
Abstract
One of the major properties of the semi-synthetic minimal cell, as a model for early living cells, is the ability to self-reproduce itself, and the reproduction of the boundary layer or vesicle compartment is part of this process. A minimal bio-molecular mechanism based on the activity of one single enzyme, the FAS-B (Fatty Acid Synthase) Type I enzyme from Brevibacterium ammoniagenes, is encapsulated in 1-palmitoyl-2oleoyl-sn-glycero-3-phosphatidylcholine (POPC) liposomes to control lipid synthesis. Consequently molecules of palmitic acid released from the FAS catalysis, within the internal lumen, move toward the membrane compartment and become incorporated into the phospholipid bilayer. As a result the vesicle membranes change in lipid composition and liposome growth can be monitored. Here we report the first experiments showing vesicles growth by catalysis of one enzyme only that produces cell boundary from within. This is the prototype of the simplest autopoietic minimal cell.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The self-reproduction process in a cell involves both self boundary layer reproduction, which occurs by membrane replication, and core self-reproduction, that is based on genome replication. During the origin of life the initial mechanisms driving prebiotic cell formation are thought to have originated from the spontaneous self-assembly of molecules. The bilayer membrane constituting the first cell compartment may have formed originally through the self assembly of amphiphilic molecules into membrane vesicles (Hargreaves and Deamer 1978; Apel et al. 2002; Monnard and Deamer 2002). Furthermore, the prebiotic boundary layer self-reproduction process may have been the consequence of the vesicle growth, by spontaneous incorporation of lipids, until instability triggered division into smaller stable vesicles (Walde et al. 1994; Berclaz et al. 2001); other suggestive mechanisms of chemical induction of vesicle growth and division have been reported more recently by Sugawara team (Takakura et al. 2003; Takakura and Sugawara 2004). A detailed process was recently reconstructed where vesicle growth was monitored by Fluorescence Resonance Energy Transfer (FRET) assay, directly measuring the fraction of added fatty acids that become incorporated into preexisting vesicle marked with membrane localized fluorescent dyes (Hanczyc et al. 2003).
The major difficulty to propose models describing prebiotic cells, based on bio-molecules and basic metabolisms, is underlined by the absence of a historical record to suggest how the initial cell-biochemistry was organized. To overcome this problem a synthetic biology approach can be followed to build a cell model for early living cells, where extant enzymes and synthetic compartments are used to construct a semi-synthetic minimal cell in the laboratory (Luisi et al. 2006; Murtas 2007).
In this challenge the first milestone is represented by the implementation of a self-maintenance or a basic metabolism, based on protein synthesis, into a lipid or liposome compartment. Complex biochemical mechanisms or protein synthesis systems have been already reconstructed and tested in lipid vesicles (Oberholzer et al. 1995a, b; Ishikawa et al. 2004; Noireaux and Libchaber 2004). Good progress was recently achieved introducing protein synthesis in liposomes using a minimal set of enzymes (Murtas et al. 2007).
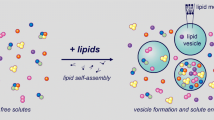
The introduction of a bio-mechanism to allow boundary layer self-reproduction would certainly deliver the second important milestone: a liposome that builds its own membrane with the help of entrapped enzymes. This represents a prototype of the simplest autopoietic minimal cell (Fig. 1), (Luisi 2006; Bachmann et al. 1992). Initial studies on membrane biogenesis, demonstrating an enzymatic driven membrane reconstitution, have been carried out in Deamer lab (Deamer and Boatman 1980; Deamer and Gavino 1983). The first attempts to propose an autopoietic system based on enzymes that produce cell boundary from within was set up in Luisi’s laboratory when two (Kuruma et al. 2009) or all four enzymes (Schmidli et al. 1991) of the salvage pathway were entrapped in liposomes to control lipid synthesis. Although lipid synthesis was demonstrated, unfortunately no vesicle growth was clearly proven in both cases.

A liposome that builds its own membrane based on entrapped enzymes is the prototype of the simplest autopoietic minimal cell. P Precursors are the reagents required for an enzymatic catalysis, E enzyme/s controlling lipid catalysis, L lipids are released within the liposome lumen and spontaneously become incorporated into the lipid bilayer and this results in vesicle growth. The enzymatic pathway produces the palmitate represented as the dark shaded lipids
In order to introduce a biological mechanism to control boundary layer self-reproduction in liposomes, biochemical synthesis of fatty acids based on a FAS type I enzyme catalysis (Schweizer and Hofmann 2004) was selected for this work. This was the mechanism of choice because fatty acid synthesis is simply controlled by only one enzyme, in agreement with the low complexity of our minimal cell model system. In particular we used the FAS-B type I enzyme from Brevibacterium ammoniagenes already biochemically characterized, expressed and purified from the heterologous system Escherichia coli (Stuible et al. 1997).
Moreover the main product of this catalysis, palmitate, is one of the surfactant synthesized in the laboratory under presumably prebiotic conditions (Walde 2006; Orò 1994; Simoneit 2004). It is chemically less reactive, therefore stable in comparison to unsatured fatty acids such as oleate, due to the absence of double bonds between the carbon atoms of the fatty acid chains. We propose that a similar and minimal enzymatic catalysis may have occurred in vesicle compartments to control the first biological boundary layer self-reproduction process (Fig. 1).
Results
Palmitate synthesis by FAS type I catalysis in liposomes
The biochemical components for the synthesis of lipids are the bacterial FAS Type I enzyme FAS-B from B. ammoniagenes (Stuible et al. 1996) as the catalyst and the precursors or reagents required for the fatty acid synthesis of palmitate as shown in Fig. 2a. This FAS type I is a water soluble multifunctional enzyme consisting of a dimmer with a single long multifunctional polypeptide encoded by a single c-DNA. The FAS-B protein has already been expressed in heterologous system, purified and characterized by Stuible et al. (1997) with an E. coli transformant based on the fasB gene isolated from B. ammoniagenes. The same E. coli clone was generously provided from the Schweizer team (University of Erlangen-Nürnberg, Germany) and used to express and purify the FAS-B enzyme following the methods for FAS-B purification reported by Stuible et al. (1997). Since endogenous E. coli FAS is a non aggregated type II system, based on the activity of 6 independent genes, protein contamination of the heterologous type I FAS by any of the endogenous type II FAS is considered unlikely.

FAS-B catalysis and assay. a The Stoichiometry of the synthesis of palmitate controlled by the FAS-B enzyme from Brevibacterium ammoniagenes. The acetyl-CoA primes the reaction, while the elongation steps are sustained by malonyl-CoA. The free energy needed for the FAS functions is provided by the decarboxilation of the malonyl-CoA. Reducing power is provided by the oxidation of 14 molecules of NADPH for each molecule of palmitate. b FAS-B enzyme assay measuring the NADPH disappearance (oxidation) spectrophotometrically. The decline of absorbance of NADPH, when the FAS-B catalysis is active, (square) in comparison with the absorbance measured when malonyl-CoA was omitted, (circle). Each value reported in the graph equal the delta between the absorbance at 340 and 600 nm wavelength. The error bars reported in the graph represent the range of values of experiments performed in triplicate
After purification, an SDS–PAGE gel was loaded with aliquots of the purified FAS-B protein showing only one high molecular weight band of 325 kDa and corresponding to the expected size of the FAS-B enzyme, as reported by Stuible et al. (1997) (data not shown). The FAS-B enzyme activity, based on the biochemical reaction shown in Fig. 2a, was first assayed in vitro monitoring the NADPH oxidation, by spectrophotometric analysis, in the presence or in the absence of the prime precursor malonyl-CoA (Fig. 2b). The FAS activity reported in Fig. 2b was assayed according to the method of Lynen (1980), and corresponding to 0.37 μmol/min/mg, and described in “Materials and Methods”.
Once the activity of the lipid synthesis pathway was confirmed in vitro, the system (Fig. 2a) was introduced into POPC liposomes. The protocol followed the FAS-B assay conditions described in detail in “Materials and Methods”, with the reaction incubated at 30°C for 3 h. In order to examine the products of the FAS-B catalysis either in bulk solution or in liposomes, the synthesis was performed with in vitro incorporation of 14C radioactively labeled malonyl-CoA, and the resulting fatty acids analyzed by Thin Layer Chromatography (TLC). As reported in Fig. 3, the autoradiography of the TLC chromatogram indicates that with the FAS-B reaction the radioactivity is exclusively incorporated into long chain fatty acids and palmitic acid was produced under these conditions.

Palmitate synthesis. Palmitate is synthesised in bulk solution or in liposomes, based on the FAS-B catalysis, and analysed by TLC. The palmitate released is 14C-labeled by [2-14C] malonyl-CoA incorporation. The following samples are separated by TLC after synthesis: line 1, palmitate synthesis (p.s.) entrapped in liposomes supplying Proteinase K externally (sample applied twice more concentrated); line 2, p.s. encapsulated in liposomes and no Proteinase K added externally; line 3, p.s. in liposomes without FAS-B enzyme; line 4, p.s. in liposomes without acetyl-CoA; line 5, p.s. in bulk solution and no Proteinase K supplied; line 6, p.s. in bulk solution with Proteinase K; line 7, [2-14C] malonyl-CoA (0.74 nmoles). The arrow shows the position expected for palmitate as determined in a separate TLC (same conditions) experiment with non-radioactive palmitic acid standard (not shown)
This experiment shows that the fatty acid palmitate can be synthesized by the FAS-B enzyme activity within vesicles. In particular, to demonstrate that this reaction can take place exclusively within the liposome lumen, therefore in the absence of any external FAS activity, Proteinase K was added to the outside of liposomes after palmitate pathway entrapment. As expected, when Proteinase K is added palmitate synthesis is weaker than the signal produced by palmitate synthesis in both the liposome lumen and the external volume (Fig. 3). In the same experiment, the specific activity of the FAS-B enzyme and the essential requirement of the reagents in FAS (Fig. 2a) were tested. Clearly, only in the absence of precursors such as acetyl-CoA or in the absence of the enzyme FAS-B there is no lipid synthesis, confirming the lack of any contaminant that promote palmitate synthesis. Finally, the proteolytic activity of the Proteinase K on the FAS enzyme was tested by adding Proteinase K to the FAS-B reaction in bulk solution; as a result no palmitate is detected on TLC.
Liposome growth by internal FAS type I palmitate synthesis and incorporation into membrane
We initially examined how the surfactant palmitic acid behaves in the chemical induction of vesicle membrane growth by fatty acids incorporation as spontaneous fraction of added palmitate micelles into membrane of preformed POPC vesicles. Lipid bilayer growth was monitored by FRET assay based on membrane-localized lipid fluorescent dyes (Struck et al. 1981; Malinin et al. 2001). For this purpose pre-formed vesicles were prepared with POPC and 1% of the donor fluorescent dye, the NBD-PE (N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3 phosphoethanolamine, triethylammonium salt), and the acceptor fluorescent dye, rhodamine DHPE (Lissamine rhodamine B 1,2 dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt) that become integrated within the bilayer. The vesicles with both donor and acceptor dyes, integrated within the bilayer, grow by incorporating palmitic acid chains and the average distance between donor and acceptor molecules increases with a consequent decrease in FRET efficiency. The FRET test was carried out by fluorometric analysis measuring the donor/acceptor fluorescence ratio (F don/F acc), normalized to the F don/F acc after detergent lysis and was set up on four samples of pre-formed vesicles diluted 300 times (10 ul sample diluted in 3 ml buffer), where palmitic acid was added in different concentration as reported in “Materials and Methods” . The dyes used for these experiments exchange extremely slowly between phospholipid vesicles (Hanczyc et al. 2003) and consequently even less after 300 X dilution. Therefore, a decrease of FRET efficiency in this experiment can only result from the growth of the preformed vesicle membranes due to palmitic acid incorporation (See “Fret analysis” in “Materials and Methods”). In the same way, as already demonstrated for the oleate addition and incorporation into membrane of preformed vesicles (Hanczyc et al. 2003), palmitate can induce vesicle membrane growth as shown in Fig. 4a. The preformed vesicles used for this experiment were extruded again after rehydration in order to make them more homogenous (Fig. 4b), and the FRET analysis shows an increase in F don/F acc ratio within the range of values reported in Fig. 4a.

FRET assay for vesicle membrane growth. a Curve of normalised F don/F acc versus aliquots of palmitate micelles added on preformed POPC vesicles. b number weighted DLS size distribution of preformed vesicles re-extruded after re-hydration, the DLS analysis was performed before the incubation with palmitate micelles, c curve of normalised F don/F acc versus time (hours) as result of palmitate synthesis in the inner water pool of liposomes and incorporation in boundary layer. FRET assay for vesicle with internal FAS catalysis and Proteinase K added externally (diamonds), without precursor malonyl-CoA (squares), with no FAS-B enzyme in reaction (triangles), d number weighted DLS size distribution of vesicles re-extruded after re-hydration of the freeze-dried vesicles with the FAS-B catalysis solution, the DLS analysis was performed before the incubation period. The error bars reported in the graphs represent the range of values of experiments performed in triplicate
To test whether the FAS Type I catalysis of palmitate synthesis within liposomes can induce vesicle growth by fatty acid insertion into the lipid bilayer, the biochemical components of the reaction reported in Fig. 2a were entrapped in POPC liposomes. A film of POPC mixed with 1% of the donor fluorescent dye NBD-PE and the acceptor fluorescent dye DHPE was prepared in eppendorf tubes. Precursors for palmitate synthesis were mixed together with the FAS-B enzyme (Fig. 2a) and added to the lipid film for rehydration and vesicle formation. In order to prevent lipid synthesis outside of the liposomes, the Proteinase K enzyme was added externally.
The samples were kept at 30°C for 3, 5, and 24 h to allow lipid synthesis to occur within vesicles (see “Materials and Methods”: “Synthesis of F.A. in liposomes”). To test vesicle membrane growth, by lipid synthesis and insertion, a FRET assay, based on membrane-localized lipid fluorescent dyes, was set up for those samples diluted 300 times (10 ul sample diluted in 3 ml buffer). As reported for the chemical induction experiment, the FRET test was carried out by fluorometric analysis measuring the F don/F acc ratio, normalized to the F don/F acc after detergent lysis.
The vesicles with both donor and acceptor dyes, integrated within the bilayer, grow by incorporating palmitic acid chains soon after FAS-B catalysis and the average distance between donor and acceptor molecules increases with a consequent decrease in FRET efficiency. As shown in Fig. 4c, when FASB enzyme, together with precursors, is present within vesicles the FRET efficiency clearly decreases. On the contrary when malonyl-CoA or the FASB enzyme is omitted from the reaction mix the FRET performance is higher demonstrating that in the absence of palmitate synthesis no fatty acid molecule is inserted in liposome membrane and no liposome growth is detected. When the vesicles used for this experiment were extruded again after the FAS reaction entrapment, in order to make them homogenous in size (Fig. 4d), the FRET analysis reported an increase in F don/F acc ratio within the range of values reported in Fig. 4c.
The palmitate synthesis was analyzed in detail, measuring the integrated density of each radioactive signal reported on the TLC autoradiograph (Fig. 3), and estimating the ratio in between these values and the concentration of 14C radioactively labeled malonyl-CoA incorporated into the radioactive palmitate. The palmitate synthesized within liposome is limited to 30% in yield in comparison to the palmitate released in and out of liposomes (100%). As a result the palmitate produced within liposomes, after 24 h of FAS catalysis, corresponds to an estimated 2 nanomoles of palmitate released in the internal lumen of the liposomes for 200 μl of FAS catalysis assay (see “Synthesis of fatty acids in liposomes” in “Materials and Methods”), that is, on average, one molecule of palmitic acid for one thousand POPC molecules or one palmitate chain every ten molecules of fluorescent dyes present in the vesicles.
Discussion
In order to reconstruct a boundary layer self-reproduction process based on enzyme catalysis as a model for the first prebiotic cells, a FAS Type I catalysis was introduced in POPC vesicles. POPC are the vesicles of choice because of their stability, robustness and capability of hosting complex biochemical reactions although the vesicle forming surfactant cannot be considered prebiotic compound. As palmitate is one of the surfactant synthesized in the laboratory under presumably prebiotic conditions, palmitate vesicles could have been the lipid compartments for our experiments, on the other hand those vesicles are not easily manipulated due to their high melting temperature and stability in a limited pH range. However, it is believed that the prebiotic cell compartments were likely to be of heterogeneous lipid composition (Apel et al. 2002; Luisi 2006; Peretò et al. 2004; Walde 2006).
The FAS-B type I enzyme, from B. ammoniagenes, was chosen for our semi-synthetic protocell and introduced in liposomes together with the chemical precursors to produce palmitate. Although the FAS-B enzyme, as all the FAS Type I enzymes, is a multienzymes complex, characterized by a very large polypeptide chain, is the only extant enzyme that works autonomously and free in the cytoplasm. Moreover the fatty acids synthesized are released free from catalysis and ready to be integrated within the vesicle bilayer spontaneously. It is reasonable to imagine that during the origin of early cells, the boundary layer self-reproduction process may have been controlled by a simpler ancient FAS enzyme.
FRET experiments, where FAS catalysis is the only metabolism within our protocell system, have detected a surface area change of the liposome membranes due to palmitate inclusion in between POPC anphiphilic chains. Considering that Proteinase K was added to eliminate external lipid synthesis, we can presume that internal palmitate synthesis has contributed toward liposome growth. In fact, the only source of palmitate is represented by the FAS-B activity within liposomes. On the other hand the FAS-B activity is the only one responsible for palmitate synthesis, because no synthesis is detected if FAS-B enzyme is absent or degraded by Proteinase K (Fig. 3) and if any of the chemical precursors, such as malonyl-CoA or acetyl-CoA (Figs. 3, 4c), are missing.
In order to measure the vesicle growth we examined the liposome sizes by dynamic light scattering (DLS), before and after FAS catalysis and palmitate incorporation, but a change in size is not detectable or too small to be detected (data not shown). This result is not surprising if we consider the small amount of palmitate synthesized within liposomes. We therefore argue that while a liposome surface area change, induced by FAS synthesis, is detectable by a sensitive FRET assay, the change in vesicle size is too small to be detected in aggregate by DLS.
In particular, the FAS-B enzyme has a very short lifetime (around 3 h as shown in Fig. 2b) likely due to oxidation mechanisms and, as experienced for many enzymatic catalysis and/or cell-free protein synthesis, activity stops after a few hours because of energy and nutrient consumption (Kawarasaki et al. 1994; Noireaux et al. 2003; Noireaux and Libchaber 2004). The synthesis of palmitate is little within liposomes in comparison to the FAS-B synthesis in solution (Fig. 3) and this is in part due to the reduced inner water pools produced during liposome preparation (lipid aggregates are formed), and to the Proteinase K inhibitory activity in the outer water pools. Therefore, the resulting lipid membrane growth by FAS-B catalysis is definitely reduced in comparison to the vesicle growth reported by the FRET analysis after palmitic acid chemical induction (Fig. 4a, c). During the chemical induction process the palmitic acid is released onto preformed POPC vesicles at a temperature above the melting point of palmitate (63–64°), where the palmitic acid synthesized by FAS-B within the liposome lumen is released at 30°C and this may cause molecular aggregation and affect the efficiency of lipid incorporation into membrane.
In order to overcome some of these limitations and demonstrate a complete process of boundary layer self-reproduction, a trans-membrane/channel pore system should be introduced in the liposome lipid bilayer. As an example the α-hemolysin toxin is a selective permeability system that allows exchange of nutrients, energy and anti-oxidants and can be used as a trans-membrane protein channel in lipid vesicles. This type of pore can create a selective portal for low molecular weight components in lipid compartments and is capable of boosting protein expression up to 4 days through repeated feeding of the system externally (Noireaux and Libchaber 2004).
For the first time we monitor liposome vesicle growth based on the entrapment and activity of only one enzyme capable of building its own membrane from within boundary. This is a prototype of the simplest autopoietic minimal cell (Fig. 1), (Luisi 2006).
Materials and methods
Materials
Palmitoyl-2-oleoyl-sn-glycero-3phosphatidylcholine (POPC) was purchased from Avanti Pola Lipids (Canada). The two membrane localized fluorescent dyes used for the FRET analysis, the Lissamine rhodamine B 1,2 dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (rhodamine DHPE) and the N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3 phosphoethanolamine, triethylammonium salt (NBD-PE) were purchased from invitrogen. The E. coli strain DH5α (Gibco) was used in this study for the FAS-B heterologous expression. The plasmid pGM44 is a pBlueScript derivative containing the fasB gene together with a 0.8 kb of both its 5′-flanking and 3′-flanking DNA sequences and was kindly provided by Eckhart Shweizer, Universitat Erlangen. Nurnberg, Germany. The B. ammoniagenes DNA inserted into this vector is limited by ClaI and NotI restriction sites. For heterologous expression of the B. ammoniagenes FAS proteins, the E. coli DH5α transformants were grown at 30°C in a medium containing 20 g/l casamino acids, 10 g/l yeast extract and 10 g/l Nacl, together with 50 mg/l ampicillin at pH 7.5.
DNA manipulation techniques
Transformants were routinely checked by pGM44 plasmid isolation and restriction digestion based on its restriction map. Plasmid isolation was carried out based on the method of Birnboim and Doly (1979). The DNA manipulation methods were performed as described elsewhere (Sambrook et al. 1989).
FAS-B enzyme purification and assay
Escherichia coli heterologously expressed B. ammoniagenes FAS-B enzyme was purified according to the procedure described by Stuible et al. (1997). FAS-B enzyme assay, FAS activity was assayed according to the method of Lynen (1980) with modifications (Stuible et al. 1997): NADPH disappearance, oxidation, was measured spectrophotometrically and carried out in a total volume of 1 ml containing 0.4 M potassium phosphate at 25°C, pH 7.3, 3 mM dithiothreitol, 0.5 mg Fatty acid free bovine serum albumin (SIGMA), 700 μM NADPH, 200 μM malonyl-CoA (Sigma) and 150 μM acetyl-CoA (Sigma) adding 2 μg of purified enzyme FAS-B. The absorbance of samples was recorded every 1-h interval at 340 and 600 nm and the values reported on the graph equal the delta between the values at 340 and 600 nm.
Vesicle preparation
Liposomes were prepared by the dehydration and rehydration method as described by Yomo and coworkers (Ishikawa et al. 2004). Briefly, POPC liposomes were prepared by hydrating a thin POPC film with ultra-pure water (Milli-Q, Millipore), followed by extrusion (10 passages) through two stacked 400 nm polycarbonate membranes (Nucleopore Track-Etch Membrane, Whatman) and freeze-drying. The resultant freeze-dried liposomes were therefore hydrated with Bicine buffer (200 mM pH 8.5) to prepare 100 mM POPC preformed vesicles The vesicles for FRET analysis were modified dissolving in chloroform two membrane localized fluorescent dyes, rhodamine DHPE and NBD-PE, together with freeze dried POPC liposomes in a 1% ratio (fluorescent dyes/POPC) and dried under vacuum centrifuge for 2 h. The same FRET experiments carried out with freeze dried POPC liposomes and a 1% ratio (fluorescent dyes/POPC) have been repeated with the vesicles extruded through 0.2 μm filters (four times) after rehydration with Bicine buffer or after entrapment of the 200 μl FAS-B catalysis solution at 4°C.
Palmitate supplied as micelles on preformed POPC vesicles
Palmitic acid 20 mM was dissolved in a solution of 20 mM NaOH to form sodium palmitate at 60°C for 15 min. Palmitate was added on POPC preformed vesicles, marked with membrane localized fluorescent dyes (see vesicle preparation), in a set of four samples at varying concentration. Each sample was characterized by 3 ml of buffered solution (200 mM bicine pH 8.5) where the preformed vesicles were diluted 300 times resulting in 0.3 mM POPC vesicles (10 μl of the preformed vesicles in 3 ml, final volume, of Bicine buffer). Palmitate was added soon after melting treatment at 60°C in a four set of samples as 0, 100, 200, 300 and 400 ul of 20 mM palmitic acid. The sample were analysed 30 min after palmitate addition.
Dynamic light scattering (DLS)
Measurements were carried out with an ALV home-assembled light scattering photometer made of a 25 mW He–Ne laser (Model 127, Spectra-Physics Lasers, Mountain View, Canada), an ALV DLS/SLS-5000 Compact Goniometer System (ALV, Langen, Germany), two SPCM-AQR avalanche photodiodes (PerkinElmer Optoelectronics, Vaudreuil, Canada) and an ALV-5000 Multiple-tau Digital Correlator (ALV, Langen, Germany). The cylindrical scattering cells were immersed in a fuzzy-thermostated decaline bath (ALV), which was kept at 25.0°C. All experiments were performed at the scattering angle of 90°; other settings were: solvent viscosity 0.899 MPas, solvent refractive index 1.330. Great care was taken at every step to avoid the presence of dust in the liposome preparations. Liposomes (after the incubation period) were diluted (tenfold) in isotonic buffer and measured without any pretreatment. Particle-size distribution was computed by using two different algorithms (ILT and CONTIN), which gave similar results. Number-weighted size distributions were calculated within the Rayleigh–Gans–Debye approximation.
Synthesis of fatty acids in vitro and in liposomes
The FAS-B catalysis (in vitro) as reported in Fig. 2a was set up in 200 μl solution with the following stochiometry: 0.4 M potassium phosphate pH 7.3, 3 mM dithiothreitol, 0.5 mg Fatty acid free bovine serum albumin (SIGMA), 700 μM NADPH, 200 μM malonyl-CoA (Sigma), 150 μM acetyl-CoA (Sigma) adding 2 μg of purified enzyme FAS-B. When the FAS-B catalysis was entraped in liposomes, the 200 ul of the FAS-B stoichiometry was resuspended in eppendorfs with the POPC freeze-dried liposomes and the liposomes formed by vortexing and pipetting for a few seconds to form 25 mM liposomes. For FRET analysis, the same method was used to encapsulate a FAS-B reaction but using freeze dried POPC liposomes modified with fluorescent dyes as prepared and reported in vesicle preparation. Proteinase K (5 μl, 5 mg/ml) was added soon after liposome entrapment to prevent FAS-B activity at the outside of ready formed vesicles unless explicitly stated. The reactions were in all cases incubated at 30°C for 3 h. TLC experiment: Synthesis of 14C-labeled fatty acids was performed as reported for the FASB- catalysis in vitro with the following modifications: 0.5 μCi of [2-14C] malonyl-CoA (Amersham) was added to the non-radioactive malonyl-CoA. The fatty acyl-CoA derivatives synthesized were hydrolysed using 10 μl 5 M NaOH at 50°C for 3 h followed by the addition of 15 μl 6 M H2SO4. When synthesis of 14C-labeled fatty acids was performed in liposomes the same conditions were applied as described above entrapping the solution reaction in 25 mM POPC vesicles and, after incubation 3 h at 30°C, the liposomes were broken by freeze and thaw, 2 min at −70°C three times. After addition of 10 μg of palmitic acid the free fatty acids were extracted twice with 80 μl ethyl acetate. The ethyl acetate extracts were dried under vacuum and resuspended in 50 μl of acetone. For fatty acid separation Silica-gel 60 thin layer plates were used. The mobile phase used for fatty acids separation was a 90:24:1 (by vol.) mixture of toluene, dioxane and acetic acid. The TLC plates were spotted on one edge with 10 μl out of 50 ul of the acetone resuspension for each sample, dried and the same edge exposed onto the mobile phase soaking only 1 mm of the silica-gel side. The chromatogram was exposed to Kodak BioMax XAR Film O/N (−70°C) and the film developed the day after.
FRET analysis
About 10 μl out of 200 μl FAS-B catalysis in liposomes were diluted 300 times in a 3 ml final volume of 0.4 M potassium phosphate buffer pH 7.3 and analyzed. Fluorescence measurements were made on a Jasco.
Spectrofluorometer FP-6200 Fluorescence resonance energy transfer (FRET) experiments were done using NBD-PE as the donor and rhodamine DHPE as the acceptor using an excitation wavelength of 430 nm, and emission wavelength of 530 nm for NBD-PE and 586 for DHPE. Fluorescence intensities for the donor and the acceptor dyes were normalized to the fluorescence intensities after sample lysis with 1% triton X-100. All measurements were taken at sample turbidities of <0.1, and dye absorbance of <0.1 to minimize multiple scattering and re-absorption effects. All FRET experiments reported here were performed in triplicate.
References
Apel CL, Deamer DW, Mautner MN (2002) Self-assembled vesicles of monocarboxylic acids and alcohols: conditions for stability and for the encapsulation of biopolymers. Biochim Biophys Acta 1559:1–9
Bachmann PA, Luisi PL, Lang J (1992) Autocatalytic self-replicating micelles as models for prebiotic structures. Nature 357:57–59
Berclaz N, Muller M, Walde P, Luisi PL (2001) Growth and transformation of vesicles studied by ferritin labelling and cryotransmission electron microscopy. J Phys Chem B 105:1056–1064
Brirnboim HC, Doly J (1979) A rapid alkaline estraction procedure for screening recombinant plasmid DNA. Nucelic Acids Res 7:1513–1523
Deamer DW, Boatman DE (1980) An enzymatically driven membrane reconstitution from solubilised components. J Cell Biol 84:461–467
Deamer DW, Gavino V (1983) Lysophosphatidylcholine acyltransferase: purification and applications in membrane studies. NYAS 414:90–96
Hanczyc MM, Fujikawa SM, Szostak JW (2003) Experimental models of primitive cellular compartments: encapsulation, growth, and division. Science 302:618–622
Hargreaves WR, Deamer DW (1978) Liposomes from ionic, single-chain amphiphiles. Biochemistry 17:3759–3768
Ishikawa K, Sato K, Shima Y, Urabe I, Yomo T (2004) Expression of a cascading genetic network within liposomes. FEBS Lett 576:387–390
Kawarasaki Y, Nakano H, Yamane T (1994) Prolonged cell-free protein synthesis in a batch system using wheat germ extract. Biosci Biotechnol Biochem 58:1911–1913
Kuruma Y, Stano P, Ueda T, Luisi PL (2009) A synthetic biology approach to the construction of membrane proteins in semi-synthetic minimal cells. Biochim Biophys Acta 1788:567–574
Luisi PL (2006) In: The emergence of life. Cambridge University Press, Cambridge
Luisi PL, Ferri F, Stano P (2006) Approaches to semi-synthetic minimal cells: a review. Naturwissenschaften 93:1–13
Lynen F (1980) On the structure of fatty acid synthetase of yeast. Eur J Biochem 112:431–442
Malinin VS, Haque ME, Lentz BR (2001) The rate of lipid transfer during fusion depends on the structure of fluorescent lipid probes: a new chain-labeled lipid transfer probe pair. Biochemistry 40:8292–8299
Monnard PA, Deamer DW (2002) Membrane self-assembly processes: steps toward the first cellular life. Anat Rec 268:196–207
Murtas G (2007) Construction of a semi-synthetic minimal cell: a model for early living cells. Orig Life Evol Biosph 37:419–422
Murtas G, Kuruma Y, Bianchini P, Diaspro A, Luisi PL (2007) Protein Synthesis in liposomes with a minimal set of enzymes. Biochem Biophys Res Commun 363:12–17
Noireaux V, Libchaber A (2004) A vesicle bioreactor as a step towards an artificial cell assembly. PNAS 101:17669–17674
Noireaux V, Bar-Ziv R, Libchaber A (2003) Principles of cell-free genetic circuit assembly. Proc Natl Acad Sci USA 100:12672–12677
Oberholzer T, Albrizio M, Luisi PL (1995a) Polymerase chain reaction in liposomes. Chem Biol 2:677–682
Oberholzer T, Wick R, Luisi PL, Biebricher CK (1995b) Enzymatic RNA replication in self-reproducing vesicles: an approach to a minimal cell. Biochem Biophys Res Commun 207:250–257
Orò J (1994) Chemical synthesis of lipids and the origin of life. J Biol Phys 20:135–147
Peretò Y, Lopez-Garcia P, Moreira D (2004) Ancestral lipid biosynthesis and early membrane evolution. Trends Biochem Sci 29:469–477
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Laboratory Press, Cold Spring Harbour
Schmidli PK, Schurtenberger P, Luisi PL (1991) Liposome-mediated enzymatic synthesis of phosphatidylcholine as an approach to self-replicating liposomes. J Am Chem Soc 113:8127–8130
Schweizer E, Hofmann J (2004) Microbial type I fatty acid synthases (FAS): major players in a network of cellular FAS systems. Microbiol Mol Biol Rev 68:501–517
Simoneit BRT (2004) Prebiotic organic synthesis under hydrothermal conditions: an overview. Adv Space Res 33:88–94
Struck DK, Hoekstra D, Pagano RE (1981) Use of resonance energy transfer to monitor membrane fusion. Biochemistry 20:4093–4099
Stuible HP et al (1996) Identification and functional differentiation of two type I fatty acid synthases in Brevibacterium ammoniagenes. J Bacteriol 178:4787–4793
Stuible HP, Meurer G, Schweizer E (1997) Heterologous expression and biochemical characterization of two functionally different type I fatty acid synthases from Brevibacterium ammoniagenes. Eur J Biochem 247:268–273
Takakura K, Sugawara T (2004) Membrane dynamics of a myelin-like giant multilamellar vesicle applicable to a self-reproducing system. Langmuir 20:3832–3834
Takakura K, Toyota T, Sugawara T (2003) A novel system of self-reproducing giant vesicles. J Am Chem Soc 125:8134–8140
Walde P (2006) Surfactant assemblies and their various possible roles for the origin(s) of life. Orig Life Evol Biosph 36:109–150
Walde P, Wick R, Fresta M, Mangone A, Luisi PL (1994) Autopoietic self-reproduction of fatty acid vesicles. JACS 116:7541–7547
Acknowledgments
We thank E. Schweizer, J. Hofmann and U. Hoja, University of Erlangen-Nürnberg (Germany), for providing the pGM44 plasmids (fasB gene); Dr. P. Stano and Dr. P. L. Luisi, University Rome Tre (Italy), for generous assistance with liposome technology and helpful discussion and comments on the manuscript, and Dr. M. Hanczyc, University of Southern Denmark, for valuable discussions and critical reading of the manuscript. This work was supported by Centro Enrico Fermi (Rome, Italy), providing a Senior Grant to G. M, and by Grant RGP0033/2007-C from Human Frontier Science Program. “The experiments carried out for this manuscript comply with the current laws in force in Italy in which they were performed.”
Conflict of interest statement
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Murtas, G. Internal lipid synthesis and vesicle growth as a step toward self-reproduction of the minimal cell. Syst Synth Biol 4, 85–93 (2010). https://doi.org/10.1007/s11693-009-9048-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11693-009-9048-1