
Abstract
The main goal of this study was to select micro- and ultrafiltration membranes that can be used for the purification of mushroom tyrosinase, replacing salting-out dual-step processes followed by centrifugations. In experiments, a raw extract from white mushrooms was used with high level of ballast proteins and brownish impurities. Four microfiltration membranes for the removal of undesired high molecular weight compounds were screened and that made of nitrocellulose was selected due to high recovery of enzymatic activity. Then diafiltration and concentration on the membrane made of polyethersulphone (300 kDa) was selected to recover 8% of proteins and 58% of tyrosinase activity with five- to seven purification fold, 10% of proteases, and 8% of brown impurities. It was shown that tyrosinase can be pre-purified by selected membranes yielding the enzyme quality at least comparable to that after double salting-out method but in one device. In both cases, subsequent purification by ion-exchange chromatography slightly increased purification degree of the enzyme and brown impurity removal. The surplus of membrane pre-purification is substantially higher thermal stability of the enzyme, enlarged after the chromatographic step, due to very low content of proteolytic enzymes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tyrosinases (EC 1.14.18.1) are copper-dependent enzymes that are capable to oxidize various phenolic substrates in the presence of molecular oxygen (Gąsowska et al. 2004; Garcia-Jimenez et al. 2016). The enzyme catalyses hydroxylation of monophenols to o-diphenols (monophenolase activity, M) and oxidation of o-diphenols to o-quinones (diphenolase activity, D) (Agarwal et al. 2019; Cieńska et al. 2016). Both activities have found applications in several biotechnological areas, including detoxification of waste water or soil contaminated with phenolic compounds, and/or aromatic amines (e.g. Duran and Esposito, 2000; Girelli et al. 2006), construction of biosensors for the detection of phenolic compounds (e.g. Da Silva et al. 2019; Kochana et al. 2015), cross-linking reactions (e.g. Le Roes-Hill et al. 2015); biosynthesis of antioxidants (e.g. hydroxytyrosol) (Botta et al. 2015), production of prodrugs (e.g. L-DOPA) (Kampmann et al. 2016) or compounds suppressing melanoma (Gąsowska-Bajger et al. 2009).
The number of tyrosinase applications caused growing interest in cheap sources of this enzyme. Although tyrosinases are widespread in nature (Halaouli et al. 2006; Nawaz et al. 2017), the main problem is its presence in low quantities as an intracellular protein. It seems that Agaricus bisporus tyrosinase should be regarded as the source of choice, mainly due to its availability (Seo et al. 2003). Moreover, there are several reports related to the isolation and purification of tyrosinase from A. bisporus (Bouchilloux et al. 1963; Duckworth and Coleman, 1970; Fan and Flurkey 2004; Gąsowska et al. 2004; Gouzi and Benmansour 2007; Khan et al. 2005; Lopez-Tejedor and Palomo 2018; Papa et al. 1994; Simsek and Yemenicioglu 2007; Smith and Krueger 1962; Wichers et al. 1996). Each method of tyrosinase purification involved three main steps: (i) cell disruption, (ii) removal of undesired low and high molecular weight compounds, and (iii) several kinds of chromatography. Cell disruption is commonly realized by tissue homogenization in phosphate buffer or chilled acetone or grinding in liquid nitrogen and then liquid phase is separated by passing suspension through cheesecloth or by centrifugation. The initial purification of crude extract is usually carried out by salting out or precipitation with acetone. In further purification steps, there are two or more chromatographic separations with several kinds of additional operations, such as concentration, dialysis, centrifugations, and adsorption of coloured impurities on a variety of gel matrices. However, very pure enzyme preparation is too expensive to be used in industry. Thus, the number of separation techniques should be minimized in relation to their environmental impact, cost and easiness. Membrane separations meet these criteria although their additional advantages such as flexibility and integrity are rarely presented with real mixtures of proteins. The main advantages of micro- (MF) and ultrafiltration (UF) are lack of phase change during the process, the absence of organic solvents, lack of extreme pH values and relatively slight effect of phase interface on the conformational structure of proteins (Charcosset 2006; Kołtuniewicz and Drioli 2008; Drioli and Giorno 2009).
Taking into account the 110–120 kDa molecular weight of mushroom tyrosinase (Bouchilloux et al. 1963; Jolley et al. 1969), the key step of its membrane purification is UF, during which the enzyme could be retained in the retentate while the most of ballast proteins (especially proteases) and brown impurities should be expelled from the enzyme solution. Obviously, UF should be preceded by MF process to remove cells, their fragments and other water-insoluble contaminants, including mucus-forming compounds.
The main goal of this study was to select membranes that can be used for the purification of tyrosinase to replace common salting-out dual-step process with technique based on membranes. In the case presented here, the feed solution is non-entirely defined and dark-brown impurities are in high concentrations. For that reason, four components have been chosen to represent the quality of separation: (i) total protein content; (ii) catalytic activity of tyrosinase (D); (iii) total proteases activity which can cause loss of the enzyme activity; (iv) level of undefined brown impurities, mainly originated from polymerization of phenolic compounds by tyrosinase. As MF is a prerequisite step before UF, four membranes were screened for obtaining tyrosinase solution with the highest catalytic activity. Then two UF membranes with different pore sizes were tested to obtain retentate with the high enzyme recovery, low proteases and brown impurity content and satisfactory permeate flux. Simultaneously, extract from mushrooms was purified by salting-out method to compare both methods for obtaining tyrosinase-enriched solution. Then ion-exchange chromatography was applied for subsequent purification of salted-out and membrane-treated enzyme solutions from brown impurities. Finally, to assess the impact of degree of purification on tyrosinase stability, inactivation of obtained preparations after all steps was tested at 40 °C.
Experimental
The Lowry reagent, l-3,4-dihydroxyphenylalanine (L-DOPA) and bovine serum albumin were obtained from Sigma-Aldrich (Germany). The other reagents, all of analytical grade, were supplied by Avantor Performance Materials (Poland). DEAE-Granocel was kindly supplied by Prof. Jolanta Liesiene from Kaunas University of Technology. Fresh white mushrooms (Agaricus bisporus) for tyrosinase isolation were obtained from a local producer (W. Idasiak, Wrocław, Poland). Microfiltration disc membranes made of nitrocellulose (0.30 μm) were from Whatman whereas made of regenerated cellulose (0.22 μm), poly(tetrafluoroethylene) (0.20 μm) and poly(fluorovinylidene) (0.22 μm) as well as ultrafiltration membranes made of polyethersulphone were from Millipore (Germany).
Tyrosinase activity was measured in the presence of 1 mM l-DOPA (D) in a 0.1 mol L−1 phosphate buffer (pH 7.0). The reaction was monitored spectrophotometrically by HELIOSα (Unicam) at 475 nm and the linear absorbance increase was used for activity calculations. An enzyme activity unit (U) was defined as the amount of the enzyme which converts l-DOPA into coloured products causing an increase in absorbance by 0.001 per min at 30 °C. The mean analytical error was lower than ± 3.3%.
Activity of proteolytic enzymes was assessed by the non-specific caseinolytic test as in previous paper (Bryjak and Rekuć 2010). One unit of proteolytic activity (U) was defined as the amount of enzyme that induced absorbance (280 nm) increase of 1.0 per 10 min at 37 °C. Mean analytical error was lower than ± 6.3%.
Protein concentration was determined spectrophotometrically according to Lowry’s method (Lowry et al. 1951) using bovine serum albumin as a standard. Mean analytical error was lower than ± 2.2%. To track brown impurities, the absorbance of 450 nm was chosen for a linear relationship between colour intensity and dilution factors of concentrated preparation.
Tyrosinase was prepared as described earlier (Zynek et al. 2010). Briefly, young and fresh mushrooms (300 g) were homogenized in cold acetone (− 26 °C), mixed for 30 min at 0–4 °C and centrifuged at 7000 rpm for 20 min. The pulp was put under a vacuum pump to remove the remaining acetone and then suspended in the phosphate buffer. After another centrifugation, the supernatant dissolved in the buffer was used as the crude preparation.
Microfiltration was carried out using an Amicon (USA) stirred cell with 400 mL working volume as presented previously (Bryjak and Rekuć, 2010). Disc membranes were of 41.8 cm2 effective membrane filtration area. Selected membranes were made of regenerated cellulose (RegCel; Millipore), poly(vinyl fluoride) (PVDF; Millipore), poly(tetrafluoroethylene) (PTFE; Millipore), and cellulose nitrate (NitroCel; Whatman). The system operated at 200 rpm and constant pressure set at 0.4 MPa. The feed (200 mL of the crude extract; 4 °C) was filtered in the dead-end mode and after that proteins loosely bound to the membrane were washed-out with the buffer. The wash-out, feed and permeate were analysed for tyrosinase, protease and protein presence.
Ultrafiltration was done on a Labscale™ TFF System (Millipore, Bedford, USA) equipped with two Pellicon® XL (Millipore, Bedford, USA) cassettes with 50 cm2 filtration area of membranes made of polyethersulphone: Biomax-100 and Biomax-300 with nominal molecular weight cut-off 100 and 300 kDa, respectively. Before experiments, the UF unit with the Pellicon membrane was rinsed intensively and then deionised water flux was measured. The permeate, obtained after microfiltration, was a feed for diafiltration (four exchange of the volume of the retentate) and concentration (four times) on one of the tested membranes. Diafiltration was carried out by maintaining a constant reservoir level with 0.1 mol L−1 phosphate buffer, pH 7.0. The concentration was started just after retentate diafiltration. The permeate volumes (100 mL—diafiltration, 40 mL—concentration) were time collected. The transmembrane pressure was set at 0.137 MPa. After the concentration step, the retentate was drained, and 50 mL of 0.1 mol L−1 phosphate buffer was added to the system to wash proteins loosely bound to the membrane. Feed, wash-out, retentate and permeate samples were analysed for tyrosinase and protease activities and the presence of proteins and brown impurities. The overall recovery was estimated with reference to the crude extract.
Crude extract of tyrosinase was salted out with ammonium sulphate (30% saturation) during stirring at 0–4 °C for 30 min, as described previously (Zynek et al. 2010). The suspension was then centrifuged (8000 rpm; 20 min; − 2 °C) and the ammonium sulfate concentration in the supernatant was adjusted to 60% m:v saturation. Protein aggregation and precipitate separation was carried out as in the previous precipitation step. The collected sediment was dissolved in 20 mL of the buffer.
Enzyme preparation (after membrane fractionation or salting out) was purified by ion-exchange chromatography using DEAE-Granocel 500 (bead particle size 100-200 μm; exclusion limit 105 Da; ion-exchange capacity 1.4 mEq g−1) as chromatographic packing (bed dimension: 120 × 10 mm), equilibrated with 0.01 M phosphate buffer with pH 7.0. The same buffer was used as eluent with 0–0.4 mol L−1 NaCl stepwise gradient. The injected amount of preparation was 4 mL (protein concentration 0.726 mg mL−1) and the flow rate was 1 mL.min−1. The separation process was monitored by measuring the protein concentration, tyrosinase and protease activities and brown impurity content in the collected 3 mL fractions.
The enzyme stability was tested by incubating the crude enzyme preparation at 40 °C as previously (Zynek et al. 2010). The samples were withdrawn in specified time intervals, cooled rapidly to 0 °C in an iced-water bath and stored in ice water prior the activity measurement (1 h). The same procedure was applied for the enzyme solutions after microfiltration and ultrafiltration, salting out and ion-exchange chromatography. In all cases, protein concentration of the enzyme solutions was aligned to 0.042 mg mL−1. Residual activity was measured using 1 mM l-DOPA as the substrate.
Results and discussion
Membrane fractionation
Extracts from A. bisporus tissues contain cell debris and other unspecified particles and they were subjected to prefiltration on MF membranes made of hydrophilic (RegCel, NitroCel) and hydrophobic (PVDF, PTFE) materials were selected. To evaluate the properties of membranes used, four elements of the feed and permeate were balanced: proteins, monophenolase and diphenolase activities of tyrosinase, and proteolytic activity. The most important results (Fig. 1) indicated that RegCel and PTFE membranes allowed all the species to pass into permeate (at least 80%). From the other hand, PVDF and NitroCel caused removal of proteases in about 40 and 30%, respectively. The state in which part of proteolytic enzymes was adsorbed on the membrane and/or self-digested was profitable from the preoperational point of view; therefore, in the next purification steps, tyrosinase will be subjected to attack by proteases to a lesser degree.

Representative data on relative amounts of protein, tyrosinase [measured in the presence of L-DOPA (D) as the substrate] and proteases in the permeate after MF (a), deposited on the membrane (b), and overall activity balance (c). Membranes: RegCel (black bars), PTFE (white bars), PVDF (stripped bars), NitroCel (grey bars). The values before MF were set as 100%
After gentle washing of the membrane after MF, one can remove loosely bound species (Fig. 1b). It was found that reversible deposition of proteins, tyrosinase and proteases is the lowest in the case of RegCel and then NitroCel membranes, both being more hydrophilic than PVDF and PTFE. It supports the common knowledge that the use of hydrophilic material prevents strong deposition of hydrophilic proteins on interfacial surfaces. Interestingly, it seemed that proteases which passed through PVDF and NitroCel were subjected to self-digestion/inactivation rather (activity balance around 75% with total protein balance 99 and 92%, respectively, Fig. 1c) than to deposition on the membrane (activity recovery 10 and 2%, respectively; Fig. 1b). It is rather unlikely that about 25% of proteases are tightly bound to the membranes, especially in the case of PVDF membrane with almost complete protein balance. However, the reason of such a behaviour in the case of both membranes is unclear.
Taking into account high recovery of tyrosinase catalytic activity, low protease content and low deposition of filtered species on the membrane surface, NitroCel membrane seems to be the material of choice. Five independent experiments gave species recovery as follows: protein − 90.2 ± 3.1% m:m; tyrosinase (D) − 94.0 ± 3.9% m:m; proteases − 73.4 ± 12.3% m:m.
In the next step two UF membranes were tested, using MF permeate as a feed solution. In this case, we assumed that good results are as follows: high tyrosinase recovery, low protease content, significant reduction of coloured by-products, and satisfactory purification fold (Table 1).
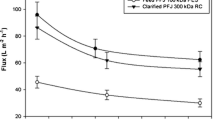
In preliminary studies, membrane with cut-off 100 kDa was excluded due to high by-products and proteolytic enzyme recovery (Fig. 2a) and relatively low purification fold (Fig. 2c); despite that, tyrosinase recovery was the highest (Fig. 2a). Moreover, a significant drop of permeate flux was noted (88.4% in comparison to deionized water flux and 28% in a reference to the flux of the first permeate fraction).

Recovery of protein, tyrosinase (measured in the presence of l-DOPA (D) as substrate), proteases and coloured impurities in retentate (a), wash-out solution from the membrane (b), purification fold of tyrosinase and proteases in the retentate after UF (c) and the exemplary results of permeate flux changes during UF process (300 kDa with initial permeate recirculation; the flux determined for clean water was considered as 100%). Cut-off of the membranes: 100 kDa (black bars), 300 kDa (white bars) and 300 kDa with initial permeate recirculation (grey bars)
Thus, membrane with 300 kDa was used but with similar drop in flux (88.5 and 19.3%, respectively). Although brown by-products and proteases were removed more efficiently (Fig. 2a) and purification factor was higher (above fivefold), tyrosinase recovery was found to be too low (42–46%). For that reason, in the next experiment, the first permeate fraction (100 mL in diafiltration mode) was continuously returned to the retentate container to stimulate polarization layer formation on the membrane that creates an additional filtration layer [dynamic layer (Kołtuniewicz and Drioli 2008)] composed of proteins. It was expected that in this case tyrosinase recovery should increase as the presence of 35% of proteins and 15% of tyrosinase activity was found in the first fraction. As seen from Fig. 2a, c, the expectation was correct; tyrosinase recovery increased by 21-26% with no changes in other retentate parameters.
Comparative study on membrane and salting out purification
To compare the processes studied, the same mushroom extract was used for tyrosinase purification by both methods. Data summarized in Table 2 show that tyrosinase, protein and brown impurity yields are comparable in both cases. However, significant difference was noted in the yield of proteases; in the case of membrane separation, the level of proteolytic enzymes was 4 times lower than that in salted-out preparation. Moreover, higher purification fold was obtained after the membrane separation.
Chromatographic step
In chromatographic step, the column with DEAE cellulose bed was applied to separate brown impurities. To avoid the problem of different protein concentrations of preparations after membrane filtration and salting out procedures, the salted-out preparation was slightly diluted up to 0.726 mg mL−1 concentration that allowed direct comparison of both preparations. It was interesting that elution of proteins was quite different (Fig. 3 and Table 3); in salted-out preparation, tyrosinase was eluted in the first fractions, similarly as described by Gąsowska et al. (2004), whereas the enzyme after membrane treatment was found in the 3rd and 4th fractions and in the fractions 24 and 25 (Fig. 3).

Ion-exchange chromatography with DEAE-Granocel applied for tyrosinase preparation after purification by membrane separation (a) or salting out (b); solid line—protein; dashed line—tyrosinase activity; points—data for particular fraction. The values of protein amount and catalytic activity obtained for tyrosinase preparation prior the chromatographic step were taken as 100%. 0.1, 0.2 and 0.4 NaCl means the sodium chloride concentrations in mol L−1
It means that both enzyme preparations obtained after chromatography differed in protein composition. In fact, almost all proteolytic enzymes were excluded from the preparation pre-treated with membranes whereas the salted-out preparation preserved about 15% of proteases. As it was mentioned, the objective of this step was to remove coloured by-products and in this case both preparations were cleared to the same extend. However, yield of tyrosinase recovery from samples after membranes pre-treatment was considerably lower, but with significantly lower content of proteases.
Tyrosinase stability
Assuming that there are unspecified potential stabilizers or destabilizers of the enzyme in the real protein extract, tyrosinase thermal tolerance can be affected by its purity level. Tyrosinase thermal tolerance was tested at 40 °C, using crude, membrane-treated, salted-out samples and preparations after ion-exchange chromatography. Inactivation rate constants were assumed to follow the first-order kinetics (Zynek et al. 2010) and are presented in Table 4 (activity decays in time are not shown). As protein concentration in the enzyme solutions was set to be the same (0.042 mg mL−1), the stability increase could be attributed to the removal of potential destabilizers. It was shown that double salting-out procedure caused increase in tyrosinase stability in about 30% but membrane separations allowed to obtain the preparation with almost three times more stable enzyme. Interestingly, salted out or membranes pre-treatment with subsequent purification by ion-exchange chromatography caused a subsequent increase in the enzyme stability. One can assume that exceptionally high tyrosinase stability after membrane and chromatographic fractionation is attributed to significantly lower content of proteolytic enzymes than after double salting-out and chromatographic procedures (Tables 2 and 3).
Conclusions
The experiments presented in this study indicated that the MF → UF resulted in an acceptable resolution and recovery of tyrosinase. In comparison with common procedure based on dual successive salting out followed by centrifugations of sediments, membrane process can be carried out in one filtration unit. Moreover, obtained enzyme preparations contained lower level of proteinases that probably caused exceptionally increased tyrosinase stability. It is worth pointing out that not only tyrosinase activity was monitored during the investigated processes. Special attention was paid to assessing the presence of brown pigments and proteolytic enzymes. According to our best knowledge, such an approach is rarely presented. On the basis of the obtained results, several conclusions have been drawn:
- 1.
MF on NitroCel membrane is profitable due to partial reduction of proteases in the permeate with the almost complete protein recovery and insignificant tyrosinase activity loss.
- 2.
UF (diafiltration and concentration) on polyethersulphone membrane of 300 kDa with partial turn the permeate back to retentate offers tyrosinase preparations with the reasonable purification level and the higher thermal stability than in the case of salting-out procedure. Moreover, one unit operational step enables minimization of tyrosinase activity loss. Obtained preparation can be used directly in most biotechnological processes. Even higher stability of tyrosinase can be obtained after subsequent chromatography but at the cost of the enzyme yield.
- 3.
In the case of processes demanding exceptionally high tyrosinase purity, it is recommended to apply double salted-out procedure of purification followed by ion-exchange chromatography to obtain the enzyme solution as the feed for further steps. In this case, the yield of tyrosinase is substantially higher than after MF and UF.
References
Agarwal P, Singh M, Singh J, Singh RP (2019).Chapter 1—microbial tyrosinases: a novel enzyme, structural features, and applications. In: Applied microbiology and bioengineering. An interdisciplinary approach, pp 3–19, https://doi.org/10.1016/b978-0-12-815407-6.00001-0
Botta G, Bizzarri M, Garozzo A, Timpanaro R, Bisignano B, Amatore D, Palamara AT, Nencioni L, Saladino R (2015) Carbon nanotubes supported tyrosinase in the synthesis of lipophilic hydroxytyrosol and dihydrocaffeoyl catechols with antiviral activity against DNA and RNA viruses. Bioorg Med Chem 23:5345–5351. https://doi.org/10.1016/j.bmc.2015.07.061
Bouchilloux S, McMahill P, Mason HS (1963) The Multiple Forms of Mushroom Tyrosinase: purification and molecular properties of the enzymes. J Biol Chem 238:1699–1707
Bryjak J, Rekuć A (2010) Effective purification of Cerrena unicolor laccase using microfiltration, ultrafiltration and acetone precipitation. Appl Biochem Biotechnol 160:2219–2235. https://doi.org/10.1007/s12010-009-8791-9
Charcosset C (2006) Membrane processes in biotechnology: an overview. Biotechnol Adv 24:482–492. https://doi.org/10.1016/j.biotechadv.2006.03.002
Cieńska M, Labus K, Lewańczuk M, Koźlecki T, Liesiene J, Bryjak J (2016) Effective l-tyrosine hydroxylation by native and immobilized tyrosinase. PLoS One 11(10):e0164213. https://doi.org/10.1371/journal.pone.0164213
Da Silva W, Ghica ME, Ajayi RF, Iwuoha EI, Brett MA (2019) Tyrosinase based amperometric biosensor for determination of tyramine in fermented food and beverages with gold nanoparticle doped poly(8-anilino1-naphthalene sulphonic acid) modified electrode. Food Chem 282:18–26. https://doi.org/10.1016/j.foodchem.2018.12.104
Drioli E and Giorno L (eds) (2009) Membrane operations: innovative separations and transformations. WILEY-VCH Verlag GmbH 7 Co. KGaA, Weinheim
Duckworth HW, Coleman JE (1970) Physicochemical and kinetic properties of mushroom tyrosinase. J Biol Chem 245:1613–1625
Duran N, Esposito E (2000) Potential applications of oxidative enzymes and phenoloxidase-like compounds in wastewater and soil treatment: a review. Appl Catal B 28:83–99. https://doi.org/10.1016/S0926-3373(00)00168-5
Fan Y, Flurkey WH (2004) Purification and characterization of tyrosinase from gill tissue of Portabella mushrooms. Phytochemistry 65:671–678. https://doi.org/10.1016/j.phytochem.2004.01.008
Garcia-Jimenez A, Teruel-Puche JA, Berna J, Ridruguez-Lopez JN, Tudela J, Garcia-Ruiz PA, Garcia-Canovas F (2016) Characterization of the action of tyrosinase on resorcinols. Bioorg Med Chem 24:4434–4443. https://doi.org/10.1016/j.bmc.2016.07.048
Gąsowska B, Kafarski P, Wojtasek H (2004) Interaction of mushroom tyrosinase with aromatic amines, o-diamines and o-aminophenols. Biochem Biophys Acta 1673:170–177. https://doi.org/10.1016/j.bbagen.2004.04.013
Gąsowska-Bajger B, Frąckowiak-Wojtasek B, Koj S, Cichoń T, Smolarczyk R, Szala S, Wojtasek H (2009) Oxidation of carbidopa by tyrosinase and its effect on murine melanoma. Bioorg Med Chem Lett 19:3507–3510. https://doi.org/10.1016/j.bmcl.2009.05.002
Girelli AM, Mattei E, Messina A (2006) Phenols removal by immobilized tyrosinase reactor in on-line high performance liquid chromatography. Anal Chim Acta 580:271–277. https://doi.org/10.1016/j.aca.2006.07.088
Gouzi H, Benmansour A (2007) Partial purification and characterization of polyphenol oxidase extracted from Agaricus bisporus (JELange) Imbach. Int J Chem React Eng 5:1–11. https://doi.org/10.2202/1542-6580.1445(A76)
Halaouli S, Asther M, Sigoillot JC, Hamdi M, Lomascolo A (2006) Fungal tyrosinases: new prospects in molecular characteristics, bioengineering and biotechnological applications. J Appl Microbiol 100:219–232. https://doi.org/10.1111/j.1365-2672.2006.02866.x
Jolley RL, Nelson RM, Robb DA (1969) The multiple forms of mushroom tyrosinase: studies of the isozymes. J Biol Chem 244:3251–3257
Kampmann M, Riedel N, Mo YL, Beckers L, Wichmann R (2016) Tyrosinase catalyzed production of 3,4-dihydroxyphenylacetic acid using immobilized mushroom (Agaricus bisporus) cells and in situ adsorption. J Mol Catal B Enzym 123:113–121. https://doi.org/10.1016/j.molcatb.2015.11.012
Khan AA, Akhtar S, Husain Q (2005) Simultaneous purification and immobilization of mushroom tyrosinase on an immunoaffinity support. Process Biochem 40:2379–2386. https://doi.org/10.1016/j.procbio.2004.09.020
Kochana J, Hnida K, Sulka G, Knihnicki P, Kozak J, Gilowska A (2015) Application of polypyrrole nanowires for the development of a tyrosinase biosensor. Chem Pap 69:1130–1135. https://doi.org/10.1515/Chempap-2015-0114
Kołtuniewicz AB and Drioli E (eds) (2008) Membranes in clean technologies—Theory and Practice. WILEY-VCH Verlag GmbH 7 Co. KGaA, Weinheim
Lopez-Tejedor D, Palomo JM (2018) Efficient purification of a highly active H-subunit of tyrosinase from Agaricus bisporus. Protein Expr Purif 145:64–70. https://doi.org/10.1016/j.pep.2018.01.001
Le Roes-Hill M, Palmer Z, Rohland J, Kirby BM, Burton SG (2015) Partial purification and characterisation of two actinomycete tyrosinases and their application in cross-linking reactions. J Mol Catal B Enzym 122:353–364. https://doi.org/10.1016/j.molcatb.2015.10.012
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
Nawaz A, Shafi T, Khsliq A, Mukhtar H, Ul Haq I (2017) Tyrosinase: sources, structure and applications. Int J Biotechnol Bioeng 3(5):142–148. https://doi.org/10.25141/2475-3432-2017-5.0135
Papa G, Pessione E, Leone V, Giunta C (1994) Agaricus bisporus tyrosinase—I. progress made in preparative methods. Int J Biochem 26:215–221. https://doi.org/10.1016/0020-711X(94)90148-1
Seo S, Sharma VK, Sharma N (2003) Mushroom tyrosinase: recent prospects. J Agric Food Chem 51:2837–2853. https://doi.org/10.1021/jf020826f
Simsek S, Yemenicioglu A (2007) Partial purification and kinetic characterization of mushroom stem polyphenoloxidase and determination of its storage stability in different lyophilized forms. Process Biochem 47:943–950. https://doi.org/10.1016/j.procbio.2007.03.005
Smith JL, Krueger RC (1962) Separation and purification of the phenolases of the common mushroom. J Biol Chem 237:1121–1128
Wichers H, Gerritsen YAM, Chapelon ChGJ (1996) Tyrosinase isoforms from the fruit bodies of Agaricus bisporus. Phytochemistry 43:333–337. https://doi.org/10.1016/0031-9422(96)00309-3
Zynek K, Bryjak J, Polakovič M (2010) Effect of separation on thermal stability of tyrosinase from Agaricus bisporus. J Mol Catal B Enzym 66:172–176. https://doi.org/10.1016/j.molcatb.2010.05.003
Acknowledgements
This study was supported by the project “Biotransformations for pharmaceutical and cosmetics industry” No. POIG.01.03.01-00-158/09-01 partly financed by the European Union within the European Regional Development Fund.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Labus, K., Wiśniewski, Ł., Cieńska, M. et al. Effectivity of tyrosinase purification by membrane techniques versus fractionation by salting out. Chem. Pap. 74, 2267–2275 (2020). https://doi.org/10.1007/s11696-020-01060-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-020-01060-1