molecule, the electron density of the bond C5–C6 is comparatively less (1.519 e/Å3), and this might be because of the sharing of excess electrons of C6 atom with O4 atom.

a Residual density in the plane of the bromanilate moiety. Blue solid lines indicate the positive contours, negative contours are the red lines, and zero contours are yellow dashed lines. The contour interval is 0.05 e/Å3. The experimental static deformation density maps. b Bromanilate and c 2,5-lutidine. d Three-dimensional experimental deformation density map of the 2,5-LB molecule. The contour maps are drawn at ± 0.1 e/Å3
Notably, the electron density of the C=O bonds is in the range of 2.404–2.688 e/Å3, and this value is closer to similar bonds reported in l-glutamine molecule (Wagner and Luger 2001). The shortest O1–H1 and N1–H1A bonds reveal the higher magnitude of electron density at the critical point sites and are found to be 2.021 e/Å3 and 2.063 e/Å3, respectively. The ρBCP(r) of the C–N bonds is in the range 2.012–2.199 e/Å3; a similar behavior is found in the C–N bond of the dl-histidine molecule (Coppens et al. 1999). For the methyl groups, the electron density values at the BCP of the C–H bonds with the range of 1.578–1.826 e/Å3 are found to be much less than those of the N–H bond. In addition to the values reported in Table S3, a ring CP (3, + 1) was found at the center of the pyridine and benzene ring between the atoms C8 and C11 and between the atoms C6 and C3 with ρBCP(r) 0.1269 e/Å3 and 0.1675 e/Å3, respectively, which is clearly visible in the molecular graph (Fig. 5). The bond strength has been identified with the help of bond order calculation, and the values are listed in Table S3.

The molecular graph showing the (3, − 1) bond and (3, + 1) ring critical points of the 2,5-LB molecule. The large circles represent the atomic positions, and the small red and blue circles indicate the bond and ring critical points, respectively
In order to determine the polarization of the bonds, a bond path analysis has been performed using MOPRO (Jelsch et al. 2005). As far as 2,5-LB is concerned, all the heteroatomic bonds are polarized, which leads to the shifting of BCP positions nearer to the C atoms. From the molecular graph (Fig. 5), it is understood that the C=O bonds are well polarized and their BCP positions are present at a distance of 14.1% from the midpoint. Similarly, C–N bonds of the pyridine ring are also well polarized as the BCPs are shifted close to C7 and C11 atoms having the distance of CP to middle 0.1048 Å and 0.1199 Å, respectively. Among all the bonds, the N1–H1A bond is highly polarized as the BCP is shifted (nearly 22.7%) away more from the midpoint of the bond.
The average of experimental ellipticities (ε) of the aromatic bonds in the phenyl ring is 0.28 which is closer to the one found by Bader (1985, 1991) obtained for benzene (0.23) with a 631G* basis set, signifying π-delocalization. The nature of isotropy or anisotropy of the bond is revealed by the bond ellipticity. With the help of ellipticity values, the C–C and C–N bonds are said to be anisotropic as values are above 0.2 (Bader 1985, 1991). The C–C ring clearly shows the π delocalization as the ellipticities are high with an average value of 0.28 for this complex 2,5-LB.
The chemical features of the molecule can always be illustrated by the Laplacian (\({\nabla }^{2}{\rho }_{\mathrm{B}\mathrm{C}\mathrm{P}}(r))\) of the electron density which typically provides the local charge concentration (LCC) when the Laplacian is positive and local charge depletion (LCD) when the Laplacian is negative. The average experimental \({\nabla }^{2}{\rho }_{\mathrm{B}\mathrm{C}\mathrm{P}}(r)\) of the C–C bonds of the benzene ring of bromanilate is − 13.865 e/Å5 and of the pyridine ring of lutidinium is − 12.041 e/Å5. The value of \({\nabla }^{2}{\rho }_{\mathrm{B}\mathrm{C}\mathrm{P}}(r)\) of the bond C10–C13 (− 8.845 e/Å5) shows significantly low charge concentration among all the C–C bonds. The Laplacian of the O4–C6 bond is − 12.19 e/Å5; the electron density in the bond is less concentrated than in the other O=C bonds where the range of O=C bond is − 12.19 e/Å5 to − 25.40 e/Å5 and the average value is − 21.647 e/Å5. Notably, \({\nabla }^{2}{\rho }_{\mathrm{B}\mathrm{C}\mathrm{P}}(r)\) of the bonds Br1–C4 and Br2–C1 is found to be 3.986 e/Å5 and 4.268 e/Å5, respectively; the positive value indicates that the electron density of the bond is highly depleted, and the bond is a closed-shell interaction (Fig. 6a). It is a strange behavior of Br not making any covalent bond with C atoms and remained alone with its lone pairs making a closed shell, which is evident from Fig. 6a. This is pronounced as a weaker interaction than a covalent or shared-shell interaction and can be bromine lone-pair π interaction.

Laplacian of the electron density of the 2,5-LB. Solid blue lines and dotted red lines represent the positive and negative contours, respectively. a Bromanilate, b 2,5-lutidine and c view of the bifurcated hydrogen bond
The \({\nabla }^{2}{\rho }_{\mathrm{B}\mathrm{C}\mathrm{P}}(r)\) value of C–N bonds of the pyridine ring at the BCP varies from − 11.11 to − 15.39 e/Å5, whereas the \({\nabla }^{2}{\rho }_{\mathrm{B}\mathrm{C}\mathrm{P}}(r)\) value of the bond N1–H1A of the ring is highly concentrated (− 25.44 e/Å5) than that of the other two N–C bonds and is quite similar to the one obtained for the C–N bond in pyrazinamide (Rajalakshmi et al. 2014). The high negative value of the N1–H1A bond shows that the charges of the bond are highly concentrated, which are relatively higher than those of the C–H bonds found in the molecule (Fig. 6b). Among all the interactions, the \({\nabla }^{2}{\rho }_{\mathrm{B}\mathrm{C}\mathrm{P}}(r)\) value of the bond O1–H1 is − 34.64 e/Å5 having a high accumulation of charges as the bond length is very short compared to all the other bonds in 2,5-LB.
The topology of bifurcated hydrogen bond
Special attention is given to the topological properties of intermolecular hydrogen-bonding interaction, and the critical point search on the listed hydrogen bonds is carried out and listed in Table S4. The corresponding topological properties and the total electrostatic interaction energies of these intermolecular interactions are also calculated for the complete understanding of these bonds. In addition to these, the analysis of kinetic energy density G(r), potential energy density V(r) and total energy density H(r) at the BCP of hydrogen bonds is performed to authenticate the strength of the bonds.
The Laplacian of electron density and potential, kinetic and total energy density are connected by virial theorem. Potential energy dominates whenever the negative Laplacian of the electronic charge density is concentrated as in the cases of covalent bonds and lone pairs. The O3–H1A hydrogen bond has a small electron density, 0.0483 e/Å3, and a positive Laplacian, with |V|/G < 2 and H(r) < 0, which indicates its partial covalent-type character (Fig. 6c). This is the strong hydrogen bond in the crystal, and the corresponding calculated bond dissociation energy also testifies the strength of the bond. Moreover, the electron density at the BCP of the O…H bonds is below 0.1 e/Å3, where the hydrogen-bonding energies are around − 0.004 a.u., and the ratio of the potential and the kinetic energy densities is close to 0.6. The Laplacian \({\nabla }^{2}{\rho }_{\mathrm{B}\mathrm{C}\mathrm{P}}(r)\) of the O…H bonds lies in the range 1.3–1.5 e/Å5, which clearly shows the closed-shell interaction of the bonds.
Gradient vector field \(\nabla \rho (r)\)
The gradient vector field of the electron density (\(\nabla {\rho }_{\mathrm{B}\mathrm{C}\mathrm{P}}(r))\) is plotted and shown in Fig. 7, which shows the zero-flux surface of atoms in the molecule as well as defines the boundary of the atomic basin. The basins of N and C atoms seem like a prismatic shape, and the O atoms exhibit larger Bader atomic basins in comparison with the C and N atoms (Fig. 7a). In Fig. 7b, the atoms Br1 and Br2 which are high electronegative occupy larger volume. The main contribution of gradient lines inside the atomic basin comes from the positive nuclear charge (Rajalakshmi et al. 2014; Stephen et al. 2011).

A plot of the gradient vector field of the electron density (\(\nabla {\rho }_{\mathrm{B}\mathrm{C}\mathrm{P}}(r))\) and trajectory path of a 2,5-lutidine and b bromanilate
Atomic charges
Analysis of the experimental valence population was performed, and atomic charges are derived from them on the basis of the multipolar refinement result. The charges of three oxygen atoms (O2, O3 and O4) are worth comparing and are having the same charges − 0.124e, while the atom O1 has comparatively less charge (− 0.058e). The bromine atoms are negatively charged and are higher than all the other negatively charged atoms, and both bromine atoms have a similar charge (− 1.109e). The atom C13 holding the three hydrogen atoms (H13A, H13B and H13C) bears a large positive charge (0.228e). The carbon atoms (C8, C9) which are attached to the positively charged H atoms (H8, H9) have negative charges, showing the polarity of the bonds.
Electrostatic potential
The experimental molecular electrostatic potential maps are valuable to visualize the size, shape, charge distributions and charge-related properties of the molecule, which also affords us to identify the reactive sites of a molecule as well as the change in a reactive site when molecule acquires and loses different functional groups (Espinosa and Molins 2000). Through the reactive sites, one can predict the reactive behavior of nucleophilic (proton-loving) and electrophilic (electron-loving) molecules. As the 2,5-LB compound is an ion pair, the bromanilate has an overall negative electrostatic potential (ESP) which acts as a Lewis base and the 2,5-lutidinium moiety has an overall positive ESP which acts as a Lewis acid (Fig. 8a). In the two-dimensional ESP, red lines around the bromanilate show the negative potential, while the blue lines around the 2,5-lutidine show the positive potential (Fig. 8b, c). The color gradient from blue to red shows the positive and negative regions of MESP enunciate the electrophilic and nucleophilic regions in the molecule.

Isosurface representation of the electrostatic potential of the 2,5-LB molecule in the crystal, displaying a the encapsulation of negative electrostatic potential surface over bromanilate moiety. Two-dimensional view of ESP of b bromanilate and c 2,5-lutidine. The blue and red areas indicate positive and negative surfaces, respectively
Conclusion
An experimental charge density analysis of the compound 2,5-LB was carried out to understand the topological and electrostatic properties of the complex. The bifurcated hydrogen bond has been discussed in detail. The calculated C–C–H bond angles for the two different methyl (CH3) groups of 2,5-LB show the tetrahedral shape and the hybridization associated with the carbon atoms C12 and C13 (sp3 hybridization). The Hirshfeld surface analysis visualizes the strong O–H…O interactions, and the fingerprint plot shows that Br…H interactions give major contribution (25.0%) to the total Hirshfeld surface. The topological analysis reveals that the electron density of the bond C5–C6 is comparatively less (1.519 e/Å3) among all C–C bonds, and this might be because of the sharing of excess electrons of C6 atom with O4 atom. The electrostatic potential at the molecular surface shows the strong electronegative region around the bromanilate compound which is favorable to attract an electrophile acid. The present charge density study provides fine details of structural information, charge density distribution and intermolecular interactions of the 2,5-LB compound at the electronic level.
References
Aakeröy CB, Wijethunga TK, Desper J, Đaković M (2016) Electrostatic potential differences and halogen-bond selectivity. Cryst Growth Des 16:2662–2670
Allen FH, Bruno IJ (2010) Bond lengths in organic and metal-organic compounds revisited: X–H bond lengths from neutron diffraction data. Acta Crystallographica Sect B Struct Sci 66:380–386
Allen FH, Kennard O, Watson DG, Brammer L, Orpen AG, Taylor R (1987) Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J Chem Soc Perkin Trans 2:S1–S19
Bader RFW (1985) Atoms in molecules. Acc Chem Res 18:9–15. https://doi.org/10.1021/ar00109a003
Bader RFW (1991) A quantum theory of molecular structure and its applications. Chem Rev 91:893–928. https://doi.org/10.1021/cr00005a013
Braga D, Grepioni F, Desiraju GR (1998) Crystal engineering and organometallic architecture. Chem Rev 98:1375–1406
Cleland W (1997) Science1994, 264, 1887. Gerlt, JA; Kreevoy, MM; Cleland, WW; Frey, PA. Chem Biol 4:259
Coppens P, Abramov Y, Carducci M, Korjov B, Novozhilova I, Alhambra C, Pressprich MR (1999) Experimental charge densities and intermolecular interactions: electrostatic and topological analysis of dl-histidine. J Am Chem Soc 121:2585–2593
Desiraju GR, Steiner T (1999) The weak hydrogen bond: in Structural chemistry and biology. Oxford University Press, Oxford
Dolomanov OVB, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) OLEX2: a complete structure solution, refinement, and analysis program. J Appl Cryst 42:339–341
Espinosa E, Molins E (2000) Retrieving interaction potentials from the topology of the electron density distribution: the case of hydrogen bonds. J Chem Phys 113:5686–5694. https://doi.org/10.1063/1.1290612
Guillot B, Viry L, Guillot R, Lecomte C, Jelsch C (2001) Refinement of proteins at subatomic resolution with MOPRO. J Appl Crystallogr 34:214–223
Hansen NK, Coppens P (1978) Testing aspherical atom refinements on small-molecule data sets. Acta Crystallographica Sect A Cryst Phys Diffr Theor Gen Crystallogr 34:909–921
Hirshfeld F (1977a) XVII. Spatial partitioning of charge density. Isr J Chem 16:198–201
Hirshfeld FL (1977b) Bonded-atom fragments for describing molecular charge densities. Theoret Chim Acta 44:129–138
Jeffrey GA, Jeffrey GA (1997) An introduction to hydrogen bonding, vol 32. Oxford University Press, New York
Jelsch C, Guillot B, Lagoutte A, Lecomte C (2005) Advances in protein and small-molecule charge-density refinement methods using MoPro. J Appl Crystallogr 38:38–54
Laidig KE, Bader RF (1990) Properties of atoms in molecules: atomic polarizabilities. J Chem Phys 93:7213–7224
Madsen AØ (2006) SHADE web server for estimation of hydrogen anisotropic displacement parameters. J Appl Crystallogr 39:757–758
Martin CRL (2012) Crystal engineering approaches to controlling the formation of molecular complexes and their polymorphs. University of Glasgow, Glasgow
McKinnon JJ, Spackman MA, Mitchell AS (2004) Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr B 60:627–668
Perrin CL, Nielson JB (1997) “Strong” hydrogen bonds in chemistry and biology. Annu Rev Phys Chem 48:511–544
Prince E (2004) International Tables for Crystallography Volume C: Mathematical, physical and chemical tables, 3rd edn. Springer, Dordrecht, p 1020
Rajalakshmi G, Hathwar VR, Kumaradhas P (2014) Intermolecular interactions, charge-density distribution and the electrostatic properties of pyrazinamide anti-TB drug molecule: an experimental and theoretical charge-density study. Acta Crystallographica Sect B 70:568–579. https://doi.org/10.1107/s205252061303388x
Rozas I, Alkorta I, Elguero J (1998) Bifurcated hydrogen bonds: three-centered interactions. J Phys Chem A 102:9925–9932
Scheiner S (1997) Molecular interactions: from van der Waals to strongly bound complexes. Wiley, New York
Sobczyk L, Grabowski SJ, Krygowski TM (2005) Interrelation between H-bond and Pi-electron delocalization. Chem Rev 105:3513–3560
Spackman M, McKinnon JJ (2002) Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 4:378–392
Stephen AD, Thomas R, Srinivasan P, Narayayanasamy V, Kumaradhas P (2011) Exploring the bond topological and electrostatic properties of benzimidazole molecule via experimental and theoretical charge density study. J Mol Struct 989:122–130. https://doi.org/10.1016/j.molstruc.2011.01.014
Su Z, Coppens P (1997) Relativistic X-ray elastic scattering factors for neutral atoms Z = 1–54 from multiconfiguration Dirac–Fock wavefunctions in the 0–12 Å−1 sinθ/λ range, and six-Gaussian analytical expressions in the 0–6 Å−1 range. Acta Crystallographica Sect A 53:749–762. https://doi.org/10.1107/s0108767397004558
Taylor R, Kennard O, Versichel W (1984) Geometry of the nitrogen–hydrogen… oxygen–carbon (NH…O: C) hydrogen bond. 2. Three-center (bifurcated) and four-center (trifurcated) bonds. J Am Chem Soc 106:244–248
Thomas LH, Adam MS, O'Neill A, Wilson CC (2013) Utilizing proton transfer to produce molecular salts in bromanilic acid substituted-pyridine molecular complexes—predictable synthons? Acta Crystallogr C 69:1279–1288
Wagner A, Luger P (2001) Charge density and topological analysis of l-glutamine. J Mol Struct 595:39–46
Wolff S, Grimwood D, McKinnon J, Turner M, Jayatilaka D, Spackman M (2012) Crystal explorer. University of Western Australia, Crawley
Acknowledgements
The authors are grateful to the Principal and Secretary and the Head of PG Department of Physics, The American College, Madurai, India, for their continued encouragement and support during the progress of this work. The authors (R. N. and S. I.) are grateful for the grant provided by the University Grants Commission, India, in the form of Major Research Project (F.No. 41-848/2012(SR)).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
There are no conflicts to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rajendran, N., Mookan, N., Samuel, I. et al. Experimental validation of bifurcated hydrogen bond of 2,5-lutidinium bromanilate and its charge density distribution. Chem. Pap. 74, 2689–2699 (2020). https://doi.org/10.1007/s11696-020-01107-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-020-01107-3