
Abstract
Favipiravir was first synthesized from an inexpensive and commercially available starting material, 2-aminopyrazine. The preferred route embedded within Scheme 4 consisted of seven steps, and was highlighted by the novel and efficient synthesis of 3,6-dichloropyrazine-2-carbonitrile 8. This intermediate was prepared in four successive steps which were regioselective chlorination of the pyrazine ring, bromination, Pd-catalyzed cyanation, and Sandmeyer diazotization/chlorination. This protocol eliminated the hazardous POCl3 of previous synthetic methods and offered a better yield (48%) which was 1.3-fold higher than a recently published procedure. From intermediate 8, the subsequent nucleophilic fluorination, nitrile hydration and hydroxyl substitution efficiently afforded the target product. Another synthetic approach with the same starting material was also investigated to bypass the allergy-causing dichloro intermediate 8. However, the key step of monofluorination at the pyrazine C6 position of intermediate 19 or 22 was not achieved.
Similar content being viewed by others



Introduction
Favipiravir (T-705, 6-fluoro-3-hydroxypyrazine-2-carboxamide) is a novel anti-influenza drug which functions to selectively inhibit the RNA-dependent RNA polymerase of influenza virus (Furuta et al. 2013). T-705 ribofuranosyl triphosphate (T-705 RTP, Fig. 1) is the active form, generated from the parent drug by a series of intracellular enzymes (Sangawa et al. 2013; Naesens et al. 2013). Favipiravir also displays inhibitory activities against a number of other pathogenic RNA viral infections, such as arenavirus, bunyavirus, flavivirus, alphavirus, and norovirus (Furuta et al. 2013; Furuta et al. 2009; Jin et al. 2015). Moreover, it is believed to be a promising therapeutic candidate for Ebola virus infection (Smither et al. 2014). In a retrospective clinical study during the outbreak of Ebola virus, patients who received T-705 had a significant viral load reduction compared with the control group (Bai et al. 2016). Recently, favipiravir and related structures have attracted extensive attentions in the field of antiviral and antiparasitic research (Gao et al. 2018; Huchting et al. 2018; Huchting et al. 2017; Klejch et al. 2018; Plebanek et al. 2017; Wang et al. 2016).

Chemical structures of favipiravir (T-705) and T-705 RTP
Favipiravir is a small pyrazine compound containing three substituents (–F, –OH, –CONH2) in place. Due to the hydroxyl group, this compound exhibits acidic properties and could be tautomerized to the keto form, shown in Fig. 1 (El-Nahas and Hirao 1999). With respect to the synthesis, three routes are now available. Route 1 is the original one starting from methyl 3-amino-6-bromopyrazine-2-carboxylate 1, and consisted of five steps, diazotization-alcoholysis, Pd-catalyzed imine substitution/hydrolysis, aminolysis, Schiemann fluorination and demethylation (Scheme 1). The first step of diazotization-alcoholysis needs to be performed in concentrated sulfuric acid and only gives a yield of 35%. The following step is a sequence of two reactions, which is tedious and inefficient for the synthesis of 3. Moreover, the conversion of 4 to 5 involves the highly corrosive Olah’s reagent (Furuta and Egawa 2000). Considering these disadvantages, this route is not welcomed for the scalable production of favipiravir.
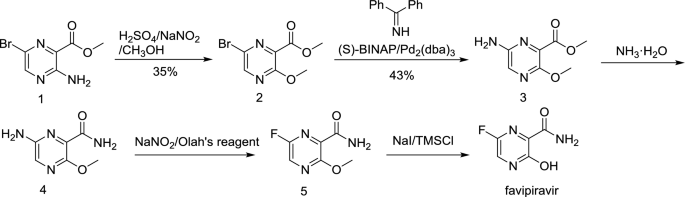
Route 1 for the synthesis of favipiravir
Route 2 and Route 3 share the same crucial intermediate, 3,6-dichloropyrazine-2-carbonitrile 8, which is prepared from 3-hydroxypyrazine-2-carboxamide 6 or 3-aminopyrazine-2-carboxylicacid 11 (Schemes 2 and 3). This dichloro-intermediate is then transformed to 9 by reacting with KF, and subsequent nitrile hydration in concentrated hydrochloric acid or treatment with an alkaline solution of H2O2 gives 10 (Beldar and Jordis 2009; Hara et al. 2010; Liu et al. 2017). The C3 fluorine of 10 is more reactive and could be easily replaced by hydroxyl group to furnish the final product. Comparatively, Route 2 is attractive with respect to step count, but not economically feasible for the preparation of 8 due to the low yield (18% yield for the nitration of 6, shown in the experiment section) and the high cost of the starting material 6. Route 3 was reported recently with cheaper 3-aminopyrazine-2-carboxylic acid 11 as the starting material, from which, five steps were required to obtain 8 in a yield of 37%. In both of the routes, a substantial excess of POCl3 and base are indispensable to completely accomplish the functional group transformations. The large quantity of POCl3 required in the procedure is a big concern with respect to both scale-up and waste disposal.

Route 2 for the synthesis of favipiravir

Route 3 for the synthesis of favipiravir
Herein, we reported an alternative approach to the synthesis of intermediate 8 without using POCl3. Starting from commercially available 2-aminopyrazine, the synthesis was achieved in 48% yield over four steps. Following the established procedures, the target compound was smoothly obtained. Another strategy for the synthesis of favipiravir not relying on the allergy-causing dichloro intermediate 8 was also investigated. However, no good results were achieved.
Experimental
Materials and methods
1H-NMR and 13C-NMR spectra were determined on a Brucker 400 Hz or Brucker 500 Hz instrument with TMS as an internal standard. Multiplicities were reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and br = broad. ESI–MS was determined on Finnigan™ LTQ™ (Thermo Fisher Scientific, Bremen, Germany) linear ion trap mass spectrometer. All the reagents were purchased from commercial suppliers and used without further purification. All reactions were monitored by thin-layer chromatography (TLC) on 25.4 × 76.2 mm silica gel plates (GF-254).
Preparation of 2-amino-5-chloropyrazine (17)
Method A. To a solution of 2-aminopyrazine (1.84 g, 19.4 mmol) in acetonitrile (15 mL) was added NCS (2.59 g, 19.4 mmol) in batches at room temperature. The reaction mixture was stirred for 5 h, and filtrated through diatomaceous earth (Celite®). The filter cake was washed with ethyl acetate (20 mL × 2). The combined filtrate was concentrated under vacuum, and then saturated aqueous Na2CO3 solution (20 mL) was added. The mixture was extracted with ethyl acetate (100 mL). The organic layer was dried over anhydrous Na2SO4, decolorized with activated charcoal (0.5 g), and concentrated. The residue was purified by chromatography on silica gel eluting with petroleum ether (PE)-ethyl acetate (EA) (10:1–2:1) to give a light-yellow solid (1.38 g, yield 55%).
Method B. To a suspension of 2-aminopyrazine (3 g, 31.6 mmol) in acetonitrile (50 mL) was added N-chloro-N-methoxy-4-methylbenzenesulfonamide (TSA, 8.2 g, 34.7 mmol) in batches at room temperature. The reaction mixture was heated at 40 °C for 4 h. Subsequently, the solvent was evaporated in vacuum and the residue was partitioned between ethyl acetate (50 mL) and saturated aqueous Na2CO3 solution (20 mL). The organic layer was dried over anhydrous Na2SO4, decolorized with activated charcoal (0.8 g), and concentrated. The crude product containing ~ 5% dichlorinated impurity judged by TLC was purified by chromatography on silica gel eluting with PE/EA (10:1–2:1) to give a light-yellow solid (3.3 g, yield 80%).
Method C. Reaction of 2-aminopyrazine (3 g, 31.6 mmol) with N-chloro-N-methoxybenzenesulfonamide (BSA, 7.7 g, 34.7 mmol) following the same procedure of method B provided 3.1 g of 17 in a yield of 77%.
2-Amino-5-chloropyrazine (17)
Yield: 80% (method B), light-yellow solid, M.p.:128-130 °C (Lit. 129–130 °C, Palamidessi et al. 1964). 1H NMR (400 MHz, CDCl3): δ 8.01 (d, 1H, J = 1.4 Hz), 7.76 (d, 1H, J = 1.4 Hz), 4.57 (s, 2H). 13C NMR (125 MHz, CDCl3): δ 153.19, 141.32, 137.48, 130.82. EI-MS m/z: 129 (M+, Cl35 100), 131 (M+, Cl37, 31), 102 (M+, Cl35, –H, –HCN, 100), 104 (M+, Cl37, –H, –HCN, 31), 67 (M+, –H, –HCN, –HCl, 28).
Preparation of 2-amino-3-bromo-5-chloropyrazine (18a)
17 (1.1 g, 8.5 mmol) was dissolved in dichloromethane (25 mL) followed by the addition of NBS (1.52 g, 8.5 mmol) in batches over a period of 15 min at room temperature. When the reaction was completed, saturated aqueous Na2CO3 solution (10 mL) was added. After stirring for 10 min, the mixture was partitioned between dichloromethane (100 mL) and water (100 mL). The organic layer was dried over anhydrous Na2SO4, decolorized with activated charcoal (0.5 g), and concentrated to give a yellow solid (1.53 g, yield 87%).
2-Amino-3-bromo-5-chloropyrazine (18a)
Yield: 87%, yellow solid, M.p.: 108–110 °C. 1H NMR (400 MHz, CDCl3): δ 7.97 (s, 1H), 5.08 (s, 2H).1H NMR (400 MHz, DMSO-d6): δ 8.10 (s, 1H), 6.99 (s, 2H). 13C NMR (125 MHz, CDCl3): δ 151.66, 140.26, 134.75, 123.27. EI-MS m/z: 207 (M+, Cl35, Br79, 76), 209 (M+, Cl37, Br79 and Cl35, Br81, 100), 211 (M+, Cl37, Br81, 26), 128 (M+, Cl35, –Br, 75), 130 (M+, Cl37, –Br, 25), 101 (M+, Cl35, –Br, –HCN, 30), 103 (M+, Cl37, –Br, –HCN, 10).
Preparation of 2-amino-3,5-dibromopyrazine (18b)
To a suspension of 2-aminopyrazine (4 g, 42.1 mmol) in dichloromethane (100 mL) was added NBS (18.7 g, 105 mmol) in batches over a period of 1 h at room temperature. The reaction mixture was stirred at room temperature for 5 h. When the reaction was completed, saturated aqueous Na2CO3 solution (40 mL) was added. After stirring for 15 min, the mixture was partitioned between dichloromethane (200 mL) and water (100 mL). The organic layer was then dried over anhydrous Na2SO4, decolorized with activated charcoal (1 g), and concentrated to give a yellow solid (8.0 g, yield 76%).
2-Amino-3,5-dibromopyrazine (18b)
Yield: 76%, yellow solid, M.p.: 112–114 °C (Lit. 113–114 °C, Sato et al. 1990a, b). 1H NMR (400 MHz, DMSO-d6): δ 8.13 (s, 1H), 6.99 (s, 2H). 1H NMR (400 MHz, CDCl3): δ 8.04 (s, 1H), 5.09 (s, 2H). 13C NMR (125 MHz, CDCl3): δ 151.86, 143.06, 123.96, 123.65. ESI–MS m/z: 249.2 [M − H]−.
Preparation of 6-chloro-3-aminopyrazine-2-carbonitrile (19a)
The suspension of 18a (3 g, 14.5 mmol), NaCN (0.85 g, 17.4 mmol), CuI (1.3 g, 7.3 mmol) and Pd(PPh3)4 (0.17 g, 0.15 mmol) were stirred in DMF (40 mL) at 120 °C for 10 h under nitrogen protection. After cooling to room temperature, 10% aqueous solution of Na2S2O3 (10 mL), water (60 mL) and ethyl acetate (200 mL) were added. The resulting mixture was stirred for 10 min, then filtered, and the filter cake was washed with ethyl acetate (150 mL). The organic layer was separated, washed with water (3 × 50 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by chromatography on silica gel eluting with PE/EA (10:1–2:1) to give a light-yellow solid (1.9 g, yield 85%).
6-Chloro-3-aminopyrazine-2-carbonitrile (19a)
Yield: 85%, light-yellow solid, M.p.: 156–158 °C. 1H NMR (400 MHz, CDCl3): δ 8.23 (s, 1H), 5.34 (s, 2H). 13C NMR (100 MHz, CDCl3): δ 154.98, 146.84, 137.41, 114.07, 111.16. EI-MS m/z: 154 (M+, Cl35, 100), 156 (M+, Cl37, 35), 127 (M+, Cl35, –H, –CN, 48), 129 (M+, Cl37, –H, –CN, 15).
Preparation of 6-bromo-3-aminopyrazine-2-carbonitrile (19b)
The suspension of 18b (2.51 g, 10.0 mmol), NaCN (0.59 g, 12.0 mmol), CuI (0.95 g, 5.0 mmol) and Pd(PPh3)4 (0.14 g, 0.1 mmol) were stirred in DMF (20 mL) at 120 °C for 10 h under nitrogen protection. After cooling to room temperature, 10% aqueous solution of Na2S2O3 (10 mL) and water (50 mL) were added. The mixture was extracted with ethyl acetate (50 mL × 3), washed with water (3 × 50 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by chromatography on silica gel eluting with PE/EA (10:1–2:1) to give an off-white solid (1.1 g, yield 55%).
6-Bromo-3-aminopyrazine-2-carbonitrile (19b)
Yield: 55%, off-white solid, M.p.: 180–182 °C (Lit. 183–184 °C, Sato et al. 1990a, b). 1H NMR (400 MHz, DMSO-d6): δ 8.45 (s, 1H), 7.62 (s, 2H). 1H NMR (400 MHz, CDCl3): δ 8.30 (s, 1H), 5.28 (s, 2H). 13C NMR (125 MHz, DMSO-d6): δ 156.70, 150.49, 123.64, 115.45, 110.60. EI-MS m/z: 198 (M+, Br79, 100), 200 (M+, Br81, 98), 171 (M+, Br79, –H, –CN, 17), 173 (M+, Br81, –H, –CN, 17), 119(M+, –Br, 32), 92 (M+, –Br, –H, –CN, 46).
Preparation of 3,6-dichloropyrazine-2-carbonitrile (8)
To a solution of 19a (3 g, 20 mmol) in DCM (50 mL) at 0 °C was successively added TiCl4 (2.2 mL, 20 mmol) and tert-butyl nitrite (7.4 mL, 62 mmol). The reaction mixture was stirred at room temperature for 3 h. When the reaction was completed monitored by TLC, the solvent was evaporated and the residue was treated with water (50 mL) and then extracted with ethyl acetate (2 × 50 mL). The extract was dried over Na2SO4, and concentrated to give 8 as a white solid (2.8 g, yield 81%).
3,6-Dichloropyrazine-2-carbonitrile (8)
Yield: 81%, white solid, M.p.: 93–94 °C (Lit. 90–91 °C, Li 2017). 1H NMR (400 MHz, CDCl3): δ 8.60 (s, 1H). 13C NMR (125 MHz, CDCl3): δ 149.82, 147.65, 147.03, 128.90, 112.60. EI-MS m/z: 173 (M+, Cl35, Cl35, 100), 175 (M+, Cl35, Cl37, 65), 177 (M+, Cl37, Cl37, 10).
Preparation of 6-bromo-3-chloropyrazine-2-carbonitrile (20)
Reaction of 19b (2 g, 10 mmol) with TiCl4 (1.1 mL, 10 mmol) and tert-butyl nitrite (3.7 mL, 31 mmol) following the above procedure for synthesizing 8 gave a white solid (1.8 g, yield 85%).
6-Bromo-3-chloropyrazine-2-carbonitrile (20)
Yield: 85%, white solid, M.p.: 98–100 °C. 1H NMR (500 MHz, DMSO-d6): δ 9.08 (s, 1H). 1H NMR (400 MHz, CDCl3): δ 8.71 (s, 1H). 13C NMR (100 MHz, CDCl3): δ 150.45, 149.82, 138.22, 129.66, 112.58. EI-MS m/z: 217 (M+, Cl35, Br79, 45), 219 (M+, Cl37, Br79 and Cl35, Br81, 65), 221 (M+, Cl37, Br81, 15), 138 (M+, Cl35, –Br, 100), 140 (M+, Cl37, –Br, 30), 111 (M+, Cl35, –Br, –HCN, 15), 113 (M+, Cl37, –Br, –HCN, 4).
Preparation of 3,6-difluoropyrazine-2-carbonitrile (9)
A mixture of 8 (2.6 g, 15.0 mmol), KF (5.23 g, 90.0 mmol), tetrabutylammonium bromide (1.93 g, 6.0 mmol) was pre-dried under vacuum over phosphorus pentoxide. The mixture was charged in a polytetrafluoroethylene bottle followed by the addition of dried DMSO (20 mL), and heated at 60 °C for 2.5–3 h. The reaction mixture was cooled to room temperature, treated with water (80 mL) and extracted with diethyl ether (100 mL). The organic layer was washed with water (3 × 25 mL), dried over Na2SO4, and purified by chromatography on silica gel with PE/EA (50:1–10:1) as eluent to give 9 as a white solid (1.27 g, yield 60%).
3,6-Difluoropyrazine-2-carbonitrile (9)
Yield: 60%. white solid, M.p.: 58–60 °C (Lit. 56–57 °C, Li 2017). 1H NMR (400 MHz, CDCl3) δ 8.35 (dd, J = 8.1, 1.4 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 159.73-157.69 (d, J = 255 Hz), 157.64-155.62 (d, J = 252.5 Hz), 135.09 (dd, J = 41.6, 11.0 Hz), 114.04 (d, J = 35.8 Hz), 110.62 (d, J = 9.1 Hz). EI-MS m/z: 141 (M+, 100), 122 (M+, –F, 50), 96 (M+, –F, –CN, 50).
Preparation of 3,6-difluoropyrazine-2-carboxamide (10)
To a solution of 9 (1.4 g, 10 mmol) in THF (5 mL) was added concentrated hydrochloric acid (30 mL). The reaction mixture was heated at 60 °C for 1.5 h, then cooled to room temperature and evaporated under vacuum. The residue was purified by chromatography on silica gel with PE/EA as eluent to give 10 as a white solid (1.2 g, yield 75%).
3,6-Difluoropyrazine-2-carboxamide (10)
Yield: 75%, white solid, M.p.: 122–124 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.57 (dd, J = 8.2, 1.7 Hz, 1H), 8.21 (s, 1H), 7.98 (s, 1H). 13C NMR (125 MHz, DMSO-d6): δ 162.55 (d, J = 7.2 Hz), 157.05–155.08 (dd, J = 246.4, 2.5 Hz), 156.73–164.69 (dd, J = 251.7, 2.5 Hz), 133.46 (dd, J = 43.2, 12.0 Hz), 131.50 (dd, J = 26.9, 8.6 Hz). EI-MS m/z: 159 (M+, 32), 116 (M+, –CONH, 100), 115 (M+, –CONH2, 25).
Preparation of 6-fluoro-3-hydroxypyrazine-2-carboxamide (favipiravir)
10 (1.2 g, 7.5 mmol), sodium bicarbonate (3.8 g, 45.2 mmol) were added into a mixture of 1,4-dioxane (10 mL) and water (20 mL). The reaction mixture was heated at 60 °C for 8 h. When the reaction was completed, hydrochloric acid (30 mL, 3 M) was added to adjust the pH to 3–4. The precipitate was collected, dried in vacuum to give a light-yellow solid (0.83 g). The filtrate was extracted with ethyl acetate (2 × 50 mL). The organic layer was washed with brine (2 × 10 mL), dried over Na2SO4, and evaporated to give another portion (0.14 g) of the product. The total yield was 82%.
6-Fluoro-3-hydroxypyrazine-2-carboxamide (favipiravir)
Yield: 82%, light-yellow solid, M.p.: 176–178 °C (175–177 °C, Liu et al. 2017). 1H NMR (400 MHz, DMSO-d6): δ 13.40 (s, 1H), 8.74 (s, 1H), 8.52 (s, 1H), 8.50 (s, 1H). 1H NMR (400 MHz, CDCl3): δ 12.35 (br, 1H), 8.31 (d, 1H, J = 8.0 Hz), 7.43 (br, 1H), 5.89 (br, 1H). 13C NMR (125 MHz, DMSO-d6): δ 169.20, 160.21, 152.87 (d, J = 243.8 Hz), 136.58, 122.40. ESI–MS m/z: 156.1 [M − H]−.
Preparation of 3,5-dibromo-2-hydroxylpyrazine (21b)
18b (3.0 g, 12 mmol) was added into acetic acid (20 mL) and concentrated sulfuric acid (4 mL) under ice bath. To this mixture was added a pre-prepared solution of NaNO2 (1.65 g, 24 mmol) in water (10 mL). The reaction mixture was stirred at room temperature for 3 h, and then poured into ice water (100 g). The precipitate was collected by filtrated, washed with water and dried to give an off-white solid (1.8 g, yield 61%).
3,5-Dibromo-2-hydroxylpyrazine (21b)
Yield: 61%, off-white solid, M.p.: 182–184 °C. 1H NMR (400 MHz, CDCl3): δ 11.23 (br, 1H), 7.53 (s, 1H). 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 7.94 (s, 1H). 13C NMR (125 MHz, CD3OD): δ 154.19, 136.73, 131.57, 114.62. ESI–MS m/z: 250.9 [M − H]−.
Preparation of 5-chloro-3-bromo-2-hydroxylpyrazine (21a)
Reaction of 18a (3.0 g, 14.5 mmol) dissolved in acetic acid (20 mL) and concentrated sulfuric acid (4 mL) with NaNO2 (1.9 g, 29 mmol) in water (10 mL) following the above procedure for synthesizing 21a gave a yellow solid (1.6 g, yield 54%).
5-Chloro-3-bromo-2-hydroxylpyrazine (21a)
Yield: 54%, yellow solid, M.p.: 184–186 °C. 1H NMR (400 MHz, DMSO-d6): δ 13.11 (br, 1H), 7.92 (s, 1H). 1H NMR (500 MHz, CD3OD): δ 7.58 (s, 1H). 13C NMR (125 MHz, CD3OD): δ 154.21, 136.07, 128.96, 127.76. ESI–MS m/z: 206.9 [M − H]−.
Preparation of 6-chloro-3-hydroxylpyrazine-2-carbonitrile (22a)
Reaction of 21a (1.0 g, 4.8 mmol) with NaCN (280 mg, 5.8 mmol) catalyzed by CuI (460 mg, 2.4 mmol) and Pd(PPh3)4 (55 mg, 0.05 mmol) according to the procedure for synthesizing 19b gave a gray solid (0.5 g, yield 64%).
6-Chloro-3-hydroxylpyrazine-2-carbonitrile (22a)
Yield: 64%, gray solid, M.p.: 158–160 °C.1H NMR (400 MHz, DMSO-d6): δ 7.97 (s, 1H). 1H NMR (500 MHz, CD3OD): δ 8.25 (s, 1H). 13C NMR (125 MHz, CD3OD): δ 160.73, 143.18, 134.78, 118.47, 113.87. ESI–MS m/z: 153.9 [M − H]−.
Preparation of 6-bromo-3-hydroxylpyrazine-2-carbonitrile (22b)
Reaction of 21b (1 g, 4 mmol) with NaCN (230 mg, 4.8 mmol) catalyzed by CuI (380 mg, 2 mmol) and Pd(PPh3)4 (50 mg, 0.04 mmol) according to the procedure for synthesizing 19b gave a gray solid (350 mg, yield 45%).
6-Bromo-3-hydroxylpyrazine-2-carbonitrile (22b)
Yield: 45%, gray solid, M.p.: 168–170 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.46 (s, 1H). 13C NMR (125 MHz, DMSO-d6): δ 151.12, 133.03, 129.18, 114.82, 99.21. ESI–MS m/z: 197.9 [M − H]−.
Preparation of 3-hydroxy-6-nitropyrazine-2-carboxamide (7)
To a solution of 6 (4.0 g, 28.7 mmol) in concentrated sulfuric acid (20 mL) under ice-cooling was added potassium nitrate (5.8 g, 57.5 mmol) in three batches. After stirring at 40 °C for 4 h, the reaction mixture was poured into ice water (200 g). The resulting precipitate was collected by filtration. The solid was slurried in pure water (20 mL) and dried to give a light-yellow solid (1 g, yield 19%).
3-Hydroxy-6-nitropyrazine-2-carboxamide (7)
Yield: 19%, light yellow solid, M.p.: 182–184 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.96 (s, 1H), 8.32 (s, 1H), 8.05 (s, 1H). 13C NMR (125 MHz, DMSO-d6): δ 163.60, 156.92, 143.19, 138.63, 134.26. EI-MS m/z: 184 (M+, 84), 141 (M+, –CONH, 10).
Results and discussion
The new synthetic approach to favipiravir starting from 2-aminopyarzine 16 was outlined in Scheme 4. The preferred route (highlighted in red) consisted of seven steps, featured with a novel and efficient method to the synthesis of 8 in four transformations.

Our synthetic approach to favipiravir in this paper and the preferred route was highlighted in red
With respect to the first step, at the beginning, mono-chlorination of 16 using NCS in CHCl3 was plagued by a very low yield and large amounts of by-products (Cai et al. 2007). After a preliminary solvent screening, we determined acetonitrile as the optimal reaction medium. However, further condition optimizations did not afford significant improvement in product yield, shown in Table 1. A stoichiometric amount of NCS was not sufficient to react with all the material (Entry 2), and an increase in the amount of NCS would lead to an overchlorinated product (Entry 3–4). Moreover, the reaction mixture became dark brown and cloudy with time, probably due to the formation of polymers (Cai et al. 2007). Recently, Pu et al. reported a chlorinating reagent (BSA) which provided an 80% yield of 17 (Pu et al. 2016). Accordingly, we prepared BSA as well as the analog TSA, and performed the reaction using the two reagents. 1.1 Equiv of BSA gave the product in a yield of 77% consistent with the previous study (Table 1, Entry 5). However, BSA was oil, not convenient to use. TSA was a solid and used for screening of reaction conditions. It was found that an increase of TSA from 0.9 to 1.1 equiv significantly improved the yield (Entry 6–8), offering a maximum of 80%. Further increase in the amount of TSA was detrimental to the yield (Entry 9), which was exclusively attributed to the overchlorinated by-product. The optimal ratio of TSA was 1.1 equiv, also producing a small amount (~ 5%) of the overchlorinated impurity, but could be removed at the stage or downstream by chromatographic purification or recrystallization.

17 was amenable to bromination at the C3 position with NBS in a yield of 87%, and subsequent replacement of the bromine atom by the nitrile group smoothly afforded 19a. Sodium cyanide proved to be a good nitrile donor in the coupling reaction with catalytic Pd(PPh3)4. The reaction yield was influenced by several factors, so we made a preliminary investigation of the reaction condition (Table 2). This coupling reaction should be performed at a temperature of about 120 °C. Reducing the temperature to 100 °C or 80 °C remarkably decreased the yield even with a prolonged reaction time (Entry 1–2). 1 mol % of Pd(PPh3)4 were sufficient for the coupling reaction (Entry 4 and Entry 7–8), and the optimal amount of NaCN was 1.2 equiv (Entry 3–4 and Entry 6). In theory, the chlorine atom at the C6 position of 19a could also be replaced by the nitrile group in the coupling reaction, so it would be better to control the amount of NaCN in the range of 1.2–1.5 equiv. Generally, the best condition was Entry 5 with 1.2 equiv of NaCN, 1 mol % of Pd(PPh3)4 and 120 °C of reaction temperature, which provided a yield of 85% (Entry 4).

With compound 19a in hand, it was easy to prepare the key intermediate 8 via Sandmeyer reaction. t-Butyl nitrite and titanium tetrachloride (TiCl4) was applied to make this transformation on account of the simple workup and the good yield (> 80%). The residual TiCl4 would be converted to the non-toxic titanium dioxide by reacting with water in the post-processing.
Nucleophilic fluorination of 8 with KF (6 equiv) and Bu4NBr (2 equiv) gave the difluoro product 9 in 60% isolated yield. Compound 19 bearing a C5 bromine group was prepared with the same strategy, but it proved to be a very poor substrate, only giving a small amount of the desired product. For the fluorination step, glass reaction vessel should be avoided, otherwise the yield would be severely decreased. The subsequent acid-mediated nitrile hydration proceeded efficiently and the derived intermediate 10 was subjected to anhydrous NaHCO3 solution to afford the final product in a yield of 62% over two steps. The difluoro intermediate 9 had a low melting point, and the general workup procedure may easily cause yield loss. Recently, Liu et al. reported a one-pot three-step protocol for the synthesis of favipiravir from intermediate 8 in good yield (60%). Since intermediates 9 and 10 did not need to be isolated, this process could minimize the yield loss (Liu et al. 2017).
Compound 8 is a potential allergy-causing substance, which we had confirmed during the course of our studies. To avoid using this intermediate, another synthetic route was investigated. Retrosynthetically, compound 23 or 24, if available, could be a favorable precursor of favipiravir. Along this line, we made our effort to synthesize the two compounds (Scheme 5). Compound 22a and 22b were synthesized from 18 in two steps via Sandmeyer hydroxylation reaction and Pd-catalyzed cyanation of 21. However, reaction of the two compounds with KF or CsF under the same condition did not give the desired product and most of the starting material was remained in the reaction mixture. Based on the reaction results from 19, 8, 20 and 22, it could be concluded that the fluoride preferred to replace the chlorine atom instead of the bromine atom; the adjacent Cl of the nitrile group of 8 was first substituted, which would facilitate the fluorination at the C6 position; and mono-fluorination at the C6 position of 19 or 22 was very difficult.

Another synthetic approach to favipiravir proposed in this paper
Conclusion
In summary, we investigated the synthesis of favipiravir starting from an inexpensive and commercially available 2-aminopyrazine. A facile route for the synthesis of 8 was developed in four steps. This new approach was free from the hazardous POCl3 and provided a good yield. The purpose of finding a novel synthesis to favipiravir not via intermediate 8 was not achieved, but due to the allergy-causing problems of 8, alternative protocols should be considered.
References
Bai CQ, Mu JS, Kargbo D, Song YB, Niu WK, Nie WM, Kanu A, Liu WW, Wang YP, Dafae F, Yan T, Hu Y, Deng YQ, Lu HJ, Yang F, Zhang XG, Sun Y, Cao YX, Su HX, Sun Y, Liu WS, Wang CY, Qian J, Liu L, Wang H, Tong YG, Liu ZY, Chen YS, Wang HQ, Kargbo B, Gao GF, Jiang JF (2016) Clinical and virological characteristics of Ebola virus disease patients treated with favipiravir (T-705)-Sierra leone, 2014. Clin Infect Dis 6310:1288–1294. https://doi.org/10.1093/cid/ciw571
Beldar SV, Jordis U (2009) Synthetic studies towards the antiviral pyrazine derivative T-705. Institute of Applied Synthetic Chemistry, Vienna University of Technology, Vienna, p 13
Cai L, Pike V W, Innis R B (2007) (Aminophenyl)imidazo[1,2-a]pyridine derivatives useful as beta-amyloid PET imaging agents and their preparation. WO2007124345A2 (issued November 1, 2007)
El-Nahas A, Hirao K (1999) A theoretical study on 2-hydroxypyrazine and 2,3-dihydroxypyrazine: tautomerism, intramolecular hydrogen bond, solvent effects. J Mol Struct 4591:229–237. https://doi.org/10.1016/s0166-1280(98)00270-x
Furuta Y, Egawa H (2000) Nitrogenous heterocyclic carboxamide derivatives or salts thereof and antiviral agents containing both. European Patent Office WO, 00/10569 (issued March 2, 2000)
Furuta Y, Takahashi K, Shiraki K, Sakamoto K, Smee DF, Barnard DL, Gowen BB, Julander JG, Morrey JD (2009) T-705 (favipiravir) and related compounds: novel broad-spectrum inhibitors of RNA viral infections. Antivir Res 823:95–102. https://doi.org/10.1016/j.antiviral.2009.02.198
Furuta Y, Gowen BB, Takahashi K, Shiraki K, Smee DF, Barnard DL (2013) Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antivir Res 1002:446–454. https://doi.org/10.1016/j.antiviral.2013.09.015
Gao J, Luo X, Li Y, Gao R, Chen H, Ji D (2018) Synthesis and biological evaluation of 2-oxo-pyrazine-3-carboxamide-yl nucleoside analogues and their epimers as inhibitors of influenza a viruses. Chem Bio Drug Des 853:245–252. https://doi.org/10.1111/cbdd.12382
Hara T, Norimatsu N, Kurushima H, Kano T (2010) Method for producing dichloropyrazine derivative. European Patent Office WO2010087117 (issued August 2, 2012)
Huchting J, Winkler M, Nasser H, Meier C (2017) Synthesis of T-705-ribonucleoside and T-705-ribonucleotide and Studies of chemical stability. ChemMedChem 129:652–659. https://doi.org/10.1002/cmdc.201700116
Huchting J, Vanderlinden E, Winkler M, Nasser H, Naesens L, Meier C (2018) Prodrugs of the phosphoribosylated forms of hydroxypyrazinecarboxamide Pseudobase T-705 and its de-fluoro analogue T-1105 as potent influenza virus inhibitors. J Med Chem 61:6193–6210. https://doi.org/10.1021/acs.jmedchem.8b00617
Jin Z, Tucker K, Lin X, Kao CC, Shaw K, Tan H, Symons J, Behera I, Rajwanshi VK, Dyatkina N, Wang G, Beigelman L, Deval J (2015) Biochemical evaluation of the inhibition properties of favipiravir and 2′-C-methyl-cytidine triphosphates against human and mouse norovirus RNA polymerases. Antimicrob Agents Chemother 5912:7504–7516. https://doi.org/10.1128/aac.01391-15
Klejch T, Pohl R, Janeba Z, Sun M, Keough D, Guddat L, Hockova D (2018) Acyclic nucleoside phosphonates with unnatural nucleobases, favipiravir and allopurinol, designed as potential inhibitors of the human and Plasmodium falciparum 6-oxopurine phosphoribosyltransferases. Tetrahedron 74:5886–5897. https://doi.org/10.1016/j.tet.2018.08.014
Li M (2017) Synthetic method of favipiravir. State Intellectual Property Office of the P.R.C. CN107226794A (issued October 3, 2017)
Liu F-L, Li C-Q, Xiang H-Y, Feng S (2017) A practical and step-economic route to Favipiravir. Chem Pap 7111:2153–2158. https://doi.org/10.1007/s11696-017-0208-6
Naesens L, Guddat LW, Keough DT, van Kuilenburg AB, Meijer J, Vande Voorde J, Balzarini J (2013) Role of human hypoxanthine guanine phosphoribosyltransferase in activation of the antiviral agent T-705 (favipiravir). Mol Pharmacol 844:615–629. https://doi.org/10.1124/mol.113.087247
Palamidessi G, Bernardi L (1964) On 2, 5-dichloropyrazine. J Org Chem 298:2491–2492
Plebanek E, Lescrinier E, Andrei G, Snoeck R, Herdewijn P, Jonghe SD (2017) Emimycin and its nucleoside derivatives: synthesis and antiviral activity. Eur J Med Chem 144:93–103. https://doi.org/10.1016/j.ejmech.2017.12.018
Pu X, Li Q, Lu Z, Yang X (2016) N-Chloro-N-methoxybenzenesulfonamide: a chlorinating reagent. Eur J Org Chem 36:5937–5940. https://doi.org/10.1002/ejoc.201601226
Sangawa H, Komeno T, Nishikawa H, Yoshida A, Takahashi K, Nomura N, Furuta Y (2013) Mechanism of action of T-705 ribosyl triphosphate against influenza virus RNA polymerase. Antimicrob Agents Chemother 5711:5202–5208. https://doi.org/10.1128/aac.00649-13
Sato N, Takeuchi R (1990a) Studies on pyrazines; part 20.1 a simple synthesis of 5-substituted 2-amino-3-cyanopyrazines: useful intermediates for pteridine synthesis. Synthesis 199008:659–660
Sato N, Takeuchi R (1990b) Studies on pyrazines; Part 20.1 a simple synthesis of 5-substituted 2-amino-3-cyanopyrazines: useful intermediates for pteridine synthesis. Synthesis 199008:659–660
Smither SJ, Eastaugh LS, Steward JA, Nelson M, Lenk RP, Lever MS (2014) Post-exposure efficacy of oral T-705 (Favipiravir) against inhalational Ebola virus infection in a mouse model. Antivir Res 104:153–155. https://doi.org/10.1016/j.antiviral.2014.01.012
Wang G, Wan J, Hu Y, Wu X, Prhavc M, Dyatkina N, Rajwanshi VK, Smith DB, Jekle A, Kinkade A, Symons JA, Jin Z, Deval J, Zhang Q, Tam Y, Chanda S, Blatt L, Beigelman L (2016) Synthesis and anti-influenza activity of pyridine, pyridazine, and pyrimidine C-nucleosides as favipiravir (T-705) analogues. J Med Chem 5910:4611–4624. https://doi.org/10.1021/acs.jmedchem.5b01933
Acknowledgements
This work was supported by National Science Foundation for Young Scientists of China (Grant no. 21502209). The authors gratefully acknowledged Topharman Shanghai Co., Ltd for collaboration.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Guo, Q., Xu, M., Guo, S. et al. The complete synthesis of favipiravir from 2-aminopyrazine. Chem. Pap. 73, 1043–1051 (2019). https://doi.org/10.1007/s11696-018-0654-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-018-0654-9