
Abstract
Emerging evidence highlights that dysfunction of adipose tissue contributes to impaired insulin sensitivity and systemic metabolic deterioration in obese state. Of note, adipocyte hypertrophy serves as a critical event which associates closely with adipose dysfunction. An increase in cell size exacerbates hypoxia and inflammation as well as excessive collagen deposition, finally leading to metabolic dysregulation. Specific mechanisms of adipocyte hypertrophy include dysregulated differentiation and maturation of preadipocytes, enlargement of lipid droplets, and abnormal adipocyte osmolarity sensors. Also, weight loss therapies exert profound influence on adipocyte size. Here, we summarize the critical role of adipocyte hypertrophy in the development of metabolic disturbances. Future studies are required to establish a standard criterion of size measurement to better clarify the impact of adipocyte hypertrophy on changes in metabolic homeostasis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Obesity is a primary cause of metabolic disorders including type 2 diabetes (T2D), dyslipidemia, and hypertension. The prevalence of obesity has doubled in 73 countries all over the world and over 2 billion people are suffering from overweight and obesity [1]. Importantly, overnutrition predisposed individuals to insulin resistance and metabolic abnormalities [1, 2]. In obese individuals, white adipose tissue (WAT), which mainly mediates energy homeostasis, becomes dysfunctional and expands improperly to store excess energy. When lipid accumulation exceeds adipose maximum storage capacity, lipolysis increased, releasing fatty acid into circulation [1, 2]. Subsequently, ectopic fat deposits in peripheral organs such as the liver or skeletal muscles and eventually leads to abnormal glucose metabolism and systemic insulin resistance. This phenomenon has been commonly referred to as “lipotoxicity” [3, 4].
In the past few decades, fat mass content has been regarded as a key factor that influences the severity of metabolic disturbances. In recent years, numerous researches have focused on how the excessive energy is stored (hypertrophy or hyperplasia) in adipose tissue (AT) rather than the total fat mass content [5,6,7]. Physiologically, adipocytes expand or proliferate in order to store more energy as triglyceride. However, in morbid obesity, adipocytes tend to expand to the greatest extent, which is termed as “adipocyte hypertrophy”. Even within the same WAT depot, cell diameter of different adipocytes varies dramatically, ranging from less than 20 to 300 μm [8]. Accordingly, alteration in adipocyte size as well as the proportions of small or large adipocytes could manifest the development of T2D and non-alcoholic fatty liver disease (NAFLD) [9,10,11,12]. Importantly, enlargement of adipocytes triggers low-grade chronic inflammation, insufficient angiogenesis, and excessive collagen deposition, which further lead to abnormal adipokine release and impaired glucose metabolism [13].
In this review, we aim to outline a comprehensive overview of adipocyte hypertrophy in terms of metabolic disorders; we also summarize underlying mechanisms and discuss the role of adipocyte size as a potential marker in the treatment of obesity-related metabolic diseases.
Distinction Among White Adipocyte Size in Different Depots
Generally, larger adipocytes are critically associated with metabolic disorders [14]; yet, the effect of enlarged adipocytes on metabolic dysregulation varied in different adipose depots.
AT is commonly classified as visceral AT (VAT) and subcutaneous AT (SAT). Anatomically, VAT is located primarily in the mesentery and omentum while SAT is presented mainly in gluteofemoral, back, and anterior abdominal region. Metabolically, SAT is suggested to have a protective role of metabolic risk [15, 16], while excessive VAT accumulation is an independent risk factor for obesity-induced metabolic disorders [17].
Specifically, VAT contains fewer preadipocytes and more large adipocytes. On the contrary, SAT tended to contain more small adipocytes [18]. One study measured adipocyte size in both SAT and VAT in 11 morbidly obese women with normal glucose level. Mean adipocyte volume was larger in VAT than that in SAT, but these two depots did not differ in the proportion of small adipocytes [14]. Also, expressions of cell differentiation markers were greater in SAT, whereas VAT displayed greater expression of inflammatory genes [19]. These results suggest that VAT tends to be less involved in triglyceride deposition but relates more to adipose inflammation, compared with SAT.
With respect to the relationship between adipocyte size and lipid/glucose profile, some studies indicated that increased visceral adipocyte size (VAS) had a much stronger detrimental influence on plasma total cholesterol, LDL cholesterol, apolipoprotein B, and triacylglycerols. On the other hand, subjects with greater subcutaneous adipocyte size (SAS) displayed higher homeostasis model assessment of insulin resistance (HOMA-IR), blood glucose level, and plasma insulin level [12, 20, 21]. This distinction could be explained by different blood supply in VAT and SAT depots, since VAT is mainly drained by portal vein into the liver where lipoprotein and lipids are synthesized [22].
Distinction Among White Adipocyte Size in Different Genders
There are strong differences in SAT and VAT biology in male and female subjects. Sex differences in fat distribution involve both cell size and number. Traditional view suggested that gluteo-femoral adipocytes of women are larger while abdominal adipocytes are comparable between both genders, and visceral adipocytes of women are significantly smaller [23]. Recent research indicated that males have larger adipocytes in both SAT and VAT than females independent of the content of adiposity [24]. Generally, at lean state, premenopausal women have a greater amount of small adipocytes especially in the femoral depot [25, 26]. Also, as lipid accumulating, men have more hypertrophic type of adipose expansion compared with women, suggesting a gender difference in adipose tissue remodeling and fatty acid storage [27].
One potential contributor to sex differences in adipose tissue expansion is the number of adipocyte precursor cells within adipose tissue. Previous animal studies showed that compared with male mice, female C57BL/6J mice have more adipocyte precursor cells (APC) with low-fat diet and increased more APCs with high-fat diet in VAT (gonadal adipose tissue (GWAT)) and SAT (inguinal white adipose tissue (IWAT)) [28]. These differences may explain why males display a higher degree of insulin resistance although inflammatory markers in GWAT were similar [28].
Accordingly, different patterns of adipocyte proliferation were attributed to sex hormones and receptors. It has been proved that estrogens influence hyperplasia by increasing APCs number and stimulating cell proliferation [29, 30]. Estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ) are estrogen receptors on adipocytes which influence adiposity [27]. The total body ERα knockout mouse has increased adiposity, increased visceral fat accumulation, and the metabolic syndrome. Males have a relative lack of ERα in the visceral depot and are therefore primed to store more fat viscerally [31]. Future investigations are required to better clarify the effect of sex hormones on systemic adipose biology.
Relationship Between Adipocyte Size and Metabolic Disorders
Obesity and Insulin Resistance
Of note, during the acceleration of adipose content, dysfunctional AT leads to defects in insulin sensitivity and the development of insulin resistance. Most studies suggested that enlarged SAS has been implicated in the progression of impaired regional and systemic insulin sensitivity [5, 9, 32,33,34,35]. This phenomenon could further counteract insulin-promoted adipogenesis.
In a group of non-diabetic individuals, SAS negatively correlated with insulin sensitivity measured by euglycemic-hyperinsulinemic clamp (M value) [5, 9]. In line with the association, similar studies reported compared with insulin-sensitive subjects, body mass index (BMI)-matched insulin-resistant overweight/obese subjects displayed larger SAS [32, 34, 35]. Also, in the field of VAT, VAS was found to be positively related to HOMA-IR in lean patients after adjustment of age and body mass [36, 37]. In contrast, one study indicated that no association was found between VAS and HOMA-IR in non-diabetic subjects [9]. Notably, mesenteric adipocyte size was suggested to be strongly related to insulin resistance compared with the other two visceral adipose depots [38]. Inconsistencies among prior studies may be explained by the different sample size, adjustment of different metabolic variables, and distinct adipose depots.
Despite abundant cross-sectional studies, some interventional studies explored the association of adipocyte size alterations during a short-term weight gain. In one recent approach, recruited metabolically healthy obese subjects were given a hypercaloric diet to induce 3.2-kg weight gain in a period of 4 weeks. At baseline, the insulin-sensitive (IS) subjects displayed smaller SAS. Surprisingly, compared with insulin resistance (IR) group, the proportion of small cells in IS group decreased from 50 to 43% while no significant alteration was observed in IR group [34]. This result indicated the negative impact of weight gain even in subjects who were metabolically healthy. Likewise, another overfeeding study which made 29 normal weight men gain 7.6 kg in 8 weeks suggested that subjects who had greater number of small adipocytes displayed greater decrease in insulin sensitivity [39].
These results highlighted “adipogenic potential”. Specifically, when enlargement of adipocytes reached maximal capacity for fat storage and recruitment and maturation of preadipocytes were significantly impaired, accumulation of small adipocytes and concomitant adipocyte hypertrophy occurred, leading to excess lipid deposition in ectopic tissues [40, 41]. It is noteworthy that compared with the study which recruited obese subjects [34], the latter study enrolled young, normal-weight men [39]. Moreover, the definition of “small adipocytes” was different between these two studies [34, 39]. In addition, these studies emphasized cell distribution along with the absolute value of adipocyte size.
Type 2 Diabetes
The relationship between enlarged adipocyte size and deterioration of blood glucose level is well confirmed. SAS was higher in subjects with impaired glucose tolerance (IGT) and T2D than subjects with normal glucose tolerance (NGT) [5, 42, 43]. Average cell size was positively correlated with systemic glucose tolerance [5]. Consistent result was obtained in another study which showed an increasing trend of average SAS in lean, impaired fasting glucose (IFG) or IGT and T2D subjects [44]. In terms of specific cell distribution, more large adipocytes and less small adipocytes were observed in T2D patients, indicating that elevated blood glucose level is accompanied by recruitment and proliferation of adipocyte precursors, though the capability to develop mature adipocytes has been impaired [45].
Interestingly, most of the studies focused on abdominal SAT while some studies compared different SAT depots. Subcutaneous abdominal adipocyte size (AAS) was identified as a risk factor for the progression of T2D in middle-age women, whereas femoral adipocyte size (FAS) displayed weaker association with T2D only via the correlation with AAS [6]. This study highlighted the impact of SAT present in the abdominal area on normal glucose metabolism. In addition, an overfeeding study suggested upper-body adipocyte size increased significantly in response to overfeeding. However, lower-body adipocyte responded to excessive food intake by hyperplasia [46]. The inherent dynamics of preadipocytes may result in distinct characteristics of SAT from different depots.
With respect to visceral adipocyte size (VAS), much less is known about VAS alteration in T2D patients, most of the studies were similar to obese and insulin-resistant subjects. Few studies with small sample size demonstrated that type 2 diabetic patients presented greater VAS [36, 38, 42, 47]. It has been speculated that compared with SAT, the number of visceral adipocytes was notably more important than adipocyte size [21]. There is a need to better understand the relationship between VAS and hyperglycemia in future observations.
NAFLD
NAFLD, one important component of metabolic syndrome (MS), is critically involved in the progression of T2D. One study highlighted that SAS could explain 21% of liver fat deposition according to linear regression [48]. In the field of VAT, liver injury was generally assessed by alanine aminotransferase (ALT), aspartate aminotransferase (AST), and NAFLD activity score. Increased omental adipocyte diameter associated tightly with ALT, AST level, and NAS, suggesting that omental adipocyte enlargement was an independent predictor of stages of NAFLD together with liver injury level [49, 50].
Other Metabolic Diseases
Few studies found that women with polycystic ovary syndrome (PCOS) indicated enlarged SAS compared with healthy women matched for age and BMI levels [51, 52]. Similarly, VAS also increased in women with PCOS [53]. In addition, one study measured SAS in normal weight and obese pregnant women, where cell diameter larger than 150 μm was defined as “very large adipocytes”. Notably, HOMA-IR level in trimesters 3 was closely associated to proportion of very large adipocytes, which indicates the potential role of adipocyte size in predicting the development of gestational diabetes [54]. More investigations are required for further exploration of extensive change during pregnancy.
Association Between Adipocyte Hypertrophy, Pathophysiological Changes, and Metabolic Dysfunction in AT
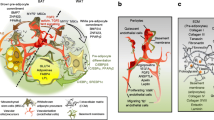
Various pathological changes occur in AT following the enlargement of adipocyte size. On the one hand, adipocyte hypertrophy triggers inflammatory response and exacerbates regional hypoxia, further making excessive collagen deposition. On the other hand, pathophysiological changes subsequently contribute to dysregulation of adipokine release and impaired glucose metabolism. Adipocyte hypertrophy serves as an important factor which connects closely to metabolic dysfunction in AT (Fig. 1).

Adipocyte hypertrophy-induced and metabolic dysregulations. With the increase of lipid accumulation, adipocyte hypertrophy is associated with, exacerbation of inflammation, adipocyte hypoxia, excessive collagen deposition, abnormal adipokine release, and impaired glucose metabolism, which finally contribute to dysregulated systemic energy metabolism
Infiltration of Inflammatory Immune Cells
Apart from adipocytes, various types of cells including adipocyte progenitors, fibroblasts, endothelial cells, and immune cells were discovered in AT from healthy individuals [55,56,57]. With the development of obesity, a high-degree of inflammation, marked by elevated infiltration of pro-inflammatory immune cells, was discovered in WAT [58,59,60,61]. Among all the pro-inflammatory immune cells, M1 macrophages, which have been reported to infiltrate into WAT in both obese human and animals, produce proinflammatory cytokines such as tumor necrosis factor α (TNF-α), interleukin-6 (IL-6), and interleukin-1b (IL-1b) [62,63,64,65,66].
On the one side, hypertrophic adipocytes exacerbate infiltration of proinflammatory immune cells and secretion of inflammatory cytokines. In obese individuals, enlargement of adipocyte size associates closely with increased recruitment of M1 macrophages and reduced anti-inflammatory M2 macrophages [19, 67,68,69,70]. Interestingly, increased M1 macrophages were regarded as a protective compensatory mechanism to limit adipocyte hypertrophy since macrophages could promote collagen accumulation in obese state [71]. When chemokine CC receptor (CCR)2, the receptor of monocyte chemoattractant protein 1 (MCP-1), was knockout, the number of macrophages was decreased, associated with elevated adipocyte diameter [65, 72, 73]. Also, in SAT, induced natural killer T (iNKT) cells, which are one subset of T cells, were depleted in obesity. When iNKT cells were transferred into obese mice, body weight and adipocyte size were observed to decrease significantly [74]. Additionally, enlarged adipocytes overexpressed inflammatory cytokines such as NF-κB [74], IL-6 [75], MCP-1 [55, 61], and TNF-α [60, 76] independent of BMI level and total body fat mass content. Also, in obese subjects, plasma C-reactive protein (CRP) level was positively correlated to the proportion of hypertrophic adipocytes [76].
On the other side, inflammatory cytokines may contribute to enlargement of adipocyte size. It has been reported that TNF-α in SAT hindered the differentiation process of preadipocytes via suppression of mesenchymal stem cell (MSC) commitment towards adipogenic differentiation [77]. Further, TNF-α reduced activity of the transcription factor EBF transcription factor 1 (EBF1), thereby promoting the development of adipocyte hypertrophy through altered expressions of key lipolytic genes [13]. One study showed that M1 macrophages were positive correlation with VAS but not with SAS and in obese T2D and obese non-T2D subjects [36]. This correlation indicates that VAS has more pronounced inflammatory phenotype, although SAT accounts for about 80% of whole-body fat mass. Further studies are required to identify whether VAS associates closer with inflammation in lean subjects.
Hypoxia and Angiogenesis
When AT undergoes insufficient oxygen supply, hypoxia-inducible factor-1α (HIF-1α) is activated. Subsequently, increased fiber deposition remodeling and proinflammatory phenotype are triggered, contributing to the progression of adipose dysfunction and insulin resistance [78, 79]. It has long been suggested that enlargement of adipocyte size limits oxygen diffusion and triggers regional hypoxia [78,79,80,81]. Generally, the hypoxic state of hypertrophic adipocyte could be ascribed to two factors. First, compared with small cells (< 66 μm), cells with large size (> 88 μm) consumed more oxygen [82]. Also, when diameters of hypertrophic adipocytes exceed 100 μm, the diffusion rate of oxygen declines profoundly [64, 83].
In recent decades, some studies suggested that the contribution of adipocyte hypertrophy to the hypoxia response becomes less important [84, 85]. One study found that in obese patients, only a small proportion of adipocytes diameter exceed 100 μm, suggesting that the hypoxic condition within hypertrophic adipocytes may also have been caused by other factors [86]. Notably, in obese state, AT blood flow is about 30–40% lower than subjects with normal weight [87]. It was noteworthy that glycation-induced inability of angiogenesis decreased blood flow [85]. Moreover, imbalance of VEGF/Ang-2 ratio inhibits endothelial cell proliferation and prevents capillarization, the disarrangement of vascular formation, and reduced blood flow exacerbate hypoxia [85], further leading to impairment of AT insulin sensitivity.
In parallel, hypoxic condition has been regarded to be detrimental to adipocyte differentiation in hypertrophic AT. In both preadipocytes and adipocytes, hypoxia induces decreased acetylation level of histone H3 and H4 of peroxisome proliferator-activated receptor γ (PPARγ) promoter [81, 87]. However, varied degrees of hypoxia directly contribute to distinct effects on adipocyte differentiation. Improved adipogenesis after short period of hypoxic exposure was reported in previous studies. In one animal study, Sprague-Dawley rats were treated with 10 hypoxia cycles/h and 6 h/day, where one hypoxia cycle was defined as 240 s for 10% O2 and 120 s for 21% O2. After 6-week exposure, the hypoxia condition accelerated expressions of adipogenesis transcription factors CCAAT-enhancer-binding proteins α (C/EBPα), C/EBPβ, PPARγ, as well as insulin-like growth factor 1(IGF-1)/Akt signaling pathway in SAT [88]. Similar result was reported by another study in which C57Bl/6J mice were exposed to chronic hypoxia (8% O2). Decreased adipocyte size and improved adipose function were observed after 3-week treatment [87]. Since there is no standard duration of hypoxic exposure at present, further studies are needed to better clarify the range of hypoxia degree which could improve adipocyte differentiation.
Accumulation of Extracellular Matrix
With the accumulation of lipid, increased deposition of collagen and reduced extracellular matrix (ECM) flexibility contribute to adipose dysfunction [89]. AT, as a connective tissue, contains a “collagen microenvironment”, which is composed of various types of collagen. Different types of collagen promote oriented differentiation of mesenchymal stem cells. Collagen I, III, and V are secreted mainly by fibroblasts, while collagen IV, VI, and XVIII (sparse) are secreted mainly by adipocytes [90, 91]. The former provides a “stiff” environment for a well-developed cytoskeleton in cells binding to these molecules. In contrast, the latter mediates changes in the cytoskeleton’s reorganization for adipocyte differentiation [90]. Physiologically, collagen is distributed in a balanced manner [90]. However, in obese state, collagen I and VI are the most common matrix prone to excessive synthesis [92]. One study showed negative correlation between total collagen content and SAS [93]. Another study confirmed this relationship that obese patients whose SAT contained greater collagen VI displayed small and medium adipocytes [94]. Likewise, VAT fibrosis level was negatively correlated with cell size and plasma triglyceride level [89]. Augmented collagen synthesis may be regarded as an adaptation mechanism in response to ameliorate adipocyte hypertrophy and limit adipose expansion.
Moreover, increased level of collagen in AT upregulates expression of FABP4 and partially upregulates PPAR-γ in adipocytes, resulting in alteration of metabolism of adipocytes [95]. These studies indicate that ECM redistribution in AT changed the morphology and metabolic function of adipocytes successively. Detailed response in cellular level will be discussed in the next section.
Collectively, adipocyte fibrosis seems to associate with limited adipocyte size and altered AT metabolism. Whether AT fibrosis serves as a more sensitive biomarker in reflection of adipose dysfunction still requires further investigations.
Dysregulated Adipokine Release
Besides fat storage, AT also plays a key role in regulating systemic homeostasis through secretion of massive proteins. These peptide or non-peptide hormones are generally referred to as adipokines [61, 96]. During the progression of obesity, adipokine concentration and phenotype altered in response to sustained energy excess.
Leptin and adiponectin are well-studied adipokines that modulate appetite and energy metabolism respectively [96]. It was reported that SAS positively was associated with plasma leptin level in 83 over-weight subjects [9]. In addition, SAS was an independent predictor of plasma leptin level [97]. Similar study conducted in T2D patients suggested that within SAT, mRNA and protein levels of adipokines, such as leptin, serum amyloid A (SAA), and transmembrane 4 L six family member 1 (TM4SF1), were elevated in very large subcutaneous adipocytes compared with small adipocytes [60]. By contrast, patients characterized by adipocyte hypertrophy in both subcutaneous and omental AT had significantly lower plasma adiponectin level, though this impact was more evident in subcutaneous adipose compartment [75]. Also, in cell cultures, production of adiponectin is negatively linked to adipocyte size [98]. In general, hypertrophic adipocytes are accompanied by concomitant enrichment of proinflammatory, pro-diabetic adipokines, and decreased production of adipokines that exert beneficial effect on regional homeostasis.
Impaired Glucose Metabolism
Although AT merely accounts for a small proportion of systemic insulin-induced glucose uptake, adipose GLUT4 specifically knockout mice displayed profound glucose intolerance, indicating that AT plays a crucial role in mediating glucose metabolism [99].
In normal state, the insulin receptor (IR) is located in caveolae. When insulin binds to the transmembrane receptor and activates protein tyrosine kinase, thus, intracellular docking proteins, including the IRS proteins, are recruited. Subsequently, PI3-kinase (PI3K) activation induces the activation of phosphoinositide-dependent kinase 1 (PDK1) [100], which then induces the activation of serine/threonine kinase (Akt)/ protein kinase B(PKB) and allows the translocation of glucose transporter type 4 (GLUT4). Importantly, within AT, insulin mainly induces glucose transport through intracellular localization glucose transporter GLUT4 [101]. Insulin increased exocytosis of GLUT4 through insulin-responsive vesicle pool. Then, increasing GLUT4 is transported to the plasma membrane and the rate of adipocyte glucose uptake was also markedly increased [101].
With the accumulation of adipose, the glucose uptake rate of hypertrophic adipocytes has changed significantly. Of note, previous study showed that, with the enlargement of adipocytes, their response to insulin in the instance of glucose utilization may decline persistently [102]. When patients underwent weight loss, their SAS was significantly decreased, with a parallel improvement of AT insulin sensitivity and plasma insulin level [102]. Likewise, SAS was proved negatively correlated with glucose uptake rate in SAT after adjustment for age, BMI, and sex [9, 103, 104]. One follow-up study reported no correlation observed between the measured GLUT4 storage vesicle (GSV) trafficking and adipocyte size isolated from different subjects [105]. Another study suggested that it might be inaccurate to compare the SAS from different individuals with distinct blood glucose level. This study sorted small and large SAT cells from the same individual and found that under the stimulation of insulin, an increase in GLUT4 located at the plasma membrane was only observed in small SAT cells, but not in large cells [106]. This interesting finding indicated that smaller adipocytes displayed increased glucose uptake rate while enlarged adipocytes associated closely with reduced insulin sensitivity in obese subjects.
In terms of VAS, no correlation was found between omental fat cell size and glucose uptake rate [9]. It was reported that insulin receptor binding affinity was greater in SAT than in VAT [107]. Thus, SAT may have a closer association to glucose uptake than VAT.
Cellular Mechanisms Hypothesized to Regulate Hypertrophic Adipocytes
Dysregulated Differentiation and Maturation of Preadipocytes in Obesity
The adipocytes originated from the potent fibroblast-like progenitor cells, such as MSCs. Generally, the process from MSCs to mature adipocytes involves two steps, whose impairment contributes to anomaly of adipocyte morphology.
In the first stage, adipocyte hypertrophy associates closely with inability to recruit and restrict MSC fate to adipogenic lineage during weight gain [108]. Previous studies have fully proved the canonical and uncanonical wingless-type mouse mammary tumor virus integration site family (WNT) signaling play critical role in regulating adipocyte commitment of precursor cells [109].
WNT1 inducible signaling pathway protein 2 (WISP2), an important WNT-associated molecular, has effect on both adipocyte commitment and differentiation. WISP2 commonly locates in cytosol. WISP2 presented in the cytosol formed a complex with PPARγ transcriptional activator zinc finger protein-423 (ZFP423). This complex is sequestered in the cytosol and the transcriptional activity of ZFP423 is repressed [110,111,112]. On the other side, extracellular WISP2 has been regarded as an atypical Wnt ligand [113, 114], which promotes the transcriptional activation of T cell factor/lymphoid enhancer-binding factor (Tcf/Lef) and induces β-catenin to the nucleus [115]. Interestingly, extracellular WISP2 (which also referred to as secreted WISP2 in some studies) promotes the proliferation of MSCs but also inhibits MSCs commitment of adipogenic lineage [112, 115]. Of note, WISP2 must be suppressed to ensure that MSCs would be restricted to adipogenic lineage [111]. Since previous study reported that WISP2 mRNA level positively correlated to SAS in 36 non-diabetic obese subjects [111], WISP2 serves as a potential target for improvement of adipocyte recruitment and differentiation.
Bone morphogenetic protein 4 (BMP4) is a ligand of the transforming growth factor-beta (TGF-β) superfamily, which recruits MSCs and commits them into adipogenic lineage. In general, BMP4 is secreted by mature adipocytes, acting as a feedback regulator to prevent abnormal adipocyte enlargement [116]. To promote adipocyte differentiation, BMP4 binds to BMP4 receptor and dissociates WISP2/ZFP423 complex by activating recombinant mothers against decapentaplegic homolog (SMAD)1/5/8 [116]. Therefore, ZFP423 enters the nucleus and initiates the transcription activity of PPARγ [111, 117]. Of note, increased BMP4 protein was found in hypertrophic adipocytes, which were regarded as “BMP4 resistance” [118]. Importantly, upregulated BMP4 inhibitor Gremlin-1 was observed in enlarged adipocytes. Reducing Gremlin-1 by specific antibodies significantly improved BMP4-driven adipocyte differentiation [113], suggesting that BMP-4 serves as a critical mediator in regulation of adipocyte differentiation.
In the second stage, preadipocytes gradually become mature adipocyte. PPARγ, a transcriptional factor, belongs to class I nuclear hormone receptor superfamily, serving as the central regulator of adipocyte differentiation [119]. When activated, PPARγ heterodimerizes with retinoid X receptor α (RXRα). These heterodimers bind to peroxisome proliferator hormone response elements (PPREs) [120] and control genes include fatty acid-binding protein (aP2), fatty acid transport protein (FATP) and fatty acid translocase (FAT/CD36), phosphoenolpyruvate carboxykinase (PEPCK), and acyl-CoA synthase, involving synthesis and lipolysis of triglyceride [120].
In addition, recent approaches emphasized several proteins which received less attention. EBF1, a transcription factor activated by C/EBPβ and C/EBPδ, could induce the activation of PPARγ. Both the activity and mRNA level of EBF1 were significantly lower in obese and hypertrophy subjects. In vitro, adipocyte volume negatively correlated with EBF1 gene expression in isolated adipocytes [13]. In addition, in line with PPARγ, C/EBPα binds on the promoter region of PPARγ and forms the self-reinforcing regulatory loop to further stimulate adipogenesis [121]. Lack of this transcriptional factor contributes to insulin resistance and obstruct WAT adipogenesis [122].
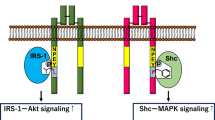
Collectively, expressions of adipogenesis genes functioned as markers of how the adipose expanded (Fig. 2) [123]. Some clinical studies observed decreased expressions of pro-adipogenic genes in patients with insulin resistance or T2D compared with NGT subjects [35, 124, 125], suggesting that expressions of adipogenesis genes could reflect adipocyte turnover and can be used to evaluate the risk of developing T2D.

Molecular mechanism of adipogenesis. During the process of mesenchymal stem cells (MSCs) commitment to adipose lineage, WNT1 inducible signaling pathway protein 2(WISP2) presented in the cytosol repressed transcriptional activator zinc finger protein-423 (ZFP423) through formation of a WISP2/ZFP423 complex in cytosol. Bone morphogenetic protein 4 (BMP4) phosphorylates SMAD1/5/8 to dissociate WISP2/ZFP423 complex, making ZFP423 enter into the nucleus and further activates peroxisome proliferator-activated receptor γ (PPARγ). BMP4 inhibitor Gremlin-1 suppresses MSCs by inhibiting BMP4. Likewise, extracellular WISP2 inhibits PPARγ by activating β-catenin. After committed differentiation to adipogenic lineage, EBF transcription factor 1(EBF1), a transcription factor activated by CCAAT-enhancer-binding proteins (C/EBPβ) and C/EBPδ, induces the activation of PPARγ. C/EBPα binds on the promoter region of PPARγ and forms the self-reinforcing regulatory loop to further stimulate adipogenesis. Activated PPARγ heterodimerizes with retinoid X receptor α (RXRα) and promotes transcription of genes involved in adipocyte differentiation and lipid transport
Development of “Supersized” Lipid Droplet
There are various adaptive changes that occurred in adipocytes in response to excessive lipid accumulation, the most evident alteration of which is the enlargement of lipid droplets (LDs). [126]. Many factors contribute to the expansion of LDs. Abnormal composition of LD membrane is one of the factors that trigger LD enlargement. LDs consist of neutral lipid embraced by a layer of phospholipids which subbed with various integral and peripheral proteins [127]. Interrupting the synthesis of phospholipids is the main approach of dysregulating LD membrane formation. Importantly, recent approaches highlighted that aberrant proteins that control phospholipids lead to “supersized” LDs [128]. Seipin is an important integral membrane protein in the endoplasmic reticulum (ER) that concentrates at junctions with LDs and serves as an important regulator of lipid storage and lipid droplet budding. Deletion of seipin directly binds glycerol-3-phosphate acyltransferase (GPAT), probably manipulates acylation of glycerol-3-P and subsequent synthesis of triacylglycerol, delays lipid formation, and further induces anomalous LD morphology [129,130,131]. Yeast mutants with impaired phosphlipid phosphatidylcholine (PC) synthesis directly induced 50-times large “supersized” LDs [132]. Also, increased phosphatidic acid (PA) promotes augmented fusion of LDs, further contributing to the enlargement of LDs [132].
Besides phospholipid dysregulation, some embedded proteins, such as fat-specific protein of 27 kDa (Fsp27) and cell death-inducing DFF45-like effector a (Cidea), play critical roles in promoting lipid accumulation in supersized LDs. Both proteins are upregulated profoundly in mice with liver steatosis [133].
Apart from the intrinsic regulation, the architecture of adipocytes was also influenced by extracellular factors. Overnutrition induced by short-term overfeeding in mice rapidly causes onset of cellular insulin resistance in adipocytes [134], marked by impaired insulin signaling at the level of insulin receptor substrate (IRS)-1 [135, 136] and Akt [137]. Besides, IQ motif containing GTPase activating protein 1 (IQGAP-1) is reduced. Due to the interaction between IQGAP-1 and caveolae, it can promote the synthesis of cytoskeleton proteins such as F-actin and inhibit the expansion of lipid droplets [138]. In addition, caveolae mediates the redistribution of GLUT4. When F-actin increases and the activity of gelosin (endogenous actin degradation protein) is inhibited, caveolae’s motor ability is enhanced, which promotes the transport of GLUT4 to the cell membrane, thus improving insulin sensitivity [138].
ECM-adipocyte interaction is also an important mechanism in adipocyte remodeling. Limited adipocyte expansion in fibrotic AT is one of the most remarkable manifestations. Silver et al. shows that αβ integrin on adipocyte cell membrane is able to bind to collagen to activate Ras, mitogen-activated protein kinase kinase (MAPKK), and inhibitor of nuclear factor kappa-B kinase (IKK), then activating MAPK and NF-κB pathway [139]. Meanwhile, as a ubiquitously expressed basement membrane proteoglycan, collagen XVIII contains a unique frizzled-like domain that binds to Wnt10b. Adipocyte-collagen XVIII interaction activates the canonical Wnt pathway, and then interferes with the lipogenic phenotype of cells through GSK3β and TGF-β pathways [140, 141], despite the low concentration of collagen XVIII in AT. Additionally, the interaction between the partially degraded collagen and adipocytes reduces the assembly of actin and enlarges fat droplets [142]; thus, mechanisms underlying this alteration still require further investigation.
Notably, there are inconsistencies among studies concerning cellular structure. Hasson et al. show that in C57BL/6J mice treated with high-fat diet for 2 weeks, F-actin increased 4-fold in VAT compared with chow-fat group in response to the intact interaction between IQGAP1 and IRS-1. Reversed feeding correspondingly restored cell size, insulin response, expression of F-actin, and its regulatory proteins [143]. Although this study indicated the critical role of actin in adipocyte remodeling, the effect of cytoskeleton on the structure of adipocytes is quite contrary to the findings in other articles [138, 144]. Considering that studies listed above used different animal models and/or analyzed different AT depots, different signaling pathways (Akt pathway, Wnt-GSK3β pathway, etc.) were probably involved in reconstruction of adipocyte cytoskeleton. More in-depth studies are required to verify this depot specificity.
Abnormal Osmolarity Sensors on Adipocytes
Generally, osmotic active compounds and extracellular tonicity exert great influence on cell volume. When encountered pathophysiological factors such as hypoxia or ischemia, cells induce generation of osmotic gradients by regulating plasma membrane ion channels and transporters [145]. Transient receptor potential cation channel subfamily V member 4 (TRPV4), an important swelling-activated channel, regulates osmotic pressure through mediation of Ca2+ influx [146]. When TRPV4 was knockout, the regulated volume decreases (RVD) were attenuated [147]. In adipocytes, TRPV4 knockout mice were protected from hypertrophy and insulin resistance [148], suggesting that TRPV4 plays an important role in promoting adipocyte hypertrophy. Another ion channel which influences cell size is swelling-activated voltage-regulated anion channel (VRAC). It was shown to initiate RVD via export of chloride ions (Cl−) [149]. Specifically, hypertrophic adipocytes displayed an increased “swell-activated” Cl− current when compared with adipocytes with smaller size. Since activation of VRAC was due to swelling protein 1 (SWELL1) [150], SWELL1 emerges as one significant cell-autonomous sensor for adipocyte size regulation.
Therapies Affecting Adipose Cell Size
Bariatric Surgery
Bariatric surgery has been demonstrated to decrease fat mass and improve insulin sensitivity [151]. In recent years, increasing reports have highlighted that the change of adipocyte size might associate closely with metabolic homeostasis after Roux-en-Y gastric bypass (RYGB). One study tested SAT adipocyte volume of 217 obese and T2D women before surgery; they defined enlarged adipocytes as the threshold for patients whose HOMA-IR was in the first quartile. Notably, subjects whose SAS greater than the thresholds were less possible to improve their insulin resistance state 6 months after surgery [39]. Similar finding was observed in another cohort of 62 obese women before and 2 years after RYGB [38]. This study was challenged by one study which examined SAS of 61 obese patients with normal blood glucose level before and 2 years after RYGB. SAS was positively correlated to the improvement of HOMA-IR, while total fat mass and BMI level did not show correlation with Δinsulin or ΔHOMA-IR [47]. Variation of these studies could be explained by whether obese patients with T2D were recruited.
Interestingly, with weight loss after surgery, adipocytes tend to proliferate through hyperplasia rather than hypertrophy, leading to decrease in mean adipocyte size instead of reduced cell number. One study reported that patients who underwent bariatric surgery had stronger amplification ability and lower DNA damage in subcutaneous adipose-derived stromal cells (ASC), which makes adipocytes more likely to store energy in a proliferative rather than hypertrophic manner [152]. Another study highlighted that angiotensin II (Ang II), the main effector of RAS, represses differentiation of precursor cells, inhibits lipolysis, and causes adipocyte hypertrophy. When adipose-derived MSCs were exposed to Ang II for 13 days, lipid droplet formation together with adipose differentiation-specific marker was significantly decreased. Surprisingly, even patients who conducted RYGB 1 year ago had higher BMI levels compared with non-obese control subjects (29.1 ± 7.8 kg/m2 vs 26.9 ± 2.6 kg/m2), their plasma Ang II levels were significantly lower (52.1 ± 4.2 pg/ml vs 85.4 ± 12.4 pg/ml) [153].
Importantly, although bariatric surgery significantly reduces body weight and improves metabolic homeostasis in obese patients, some patients will undergo weight regain and are hard to maintain ideal body weight [154]. One study explored SAS and number in obese subjects who regained weight 2 to 5 years after bariatric surgery. Surprisingly, regardless of the increase in body weight, subcutaneous abdominal adipocyte number increased significantly while no change was found in adipocyte size compared with 2 years after surgery [155]. Also, another study found a prominent increase in adipocyte progenitors in both SAT and VAT 12 to 18 months after surgery [156]. Through the long-term observation, even if patients undergo weight regain, bariatric surgery could still initiate AT into a healthier metabolic phenotype by increasing adipocyte number instead of limiting adipocyte expansion.
Calorie-Restricted Diet
Calorie restriction is also a common method besides conduction of bariatric surgery. Generally, both very low-calorie diets (VLCD) and low-calorie diets (LCD) contain essential nutrition but differ in the degree of calorie restriction [157]. Prior human study showed that after 4 weeks of VLCD, SAS was reduced by 11–12%. After 12 weeks of LCD, both abdominal and gluteal SAS were reduced by 15–20% [157]. Similarly, study in rodents suggested that when C57Bl/6 J mice underwent LCD (energy intake 70% of ad libitum intake) for 50 days, their VAS decreased significantly compared with control group [158].
In addition, some studies emphasized the change of adipocyte distribution rather than the absolute value of adipocyte diameter. Some studies in rodents found increased proportion of very small cells (< 20 μm) during LCD [98, 158]. In line with the animal study, one study recruited 57 subjects and they were randomly assigned to LCD group for 12 weeks or VLCD group for 5 weeks. Both groups displayed increased small adipocytes and decreased large adipocytes [159]. Intriguingly, one study showed that besides decreased adipocyte size, development of functional beige fat was also observed in lean C57BL/6 mice that underwent 4-week LCD [160]. These results indicated that the restriction on calorie intake, which induced negative energy balance, may lead to increased adipocyte progenitor commitment of new adipocytes and beige adipocytes, both of which suppress the unlimited expansion of adipocytes and exert favorable effects on metabolic homeostasis.
Medicine or Bioactive Compounds
Thiazolidinediones, which abbreviated as TZDs, also known as glitazones, have been used for treatment of T2Ds. This drug exerts its effect primarily through activating adipogenesis transcription factor PPARγ. Specifically, TZDs induce receptors of fatty acids to bind to the RXR and further form a complex with PPRE. Then, genes in regulation of adipogenesis and lipid metabolism were augmented, with increasing fatty acids stored in adipocytes [161, 162].
One human study using abdominal SAT obtained from 12 overweight/obese non-diabetic, insulin-resistant individuals after 12 weeks of pioglitazone treatment. Subsequently, increased proportion of small adipocytes as well as a 25% increase in the absolute number of these cells were observed [163]. Therefore, TZD-induced promotion of adipogenesis significantly improved adipocyte hypertrophy, inflammation, and insulin sensitivity.
However, TZDs, such as troglitazone, pioglitazone, and rosiglitazone, have many side effects including idiosyncratic hepatotoxicity, fluid retention, and heart failure [161]. Thus, TZDs were withdrawn from clinical use as anti-diabetic drugs. Consequently, increasing new bioactive compounds have been explored to regulate adipogenesis in addition to TZDs. It was reported that berberine suppresses the mRNA and protein levels of adipogenesis-related transcription factors PPARc and C/EBPα and their upstream regulator, C/EBPβ [164]. In addition, Acid sphingomyelinase (Asm), a member of phospholipids, plays a critical role in regulating membrane composition via transformation of membrane lipid sphingomyelin to ceramide. Knockout of Asm mice displayed the absence of adipocyte hypertrophy and increased expression of genes related to brown adipocyte differentiation [165]. In the field of free fatty acid, when C57BL/6J mice were fed with high-fat (HF) diet supplemented with eicosapentaenoic acid (EPA) (45% of energy from fat; 36 g/kg EPA; HF + EPA) for 11 weeks, their body weight, total fat mass, and adipocyte size were remarkably reduced [166]. In spite of abundant bioactive compounds in regulation of adipogenesis, further studies are still required for clinical applications in ameliorating AT insulin resistance.
Conclusion
Adipocyte size could reflect the progression of insulin resistance and T2D. In obesity, the function of adipose has been impaired, which could be reflected by several aspects including limited hyperplasic capacity of AT, inability to utilize glucose, and release of large amounts of fatty acids into circulation. We concluded the tight relationship between adipocyte size and metabolic dysregulation.
Most of the studies used absolute adipocyte size as an index for fat cell enlargement degree. Nevertheless, analysis of cell size distribution patterns seems to be another objective biomarker. Also, some researches concern the variance or minimal and maximal cell size. It is reasonable to take adipocyte variance into consideration for better understanding of cell size feature.
Up till now, large amounts of studies explored the relationship among adipocyte size, glucose or lipid metabolism, inflammation, and hypoxia in AT, since dynamic change of free fatty acids has been shown to be an important biomarker reflecting alterations of adipose function. With the accumulation of excessive energy intake, great amounts of fatty acids are released into circulation. It is thus logical to speculate that maybe enlargement of adipocytes was attributed to excessive or insufficient typical fatty acid.
Another factor that needs to be considered is gender impact on adipogenesis. We found most of the studies merely enrolled female subjects, especially studies related to bariatric surgery. Generally, female subjects have higher surgical intention. Also, it is hard to recruit male subjects in line with female ones when matched for age and BMI. However, one study highlighted that adipocyte distribution of different gender may vary profoundly as median adipocyte size in male individuals was much larger than that in female ones. Collectively, gender-specific threshold of hypertrophic adipocytes still requires further investigations.
Abbreviations
- T2D:
-
Type 2 diabetes
- WAT:
-
White adipose tissue
- AT:
-
Adipose tissue
- NAFLD:
-
Non-alcoholic fatty liver disease
- VAT:
-
Visceral AT
- SAT:
-
Subcutaneous AT
- VAS:
-
Visceral adipocyte size
- SAS:
-
Subcutaneous adipocyte size
- APC:
-
Adipocyte precursor cells
- GWAT:
-
Gonadal white adipose tissue
- IWAT:
-
Inguinal white adipose tissue
- ERα:
-
Estrogen receptor alpha
- ERβ:
-
Estrogen receptor beta
- HOMA-IR:
-
Homeostasis model assessment of insulin resistance
- IS:
-
Insulin-sensitive
- IR:
-
Insulin resistance
- IGT:
-
Impaired glucose tolerance
- NGT:
-
Normal glucose tolerance
- IFG:
-
Impaired fasting glucose
- AAS:
-
Abdominal adipocyte size
- FAS:
-
Femoral adipocyte size
- MS:
-
Metabolic syndrome
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- PCOS:
-
Polycystic ovary syndrome
- TNF-α:
-
Tumor necrosis factor α
- IL-6:
-
Interleukin-6
- IL-1b:
-
Interleukin-1b
- CCR:
-
Chemokine CC receptor
- MCP-1:
-
Monocyte chemoattractant protein 1
- iNKT:
-
Induced natural killer T
- CRP:
-
C-reactive protein
- MSCs:
-
Mesenchymal stem cells
- EBF1:
-
EBF transcription factor 1
- HIF-1α:
-
Hypoxia-inducible factor-1α
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- IGF-1:
-
Insulin-like growth factor 1
- CEBPα:
-
CCAAT-enhancer-binding proteins α
- SAA:
-
Serum amyloid A
- TM4SF1:
-
Transmembrane 4 L six family member 1
- PI3K:
-
PI3-kinase
- PDK1:
-
Phosphoinositide-dependent kinase 1
- PKB:
-
Protein kinase B
- GLUT4:
-
Glucose transporter type 4
- GSV:
-
GLUT4 storage vesicle
- WNT:
-
Wingless-type mouse mammary tumor virus integration site family
- WISP2:
-
WNT1 inducible signaling pathway protein 2
- ZFP423:
-
Zinc finger protein-423
- Tcf/Lef:
-
T cell factor/lymphoid enhancer-binding factor
- BMP4:
-
Bone morphogenetic protein 4
- TGF-β:
-
Transforming growth factor-beta
- SMAD:
-
Recombinant mothers against decapentaplegic homolog
- RXRα:
-
Retinoid X receptor α
- PPREs:
-
Peroxisome proliferator hormone response elements
- aP2:
-
Fatty acid-binding protein 2
- FATP:
-
Fatty acid transport protein
- FAT/CD36:
-
Fatty acid translocase
- PEPCK:
-
Phosphoenolpyruvate carboxykinase
- LDs:
-
Lipid droplets
- GPAT:
-
Glycerol-3-phosphate acyltransferase
- PC:
-
Phosphlipid phosphatidylcholine
- PA:
-
Phosphatidic acid
- Fsp27:
-
Fat-specific protein of 27 kDa
- Cidea:
-
Cell death-inducing DFF45-like effector a
- IRS:
-
Insulin receptor substrate
- IQGAP-1:
-
IQ motif containing GTPase activating protein 1
- MAPKK:
-
Mitogen-activated protein kinase kinase
- IKK:
-
Inhibitor of nuclear factor kappa-B kinase
- TRPV4:
-
Transient receptor potential cation channel subfamily V member 4
- RVD:
-
Regulated volume decreases
- VRAC:
-
Voltage-regulated anion channel
- SWELL1:
-
Swelling protein 1
- RYGB:
-
Roux-en-Y gastric bypass
- ASC:
-
Adipose-derived stromal cells
- Ang II:
-
Angiotensin II
- VLCD:
-
Very low-calorie diets
- LCD:
-
Low-calorie diets
- Asm:
-
Acid sphingomyelinase
- EPA:
-
Eicosapentaenoic acid
- HF:
-
High fat
References
Geserick M, Vogel M, Gausche R, et al. Acceleration of BMI in early childhood and risk of sustained obesity. N Engl J Med. 2018;379:1303–12.
Afshin A, Forouzanfar MH, Reitsma MB, et al. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. 2017;377:13–27.
Liu Y, Aron-Wisnewsky J, Marcelin G, et al. Accumulation and changes in composition of collagens in subcutaneous adipose tissue after bariatric surgery. J Clin Endocrinol Metab. 2016;101:293–304.
Lawler HM, Underkofler CM, Kern PA, et al. Adipose tissue hypoxia, inflammation, and fibrosis in obese insulin-sensitive and obese insulin-resistant subjects. J Clin Endocrinol Metab. 2016;101:1422–8.
Lonn M, Mehlig K, Bengtsson C, et al. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts type II diabetes independent of insulin resistance. Diabetologia. 2000;24:326–31.
Lonn M, Mehlig K, Bengtsson C, et al. Adipocyte size predicts incidence of type 2 diabetes in women. FASEB J. 2010;24:326–31.
Eriksson-Hogling D, Andersson DP, Bäckdahl J, et al. Adipose tissue morphology predicts improved insulin sensitivity following moderate or pronounced weight loss. Int J Obes. 2015;39:893–8.
Stenkula KG, Erlanson-Albertsson C. Adipose cell size: importance in health and disease. Am J Phys Regul Integr Comp Phys. 2018;315:R284–95.
Lundgren M, Svensson M, Lindmark S, et al. Fat cell enlargement is an independent marker of insulin resistance and ‘hyperleptinaemia’. Diabetologia. 2007;50:625–33.
Zhang Y, Zitsman JL, Hou J, et al. Fat cell size and adipokine expression in relation to gender, depot, and metabolic risk factors in morbidly obese adolescents. Obesity. 2014;22:691–7.
Acosta JR, Douagi I, Andersson DP, et al. Increased fat cell size: a major phenotype of subcutaneous white adipose tissue in non-obese individuals with type 2 diabetes. Diabetologia. 2016;59:560–70.
Veilleux A, Caron-Jobin M, Noel S, et al. Visceral adipocyte hypertrophy is associated with dyslipidemia independent of body composition and fat distribution in women. Diabetes. 2011;60:1504–11.
Gao H, Mejhert N, Fretz JA, et al. Early B cell factor 1 regulates adipocyte morphology and lipolysis in white adipose tissue. Cell Metab. 2014;19:981–92.
Drolet R, Richard C, Sniderman AD, et al. Hypertrophy and hyperplasia of abdominal adipose tissues in women. Int J Obes. 2008;32:283–91.
Gavrilova O, Marcus-Samuels B, Graham D, et al. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest. 2000;105:271–8.
Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. 2012;18:363–74.
Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014;156:20–44.
Misra A, Vikram NK. Clinical and pathophysiological consequences of abdominal adiposity and abdominal adipose tissue depots. Nutrition. 2003;19:457–66.
Liu A, McLaughlin T, Liu T, et al. Differential intra-abdominal adipose tissue profiling in obese, Insulin-resistant Women. Obes Surg. 2009;19:1564–73.
Hoffstedt J, Arner E, Wahrenberg H, et al. Regional impact of adipose tissue morphology on the metabolic profile in morbid obesity. Diabetologia. 2010;53:2496–503.
Arner P, Andersson DP, Thörne A, et al. Variations in the size of the major omentum are primarily determined by fat cell number. J Clin Endocrinol Metab. 2013;98:E897–901.
Hodson L, Bickerton AST, McQuaid SE, et al. The contribution of splanchnic fat to VLDL triglyceride is greater in insulin-resistant than insulin-sensitive men and women: studies in the postprandial state. Diabetes. 2007;56:2433–41.
Karastergiou K, Smith SR, Greenberg AS, et al. Sex differences in human adipose tissues – the biology of pear shape. Biol Sex Differ. 2012;3(1):13.
Newell-Fugate AE. The role of sex steroids in white adipose tissue adipocyte function. Reproduction. 2017;153(4):R133–49.
Tchoukalova YD, Koutsari C, Votruba SB, et al. Sex- and depot-dependent differences in adipogenesis in normal-weight humans. Obesity. 2010;18(10):1875–80.
Fried SK, Kral JG. Sex differences in regional distribution of fat cell size and lipoprotein lipase activity in morbidly obese patients. Int J Obes. 1987;11(2):129–40.
Lee M, Fried SK. Sex-dependent depot differences in adipose tissue development and function; role of sex steroids. J Obes Metab Syndr. 2017;26(3):172–80.
Wu Y, Lee MJ, Ido Y, et al. High-fat diet-induced obesity regulates MMP3 to modulate depot- and sex-dependent adipose expansion in C57BL/6J mice. Am J Physiol Endocrinol Metab. 2017;312(1):E58–71.
Palmer BF, Clegg DJ. The sexual dimorphism of obesity. Mol Cell Endocrinol. 2015;402:113–9.
Blouin K, Nadeau M, Perreault M, et al. Effects of androgens on adipocyte differentiation and adipose tissue explant metabolism in men and women. Clin Endocrinol. 2010;72(2):176–88.
Chang E, Varghese M, Singer K. Gender and sex differences in adipose tissue. Curr Diab Rep. 2018;18(9):69.
McLaughlin T, Lamendola C, Coghlan N, et al. Subcutaneous adipose cell size and distribution: relationship to insulin resistance and body fat. Obesity. 2014;22:673–80.
Cotillard A, Poitou C, Torcivia A, et al. Adipocyte size threshold matters: link with risk of type 2 diabetes and improved insulin resistance after gastric bypass. J Clin Endocrinol Metab. 2014;99:E1466–70.
McLaughlin T, Craig C, Liu LF, et al. Adipose cell size and regional fat deposition as predictors of metabolic response to overfeeding in insulin-resistant and insulin-sensitive humans. Diabetes. 2016;65:1245–54.
McLaughlin T, Sherman A, Tsao P, et al. Enhanced proportion of small adipose cells in insulin-resistant vs insulin-sensitive obese individuals implicates impaired adipogenesis. Diabetologia. 2007;50:1707–15.
Verboven K, Wouters K, Gaens K, et al. Abdominal subcutaneous and visceral adipocyte size, lipolysis and inflammation relate to insulin resistance in male obese humans. Sci Rep-UK. 2018;8
Rydén M, Andersson DP, Bergström IB, et al. Adipose tissue and metabolic alterations: regional differences in fat cell size and number matter, but differently: a cross-sectional study. J Clin Endocrinol Metab. 2014;99:E1870–6.
Kranendonk MEG, van Herwaarden JA, Stupkova T, et al. Inflammatory characteristics of distinct abdominal adipose tissue depots relate differently to metabolic risk factors for cardiovascular disease. Atherosclerosis. 2015;239:419–27.
Johannsen DL, Tchoukalova Y, Tam CS, et al. Effect of 8 weeks of overfeeding on ectopic fat deposition and insulin sensitivity: testing the "adipose tissue expandability" hypothesis. Diabetes Care. 2014;37:2789–97.
Marques BG, Hausman DB, Martin RJ. Association of fat cell size and paracrine growth factors in development of hyperplastic obesity. Am J Phys. 1998;275:R1898–908.
Kursawe R, Eszlinger M, Narayan D, et al. Cellularity and adipogenic profile of the abdominal subcutaneous adipose tissue from obese adolescents: association with insulin resistance and hepatic steatosis. Diabetes. 2010;59:2288–96.
Muir LA, Neeley CK, Meyer KA, et al. Adipose tissue fibrosis, hypertrophy, and hyperplasia: correlations with diabetes in human obesity. Obesity. 2016;24:597–605.
Fang L, Guo F, Zhou L, et al. The cell size and distribution of adipocytes from subcutaneous and visceral fat is associated with type 2 diabetes mellitus in humans. Adipocyte. 2015;4:273–9.
Belligoli A, Compagnin C, Sanna M, et al. Characterization of subcutaneous and omental adipose tissue in patients with obesity and with different degrees of glucose impairment. Sci Rep-UK. 2019;9
Pasarica M, Xie H, Hymel D, et al. Lower Total adipocyte number but no evidence for small adipocyte depletion in patients with type 2 diabetes. Diabetes Care. 2009;32:900–2.
Tchoukalova YD, Votruba SB, Tchkonia T, et al. Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proc Natl Acad Sci. 2010;107:18226–31.
Meena VP, Seenu V, Sharma MC, et al. Relationship of adipocyte size with adiposity and metabolic risk factors in Asian Indians. PLoS One. 2014;9:e108421.
Petaja EM, Sevastianova K, Hakkarainen A, et al. Adipocyte size is associated with NAFLD independent of obesity, fat distribution, and PNPLA3 genotype. Obesity (Silver Spring). 2013;21:1174–9.
Wree A, Schlattjan M, Bechmann LP, et al. Adipocyte cell size, free fatty acids and apolipoproteins are associated with non-alcoholic liver injury progression in severely obese patients. Metabolism. 2014;63:1542–52.
O'Connell J, Lynch L, Cawood TJ, et al. The relationship of omental and subcutaneous adipocyte size to metabolic disease in severe obesity. PLoS One. 2010;5:e9997.
Mannerås-Holm L, Leonhardt H, Kullberg J, et al. Adipose tissue has aberrant morphology and function in PCOS: enlarged adipocytes and low serum Adiponectin, but not circulating sex steroids, are strongly associated with insulin resistance. J Clin Endocrinol Metab. 2011;96:E304–11.
Broskey NT, Tam CS, Sutton EF, et al. Metabolic inflexibility in women with PCOS is similar to women with type 2 diabetes. Nutr Metab. 2018;15
Zheng S, Li X. Visceral adiposity index as a predictor of clinical severity and therapeutic outcome of PCOS. Gynecol Endocrinol. 2016;32:177–83.
Svensson H, Wetterling L, Bosaeus M, et al. Body fat mass and the proportion of very large adipocytes in pregnant women are associated with gestational insulin resistance. Int J Obes. 2016;40:646–53.
Guzik TJ, Skiba DS, Touyz RM, et al. The role of infiltrating immune cells in dysfunctional adipose tissue. Ecardiovasc Res. 2017;113:1009–23.
Grant RW, Dixit VD. Adipose tissue as an immunological organ. Obesity. 2015;23:512–8.
DiSpirito JR, Mathis D. Immunological contributions to adipose tissue homeostasis. Semin Immunol. 2015;27:315–21.
McLaughlin T, Ackerman SE, Shen L, et al. Role of innate and adaptive immunity in obesity-associated metabolic disease. J Clin Invest. 2017;127:5–13.
Winer S, Winer DA. The adaptive immune system as a fundamental regulator of adipose tissue inflammation and insulin resistance. Immunol Cell Biol. 2012;
Skurk T, Alberti-Huber C, Herder C, et al. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab. 2007;92:1023–33.
Longo M, Zatterale F, Naderi J, et al. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. Int J Mol Sci. 2019;20:2358.
Dalmas E, Clément K, Guerre-Millo M. Defining macrophage phenotype and function in adipose tissue. Trends Immunol. 2011;32:307–14.
Boutens L, Stienstra R. Adipose tissue macrophages: going off track during obesity. Diabetologia. 2016;59:879–94.
Thomas D, Apovian C. Macrophage functions in lean and obese adipose tissue. Metabolism. 2017;72:120–43.
Lumeng CN, DeYoung SM, Bodzin JL, et al. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2006;56:16–23.
Oh DY, Morinaga H, Talukdar S, et al. Increased macrophage migration into adipose tissue in obese mice. Diabetes. 2012;61:346–54.
Cinti S, Mitchell G, Barbatelli G, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–55.
Zuniga LA, Shen WJ, Joyce-Shaikh B, et al. IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J Immunol. 2010;185:6947–59.
Weisberg SP, McCann D, Desai M, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808.
van Bruggen N, Ouyang W. Th17 cells at the crossroads of autoimmunity, inflammation, and atherosclerosis. Immunity. 2014;40:10–2.
Kursawe R, Dixit VD, Scherer PE, et al. A role of the inflammasome in the low storage capacity of the abdominal subcutaneous adipose tissue in obese adolescents. Diabetes. 2016;65:610–8.
Weisberg SP, Hunter D, Huber R, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116:115–24.
Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–84.
Maury E, Noël L, Detry R, et al. In vitro hyperresponsiveness to tumor necrosis factor-α contributes to adipokine dysregulation in omental adipocytes of obese subjects. J Clin Endocrinol Metab. 2009;94:1393–400.
Michaud A, Boulet MM, Veilleux A, et al. Abdominal subcutaneous and omental adipocyte morphology and its relation to gene expression, lipolysis and adipocytokine levels in women. Metabolism. 2014;63:372–81.
Winkler G, Kiss S, Keszthelyi L, et al. Expression of tumor necrosis factor (TNF)-alpha protein in the subcutaneous and visceral adipose tissue in correlation with adipocyte cell volume, serum TNF-alpha, soluble serum TNF-receptor-2 concentrations and C-peptide level. Eur J Endocrinol. 2003;149:129–35.
Jimenez MA, Akerblad P, Sigvardsson M, et al. Critical role for Ebf1 and Ebf2 in the adipogenic transcriptional cascade. Mol Cell Biol. 2006;27:743–57.
Hosogai N, Fukuhara A, Oshima K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56:901–11.
Ye J, Gao Z, Yin J, et al. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue ofob/ob and dietary obese mice. Am J Physiol-endoc. 2007;293:E1118–28.
Guilherme A, Virbasius JV, Puri V, et al. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–77.
Trayhurn P, Wang B, Wood IS. Hypoxia in adipose tissue: a basis for the dysregulation of tissue function in obesity? Brit J Nutr. 2008;100:227–35.
Hallgren P, Sjostrom L, Hedlund H, et al. Influence of age, fat cell weight, and obesity on O2 consumption of human adipose tissue. Am J Phys. 1989;256:E467–74.
Brahimi-Horn MC, Pouysségur J. Oxygen, a source of life and stress. FEBS Lett. 2007;581:3582–91.
Goossens GH, Blaak EE. Adipose tissue dysfunction and impaired metabolic health in human obesity: a matter of oxygen? Front Endocrinol. 2015;6
Rodrigues T, Matafome P, Sereno J, et al. Methylglyoxal-induced glycation changes adipose tissue vascular architecture, flow and expansion, leading to insulin resistance. Sci Rep-UK. 2017;7
Goossens GH, Bizzarri A, Venteclef N, et al. Increased adipose tissue oxygen tension in obese compared with lean men is accompanied by insulin resistance, impaired adipose tissue capillarization, and inflammation. Circulation. 2011;124:67–76.
Engin A. Adipose tissue hypoxia in obesity and its impact on preadipocytes and macrophages: hypoxia hypothesis. Adv Exp Med Biol. 2017;960:305.
Wang Y, Mak JCW, Lee MYK, et al. Low-frequency intermittent hypoxia promotes subcutaneous adipogenic differentiation. Oxidative Med Cell Longev. 2018;2018:1–13.
Sun K, Tordjman J, Clément K, et al. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013;18:470–7.
Mor-Yossef Moldovan L, Lustig M, Naftaly A, et al. Cell shape alteration during adipogenesis is associated with coordinated matrix cues. J Cell Physiol. 2018:1–14.
Aikio M, Elamaa H, Vicente D, et al. Specific collagen XVIII isoforms promote adipose tissue accrual via mechanisms determining adipocyte number and affect fat deposition. Proc Natl Acad Sci U S A. 2014;111:E3043–52.
Park J, Scherer PE. Adipocyte-derived endotrophin promotes malignant tumor progression. J Clin Invest. 2012;122:4243–56.
Michaud A, Tordjman J, Pelletier M, et al. Relevance of omental pericellular adipose tissue collagen in the pathophysiology of human abdominal obesity and related cardiometabolic risk. Int J Obes. 2016;40:1823–31.
Pasarica M, Gowronska-Kozak B, Burk D, et al. Adipose tissue collagen VI in obesity. J Clin Endocrinol Metab. 2009;94:5155–62.
Grandl G, Müller S, Moest H, et al. Depot specific differences in the adipogenic potential of precursors are mediated by collagenous extracellular matrix and Flotillin 2 dependent signaling. Mol Metab. 2016;5:937–47.
Zhang Y, Proenca R, Maffei M, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–32.
Zhang Y, Guo K, Diaz PA, et al. Determinants of leptin gene expression in fat depots of lean mice. Am J Phys Regul Integr Comp Phys. 2002;282:R226–34.
Miller KN, Burhans MS, Clark JP, et al. Aging and caloric restriction impact adipose tissue, adiponectin, and circulating lipids. Aging Cell. 2017;16:497–507.
Abel ED, Peroni O, Kim JK, et al. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–33.
Peterson RT, Schreiber SL. Kinase phosphorylation: keeping it all in the family. Curr Biol. 1999;9:R521–4.
Rea S, James DE. Moving GLUT4: the biogenesis and trafficking of GLUT4 storage vesicles. Diabetes. 1997;46:1667–77.
Salans LB, Knittle JL, Hirsch J. The role of adipose cell size and adipose tissue insulin sensitivity in the carbohydrate intolerance of human obesity. J Clin Invest. 1968;47:153–65.
Lizunov VA, Lee JP, Skarulis MC, et al. Impaired tethering and fusion of GLUT4 vesicles in insulin-resistant human adipose cells. Diabetes. 2013;62:3114–9.
Farnier C, Krief S, Blache M, et al. Adipocyte functions are modulated by cell size change: potential involvement of an integrin/ERK signalling pathway. Int J Obes Relat Metab Disord. 2003;27:1178–86.
Lizunov VA, Stenkula KG, Blank PS, et al. Human adipose cells in vitro are either refractory or responsive to insulin, Reflecting Host Metabolic State. Plos One. 2015;10:e119291.
Franck N, Stenkula KG, Öst A, et al. Insulin-induced GLUT4 translocation to the plasma membrane is blunted in large compared with small primary fat cells isolated from the same individual. Diabetologia. 2007;50:1716–22.
Fawcett J, Sang H, Permana PA, et al. Insulin metabolism in human adipocytes from subcutaneous and visceral depots. Biochem Bioph Res Co. 2010;402:762–6.
Grünberg JR, Elvin J, Paul A, et al. CCN5/WISP2 and metabolic diseases. J Cell Commun Signal. 2018;12:309–18.
Ghaben AL, Scherer PE. Adipogenesis and metabolic health. Nat Rev Mol Cell Biol. 2019;
Smith U, Kahn BB. Adipose tissue regulates insulin sensitivity: role of adipogenesis, de novo lipogenesis and novel lipids. J Intern Med. 2016;280:465–75.
Hammarstedt A, Hedjazifar S, Jenndahl L, et al. WISP2 regulates preadipocyte commitment and PPARgamma activation by BMP4. Proc Natl Acad Sci U S A. 2013;110:2563–8.
Hammarstedt A, Gogg S, Hedjazifar S, et al. Impaired adipogenesis and dysfunctional adipose tissue in human hypertrophic obesity. Physiol Rev. 2018;98:1911–41.
Gustafson B, Hammarstedt A, Hedjazifar S, et al. BMP4 and BMP antagonists regulate human white and beige adipogenesis. Diabetes. 2015;64:1670–81.
Gustafson B, Smith U. Activation of canonical wingless-type MMTV integration site family (Wnt) signaling in mature adipocytes increases β-catenin levels and leads to cell dedifferentiation and insulin resistance. J Biol Chem. 2010;285:14031–41.
Grünberg JR, Hammarstedt A, Hedjazifar S, et al. The novel secreted adipokine WNT1-inducible signaling pathway protein 2 (WISP2) is a mesenchymal cell activator of canonical WNT. J Biol Chem. 2014;289:6899–907.
Huang H, Song TJ, Li X, et al. BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc Natl Acad Sci U S A. 2009;106:12670–5.
Hoffmann JM, Grünberg JR, Church C, et al. BMP4 gene therapy in mature mice reduces BAT activation but protects from obesity by Browning subcutaneous adipose tissue. Cell Rep. 2017;20:1038–49.
Gustafson B, Smith U. The WNT inhibitor Dickkopf 1 and bone morphogenetic protein 4 rescue adipogenesis in hypertrophic obesity in humans. Diabetes. 2012;61:1217–24.
Hong F, Pan S, Guo Y, et al. PPARs as nuclear receptors for nutrient and energy metabolism. Molecules. 2019;24:2545.
Grygiel-Gorniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications--a review. Nutr J. 2014;13:17.
Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARγ. Annu Rev Biochem. 2008;77:289–312.
Moseti D, Regassa A, Kim W. Molecular regulation of adipogenesis and potential anti-adipogenic bioactive molecules. Int J Mol Sci. 2016;17:124.
Isakson P, Hammarstedt A, Gustafson B, et al. Impaired preadipocyte differentiation in human abdominal obesity: role of Wnt, tumor necrosis factor-a, and inflammation. Diabetes. 2009;58:1550–7.
Yang X, Jansson P, Nagaev I, et al. Evidence of impaired adipogenesis in insulin resistance. Biochem Bioph Res Co. 2004;317:1045–51.
Dubois SG, Tchoukalova YD, Heilbronn LK, et al. Potential role of increased matrix metalloproteinase-2 (MMP2) transcription in impaired adipogenesis in type 2 diabetes mellitus. Biochem Bioph Res Co. 2008;367:725–8.
Yang H, Galea A, Sytnyk V, et al. Controlling the size of lipid droplets: lipid and protein factors. Curr Opin Cell Biol. 2012;24:509–16.
Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019;20:137–55.
Choudhary V, Ojha N, Golden A, et al. A conserved family of proteins facilitates nascent lipid droplet budding from the ER. J Cell Biol. 2015;211:261–71.
Pagac M, Cooper DE, Qi Y, et al. SEIPIN regulates lipid droplet expansion and adipocyte development by modulating the activity of glycerol-3-phosphate acyltransferase. Cell Rep. 2016;17:1546–59.
Fei W, Du X, Yang H. Seipin, adipogenesis and lipid droplets. Trends Endocrinol Metab. 2011;22:204–10.
Wang H, Becuwe M, Housden BE, et al. Seipin is required for converting nascent to mature lipid droplets. Elife. 2016;5:e16582.
Fei W, Shui G, Zhang Y, et al. A role for phosphatidic acid in the formation of "supersized" lipid droplets. PLoS Genet. 2011;7:e1002201.
Matsusue K, Kusakabe T, Noguchi T, et al. Hepatic steatosis in leptin-deficient mice is promoted by the PPARgamma target gene Fsp27. Cell Metab. 2008;7:302–11.
Hansson B, Wasserstrom S, Morén B, et al. Intact glucose uptake despite deteriorating signaling in adipocytes with high-fat feeding. J Mol Endocrinol. 2018;60:199–211.
Danielsson A, Ost A, Nystrom FH, et al. Attenuation of insulin-stimulated insulin receptor substrate-1 serine 307 phosphorylation in insulin resistance of type 2 diabetes. J Biol Chem. 2005;280:34389–92.
Ost A, Svensson K, Ruishalme I, et al. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol Med. 2010;16:235–46.
Rondinone CM, Carvalho E, Wesslau C, et al. Impaired glucose transport and protein kinase B activation by insulin, but not okadaic acid, in adipocytes from subjects with type II diabetes mellitus. Diabetologia. 1999;42:819–25.
Jufvas Å, Rajan MR, Jönsson C, et al. Scaffolding protein IQGAP1: an insulin-dependent link between caveolae and the cytoskeleton in primary human adipocytes? Biochem J. 2016;473:3177–88.
Silver FH, Siperko LM, Seehra GP. Mechanobiology of force transduction in dermal tissue. Skin Res Technol. 2003;9:3–23.
Zhou S, Eid K, Glowacki J. Cooperation between TGF-beta and Wnt pathways during chondrocyte and adipocyte differentiation of human marrow stromal cells. J Bone Miner Res. 2004;19:463–70.
Aikio M, Elamaa H, Vicente D, et al. Specific collagen XVIII isoforms promote adipose tissue accrual via mechanisms determining adipocyte number and affect fat deposition. Proc Natl Acad Sci U S A. 2014;111:E3043–52.
Qin Z, Fisher GJ, Voorhees JJ, et al. Actin cytoskeleton assembly regulates collagen production via TGF-β type II receptor in human skin fibroblasts. J Cell Mol Med. 2018;22:4085–96.
Hansson B, Morén B, Fryklund C, et al. Adipose cell size changes are associated with a drastic actin remodeling. Sci Rep-UK. 2019;9
Kim JI, Park J, Ji Y, et al. During adipocyte remodeling, lipid droplet configurations regulate insulin sensitivity through F-actin and G-actin reorganization. Mol Cell Biol. 2019;39:e00210–9.
Hoffmann EK, Lambert IH, Pedersen SF. Physiology of cell volume regulation in vertebrates. Physiol Rev. 2009;89:193–277.
Delany NS, Hurle M, Facer P, et al. Identification and characterization of a novel human vanilloid receptor-like protein, VRL-2. Physiol Genomics. 2001;4:165–74.
Liedtke W, Kim C. Functionality of the TRPV subfamily of TRP ion channels: add mechano-TRP and osmo-TRP to the lexicon! Cell Mol Life Sci. 2005;62:2985–3001.
Ye L, Kleiner S, Wu J, et al. TRPV4 is a regulator of adipose oxidative metabolism, inflammation, and energy homeostasis. Cell. 2012;151:96–110.
Jentsch TJ. VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat Rev Mol Cell Biol. 2016;17:293–307.
Zhang Y, Xie L, Gunasekar SK, et al. SWELL1 is a regulator of adipocyte size, insulin signalling and glucose homeostasis. Nat Cell Biol. 2017;19:504–17.
Inge TH, Courcoulas AP, Jenkins TM, et al. Five-year outcomes of gastric bypass in adolescents as compared with adults. N Engl J Med. 2019;380:2136–45.
Mitterberger MC, Mattesich M, Zwerschke W. Bariatric surgery and diet-induced long-term caloric restriction protect subcutaneous adipose-derived stromal/progenitor cells and prolong their life span in formerly obese humans. Exp Gerontol. 2014;56:106–13.
Chen JG, Spagnoli A, Torquati A. Adipogenic differentiation of adipose tissue-derived human mesenchymal stem cells: effect of gastric bypass surgery. Surg Endosc. 2012;26(12):3449–56.
van Baak MA, Mariman E. Mechanisms of weight regain after weight loss - the role of adipose tissue. Nat Rev Endocrinol. 2019;15:274–87.
Hoffstedt J, Andersson DP, Eriksson HD, et al. Long-term protective changes in adipose tissue after gastric bypass. Diabetes Care. 2017;40:77–84.
García-Rubio J, León J, Redruello-Romero A, et al. Cytometric analysis of adipose tissue reveals increments of adipocyte progenitor cells after weight loss induced by bariatric surgery. Sci Rep-UK. 2018;8
Rossmeislová L, Mališová L, Kračmerová J, et al. Adaptation of human adipose tissue to hypocaloric diet. Int J Obes (2015). 2013;37:640–50.
MacLean PS, Bergouignan A, Cornier M, et al. Biology's response to dieting: the impetus for weight regain. Am J Phys Regul Integr Comp Phys. 2011;301:R581–600.
Lenz M, Roumans NJT, Vink RG, et al. Estimating real cell size distribution from cross-section microscopy imaging. Bioinformatics. 2016;32:i396–404.
Fabbiano S, Suárez-Zamorano N, Rigo D, et al. Caloric restriction leads to Browning of white adipose tissue through type 2 immune signaling. Cell Metab. 2016;24:434–46.
Yasmin S, Jayaprakash V. Thiazolidinediones and PPAR orchestra as antidiabetic agents: from past to present. Eur J Med Chem. 2017;126:879–93.
Nanjan MJ, Mohammed M, Prashantha Kumar BR, et al. Thiazolidinediones as antidiabetic agents: a critical review. Bioorg Chem. 2018;77:548–67.
McLaughlin TM, Liu T, Yee G, et al. Pioglitazone increases the proportion of small cells in human abdominal subcutaneous adipose tissue. Obesity. 2009;18:926–31.
Huang C, Zhang Y, Gong Z, et al. Berberine inhibits 3T3-L1 adipocyte differentiation through the PPARγ pathway. Biochem Bioph Res Co. 2006;348:571–8.
Sydor S, Sowa JP, Megger DA, et al. Acid sphingomyelinase deficiency in Western diet-fed mice protects against adipocyte hypertrophy and diet-induced liver steatosis. Mol Metab. 2017;6:416–27.
LeMieux MJ, Kalupahana NS, Scoggin S, et al. Eicosapentaenoic acid reduces adipocyte hypertrophy and inflammation in diet-induced obese mice in an adiposity-independent manner. J Nutr. 2015;145:411–7.
Acknowledgments
The authors thank the study participants and investigators for their participation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Informed consent does not apply.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Fangcen Liu is the first author of this review.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, F., He, J., Wang, H. et al. Adipose Morphology: a Critical Factor in Regulation of Human Metabolic Diseases and Adipose Tissue Dysfunction. OBES SURG 30, 5086–5100 (2020). https://doi.org/10.1007/s11695-020-04983-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11695-020-04983-6