
Abstract
The accurate determination of the spatial trends on the variability of a species’ gene pool is essential to elucidate the underlying demographic-evolutionary events, thus helping to unravel the microevolutionary history of the population under study. Herein we present a new software called GenoCline, mainly addressed to detect genetic clines from allele, haplotype, and genome-wide data. This program package allows identifying the geographic orientation of clinal genetic variation through a system of iterative rotation of a virtual coordinate axis. Besides, GenoCline can perform complementary analyses to explore the potential origin of the genetic clines observed, including spatial autocorrelation, isolation by distance, centroid method, multidimensional scaling and Sammon projection. Among the advantages of this software is the ease in data entry and potential interconnection with other programs. Genetic and geographic data can be entered in spreadsheet table formatting (.xls), whereas genome-wide data can be imported in Eigensoft format. Genetic frequencies can also be exported in a format compatible with other programs dealing with population genetic and evolutionary biology analyses. All illustrations of results are saved in.svg format so that there will be high quality and easily editable vectorial graphs available for the researcher. Being implemented in Java, GenoCline is highly portable, thus working in different operating systems.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genetic variability of a species is not uniform all over its distribution range. When a species expands over a broad region, it most likely split into subpopulations so that mating probabilities between individuals from distinct subpopulations tend to decrease over distance (Kimura & Weiss, 1964). It is a fact that the existence of geographical barriers promotes local genetic differentiation among subpopulations. However, a small average migration distance compared to the species' distribution range may lead to a sort of seclusion called "isolation by distance" (Wright, 1943), even lacking physical barriers. The phenomenon of isolation by distance can then lead to a clear pattern of genetic differentiation over space. This phenomenon is called a cline or clinal variation, a term introduced by Huxley (1938).
The origins of a clinal variation have been associated with different circumstances: (i) the existence of genetic drift, (ii) resumed contact between formerly isolated populations, (iii) spatially discontinuous changes of the environment, and (iv) continuous environmental gradients (Endler, 1973). Genetic clines can be observed in quantitative and qualitative traits (Hoffmann & Weeks, 2006) and can affect variable proportions of the genome.
For polymorphic genetic markers, a cline is a gradual variation (also called gradient) of their genotype or gene frequencies (Endler, 1973). Once the genetic cline arises, it will become stable if alleles (or genotypes) reach their genetic equilibrium frequencies. Overall, a sizeable number of genetic clines occur by the interplay between the diversifying effects of local selection and the homogenizing effects of dispersal (Barton & Gale, 1993; Barton & Hewitt, 1985; Sotka & Palumbi, 2006). Consequently, one way of measuring the magnitude of both the selective pressures and the migratory flows in nature is by computing the rate of change in gene frequencies within a cline (Slatkin, 1973). Likewise, combination with other analytical methods such as estimates of linkage disequilibrium among different loci allows for analyzing the impact of simultaneous action of these evolutionary forces (Barton & Gale, 1993; Mallet et al., 1990; Sotka & Palumbi, 2006).
Genetic clines are often defined as the gradual variation in the frequency of a given genetic marker regarding latitude, longitude, altitude (Roy et al., 2018) or the distance up to an origin (Pérez-Losada & Fort, 2018). In this way, thorough studies of the spatial orientation of clinal genetic variation stand helpful to assist in the interpretation of potential evolutionary forces shaping the gene pool of a species (Kyriacou et al., 2008; Maes & Volckaert, 2002; Razgour, 2015).
In this paper, we present a new computer software called GenoCline, which has been devised to detect spatial trends in genetic variation from genetic marker frequencies or genome-wide databases. The program also provides several statistical tools to assist in the interpretation of clinal variation. The main novelty of Genocline lies in the ability to accurately determine the spatial orientation of a genetic cline. Other software tools for analyzing genetic clines focus on different goals than GenoCline. These are the cases, for example, of bgc (Gompert & Buerkle, 2012), aimed at detecting genomic regions involved in cline emergence, and also of HZAR (Derryberry et al., 2014), which requires prior knowledge of the gradient orientation to fit the results to a cline model. GenoCline has been already used, for example, to determine the geographic direction of a frequency gradient for the human MAPT*H2 haplotype in western Eurasia (Alfonso-Sánchez et al., 2018) or to study the movements of a small migratory falcon, the lesser kestrel (Falco naumanni), to unravel the origin of birds converging to pre-migratory sites (Bounas et al., 2018).
Methods
As GenoCline aimed to identify genetic clines and their spatial orientation, geographic and genetic are the input data. To explore spatial patterns of genetic variation, GenoCline makes a geographic coordinate axis rotate in successive one-degree steps until completing 180 degrees. At the starting point (zero-step), the axis follows the latitudinal line. Similarly, the 90-degrees-step follows the longitudinal axis. In every iteration, geographic positions of the targeted populations are orthogonally projected on the axis, thus obtaining a new coordinate vector (Fig. 1). Then, the program computes the Pearson product-moment correlation coefficient (r) between the allelic frequency vector and each of the 180 geographical coordinate vectors generated by iterations. If at least one of the geographical coordinate vectors significantly correlates with frequencies, such a finding could constitute evidence for a genetic cline. However, since correlation tests are not fully independent, this could affect the statistical significance of the P-value achieved, a fact that must be considered when interpreting. The spatial orientation of the gene frequency cline will be the direction of the significantly correlated geographical coordinate vector. Because it is not uncommon to find statistically significant associations for several consecutive degrees of rotation, the program will choose the orientation that yields the highest absolute value for the r correlation coefficient.

Graphic representation of the rotation of the Y axis to the Y' axis. The geographic position of a population is represented by p. The new coordinate is calculated as b' = c × cos (α'). For calculations, a clockwise axis rotation is set
It is also possible to run a cline analysis using the angular transformation arcsin√p (being p the allele frequency) to separate the estimate's variance from the estimate itself (Hartl & Clark, 1997). Worth mentioning here is that the heterozygosity cline can be computed on a single locus, whatever the extension of this locus may be, so it could well be a whole haplotype.
Often, genetic clines can better fit into a sigmoid function, particularly in hybrid zones (Barton & Gale, 1993; Fitzpatrick, 2013). GenoCline can implement cline analyses employing the sigmoid regression model. In this case, the program computes the significance values of all three parameters defining the sigmoid curve; however, the statistical criterion indicating that we might have found footprints of a genetic cline lies in the statistical significance of the F-ratio.
When this software is employed to assess spatial genetic patterning using genome-wide databases, analyses focus on individuals rather than populations. These studies may involve hundreds of thousands of SNP loci, so the one-by-one locus analysis becomes extremely difficult when dealing with numerous population samples. Alternatively, GenoCline detects patterns of clinal variation for the whole genome or the specific genomic region examined. In doing so, a multidimensional scaling (MDS) analysis is performed from the identity by state (IBS) distance matrix between each pair of individuals. After the MDS, the program retains (by default) the 10 leading eigenvectors for subsequent analyses. Each eigenvector is then treated like an allele frequency vector of a genetic marker to correlate with the geographical coordinates of the individuals' sampling site. As discussed above, evidence for potential genetic clines may be present if at least one coordinate vector significantly correlates with one of the eigenvectors.
In the case of GenoCline detecting the existence of clinal genetic variation, it gives the possibility of running further analyses to assist in the interpretation of the hypothetical clines, including spatial autocorrelation (Moran, 1950), isolation by distance (Malécot, 1973), centroid method (Harpending & Ward, 1982), MDS (Kruskal, 1964) or the Sammon projection (Sammon, 1969).
Spatial autocorrelation is roughly described as the property of variables of taking values, at pairs of locations a certain distance apart, more similar (positive autocorrelation) or less similar (negative autocorrelation) than expected for randomly associated pairs of observations. It poses a problem for statistical testing since autocorrelated data violate the assumption of independence of most standard statistical procedures (Legendre, 1993). For this reason, some authors have implemented specific methods to test whether the observed value of a variable at one locality is significantly dependent on the variability pattern of that variable at neighboring localities (Sokal & Oden, 1978).
GenoCline affords the possibility of running a spatial autocorrelation analysis for a set of allele or haplotype frequencies. For a targeted sampling site, spatial autocorrelation examines whether neighbouring populations show more genetic affinity than remote populations. Among the indexes devised to estimate spatial autocorrelation levels, Moran's I (Moran, 1950) has been widely accepted and used in biological studies. Variation of Moran's I can be represented against geographic distance in a graph known as autocorrelogram. According to the isolation-by-distance model, a downward trend of the Moran index occurs as geographic distance among populations increases. More recently, Epperson and Li (1996) analyzed the statistical properties of Moran's autocorrelation parameters and created estimators of gene dispersal based on existing patterns of genetic variation. According to that study, Moran's statistics proved to be highly informative and predictable, thus enabling robust hypothesis tests for neutral theory. Along these lines, GenoCline also tests the statistical significance of the autocorrelation values. Predictably, the statistical robustness of the autocorrelogram is greater when most of Moran's I values are statistically significant.
Measuring Genetic Kinship
The "isolation-by-distance" model developed by Malécot (1973) considers that the genetic kinship depends on the geographic distance, as mirrored in the mathematical function of the mean kinship coefficient \(\emptyset \left( d \right)\),
where d is the distance, a is a measure of local kinship, and b specifies the rate of exponential decline of \(\emptyset \left( d \right)\).
Another method to examine the isolation by distance is based on the analysis of the relationship between the quotient,
and the geographic distance through linear regression (considering all population pairs), thus estimating the slope and intercept of this relationship (Rousset, 1997; Wright, 1943). The differences between the Malécot and Wright approaches are between individual-based and population-based sampling. Rousset (1997) also describes a regression against the natural logarithm of geographic distance (x-axis) for all two-dimensional cases.
The centroid method was conceived to estimate the relative intensity of gene flow on a group of populations (for details see Harpending & Ward, 1982). In doing so, the centroid method represents the observed proportion of heterozygotes in each population against the distance from the centroid ri
where pik is the frequency of allele k in population i and, \(\overline{p}_{k}\) is the mean frequency of allele k.
To interpret the graph generated by the centroid method, drawing a line representing the expected heterozygosity values is vital. Those subpopulations placed above the line are considered to have experienced higher gene flow. On the contrary, a position below the line indicates some genetic isolation. The centroid is merely an exploratory analysis, as there is no way to test the statistical significance of the positioning of populations relative to the expected heterozygosity line. An example of this issue can be consulted in Gómez-Pérez et al. (2011).
Multidimensional scaling analysis, MDS (Kruskal, 1964), and the Sammon projection (Sammon, 1969) are two methods designed to obtain a small number of dimensions, thus facilitating the interpretation of complex distance matrices. In the first, GenoCline builds up the corresponding graphical representation from FST genetic distances (Reynolds et al., 1983), whereas the Sammon projection utilizes the Euclidean distance.
Input/Output Data
Data entry is user-friendly. Genetic (both allele and haplotype) frequencies and geographic coordinates can be imported in spreadsheet table formatting, that is, a.xls file. GenoCline supports the input of spatial coordinates in both sexagesimal and decimal degrees. In the latter, only two numbers need to be entered for each sampling site. To illustrate the spatial position of sampling sites in a two-dimensional space, the program generates a map with the projection of all sampling points of the database analyzed. This map is saved in.svg and.jpg image formats.
Genome-wide data can be imported in Eigensoft format. Genetic data can also be exported in a format compatible with other programs dealing with population genetics and evolutionary biology, such as Arlequin, Phylip, Poptree, and Past. As mentioned, the current version of GenoCline generates images in two different formats (.jpg and .svg), so there are high quality and easily editable vectorial graphics available for the researcher. The.jpg format allows the user a quick preview of graphs, which is helpful for exploratory analyses. The .svg, a standard format for vector graphics, enables the highest quality editing of all graphic's components. For editing.svg images, the user might choose Inkscape, LibreOffice Draw and Adobe Illustrator, among other applications.
Genetic Clines
Once identified the genetic patterns, GenoCline groups them into 10-degree intervals (10-degree circumference arcs). Subsequently, the program generates a radar plot (also called a spider plot) to represent the absolute frequency of clines per interval.
To illustrate how this software works to detect patterns of clinal variation, we employ the database from the work of Moreira et al. (2015) in their study on European populations of the white-tailed bumblebee (Bombus terrestris). Data are allele frequencies for a set of microsatellite loci or short tandem repeats (STRs) in several population samples. Figure 2 shows the results obtained with GenoCline. As can be seen, it is possible to identify many clines with north–south (N-S) and, above all, north-northwest to south-southeast (NNW-SE) orientation. According to the authors, the Irish Sea would have acted as a geographical barrier to gene flow between the Irish populations and the rest of the European samples, which could have been the probable cause for the emergence of allele frequency clines.

Orientation of the genetic clines detected in a set of microsatellites or STR markers in European populations of the buff-tailed bumblebee Bombus terrestris (Moreira et al., 2015). Concentric circles indicate the number of clines detected. Radii delineate the orientation of genetic clines in 10-degree intervals
By selecting a specific cline, GenoCline generates a two-dimensional plot with the regression line and 95% confidence limits. In such a plot, the x-axis represents the coordinates relative to the virtual axis (rotation axis), and the y-axis represents the corresponding allele frequencies/eigenvector values. With this option, Genocline also provides (i) the orientation of the genetic line, (ii) the parameters defining the regression line, (iii) the coefficient of determination of the regression line (r2), and (iv) the Pearson's r correlation coefficient with statistical significance. This analysis is available for both allele frequencies and angular transformations of allele frequencies.
Figure 3 displays the frequency cline detected for the CT13910 mutation of lactase persistence in European human populations (Itan et al., 2009). As can be noticed, there is an upward spatial trend progressing from the south-southeast to the north-northwest of the continent. Authors consider this finding as contributing a coherent and spatially explicit picture of the coevolution of lactase persistence and dairying in Europe. Their reasoning it would be unlikely that lactase persistence provided a selective advantage in communities without a supply of fresh milk (gene-culture coevolutionary model).

Frequency cline detected for CT13910 mutation of lactase persistence in European human populations. Displayed are the regression line along with 95% confidence limits (dotted lines)
In the quest for gene frequency clines, sometimes running sigmoid regression might be more appropriate than linear regression. Thus, in the study by Chlaida et al. (2009), the authors found clines' traces at the SOD locus along the North African coast in a set of samples of the European pilchard (Sardina pilchardus). They could identify two differentiated groups separated by the Agadir Bay. Figure 4 shows the sigmoid cline obtained with GenoCline for allele SOD*80. The midpoint of the threshold is at 0.61, between Sidi Ifni and Agadir populations.

Allele frequency cline for SOD*80 on the North African coast. Curve represents the statistically significant sigmoidal regression found for the study by Chlaida et al. (2009) on the European pilchard (Sardina pilchardus). The orientation of the cline, south southwest-north east (SSW-NNE), is also shown
GenoCline can also analyze patterns of clinal variation from genome-wide databases. To illustrate so, we chose a small group of individuals from the 'Human Origins' database in Eigensoft format (Lazaridis et al., 2014). Firstly, we obtained eigenvector values through multidimensional scaling analysis of 620,000 SNPs from 65 current European individuals. Then, we represented the clinal pattern found for eigenvector 1 values (Fig. 5). As can be noticed, such analysis was able to identify a significant cline (p < 0.0001) in the northeast-southwest (NE-SW) direction.

Clinal pattern detected for eigenvector 1 values, obtained through MDS over a database of 620,000 SNPs in 65 current European individuals. In the graph, each point represents an individual. Regression line along with 95% confidence limits (dotted lines) are shown
Spatial Autocorrelation
In GenoCline, the method to define distance categories or classes is relatively flexible as you may create up to 20. After defining distance classes based on population geographic coordinates (and according to the researcher criteria), GenoCline runs spatial autocorrelation analysis and calculates the Moran’s I for each one. The program also generates the autocorrelogram, displaying the Moran's I value per class and the corresponding 95% confidence limits. Figure 6 illustrates the autocorrelogram with Moran’s I values, generated from a sample set of the European pilchard, Sardina pilchardus (Chlaida et al., 2009).

Autocorrelogram obtained from a sample set of the European pilchard, Sardina pilchardus (Chlaida et al., 2009). Variations of Moran's I with the distance between populations are shown, as well as 95% confidence limits. The circle denotes a statistically significant p value (p < 0.05)
Isolation by Distance
Intending to assess whether the allele frequency distribution in a given region fits into Malécot's isolation-by-distance model, GenoCline computes both the intercept (a) and slope (b) values both in equations of linear regression models and exponential models. Isolation by distance includes the correction proposed by Weir and Goudet (2017), which sets a unified treatment of relatedness and population structure with an underlying framework of allelic dependence.
Centroid Method
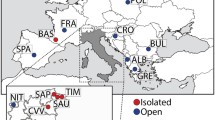
Buhler et al. (2012) surveyed the frequencies of five human HLA genes in 11 cantons of Switzerland. Figure 7 shows the graph produced by GenoCline when analyzing the database of this study using centroids. Expected heterozygosity is computed based on the variance contributed by each population to the total Wahlund variance. We found a much lower than expected gene flow in Coira, the most geographically isolated population. On the other hand, Bern and Zurich behave as hub populations for internal (within Swiss population) gene flow as per their positioning. They both placed remote from the centroid line and with a low contribution to the Wahlund variance, mirroring the homogenizing effects of gene flow. Likewise, Geneva seems to be the population receiving higher gene flow from the outside, as per distant position from the centroid and highest contribution to the Wahlund variance.

Centroid graph obtained from frequencies of five human HLA loci in 11 cantons of Switzerland. Line is the expected heterozygosity value for this group of populations, which is computed based on ri, being ri the variance contributed by each population to the total Wahlund variance
As previously discussed, multidimensional scaling analysis (Kruskal, 1964) and Sammon projection (Sammon, 1969) are further options available in GenoCline, intending to facilitate the interpretation of the genetic relationships among populations.
Download and Usage
The current version (v1.5) of the software, as well as the corresponding User’s Manual, are available at http://genocline.sourceforge.net and http://www.didac.ehu.es/genocline. GenoCline uses Java 1.8 or higher and is therefore portable to other operating systems. No installation is required. The execution of the different available options is via a graphical user interface (GUI).
References
Alfonso-Sánchez, M. A., Espinosa, I., Gómez-Pérez, L., Poveda, A., Rebato, E., & Peña, J. A. (2018). Tau haplotypes support the Asian ancestry of the Roma population settled in the Basque Country. Heredity, 120, 91–99.
Barton, N. H., & Gale, K. S. (1993). Genetic analysis of hybrid zones. In R. Harrison (Ed.), Hybrid zones and the evolutionary process. Oxford University Press.
Barton, N. H., & Hewitt, G. M. (1985). Analysis of hybrid zones. Annual Review of Ecology and Systematics, 16(1), 113–148.
Bounas, A., Tsaparis, D., Gustin, M., Mikulic, K., Sarà, M., Kotoulas, G., & Sotiropoulos, K. (2018). Using genetic markers to unravel the origin of birds converging towards pre-migratory sites. Scientific Reports, 8, 8326.
Buhler, S., Nunes, J. M., Nicoloso, G., Tiercy, J. M., & Sanchez-Mazas, A. (2012). The heterogeneous HLA genetic makeup of the Swiss population. PLoS ONE, 7, e41400.
Chlaida, M., Laurent, V., Kifani, S., Benazzou, T., Jaziri, H., & Planes, S. (2009). Evidence of a genetic cline for Sardina pilchardus along the Northwest African coast. ICES Journal of Marine Science, 66, 264–271.
Derryberry, E. P., Derryberry, G. E., Maley, J. M., & Brumfield, R. T. (2014). HZAR: Hybrid zone analysis using an R software package. Molecular Ecology Resources, 14, 652–663.
Endler, J. A. (1973). Gene flow and population differentiation: Studies of clines suggest that differentiation along environmental gradients may be independent of gene flow. Science, 179, 243–250.
Epperson, B. K., & Li, T. (1996). Measurement of genetic structure within populations using Moran’s spatial autocorrelation statistics. Proceedings of the National Academy of Science USA, 93, 10528–10532.
Fitzpatrick, B. M. (2013). Alternative forms for genomic clines. Ecology and Evolution, 3, 1951–1966.
Gómez-Pérez, L., Alfonso-Sánchez, M. A., Sánchez, D., García-Obregón, S., Espinosa, I., Martínez-Jarreta, B., de Pancorbo, M. M., & Peña, J. A. (2011). Alu polymorphisms in the Waorani tribe from the Ecuadorian Amazon reflect the effects of isolation and genetic drift. American Journal of Human Biology, 23, 790–795.
Gompert, Z., & Buerkle, C. A. (2012). bgc: Software for Bayesian estimation of genomic clines. Molecular Ecology Resources, 12, 1168–1176.
Harpending, H. C., & Ward, R. H. (1982). Biochemical systematics and human populations. In M. Nitecki (Ed.), Biochemical aspects of evolutionary biology. University of Chicago.
Hartl, D. L., & Clark, A. G. (1997). Principles of population genetics. Sinauer Associates.
Hoffmann, A. A., & Weeks, A. R. (2006). Climatic selection on genes and traits after a 100- year-old invasion: A critical look at the temperate-tropical clines in Drosophila melanogaster from eastern Australia. Genetica, 129, 133–147.
Huxley, J. S. (1938). Clines: An auxiliary taxonomic principle. Nature, 142, 219–220.
Itan, Y., Powell, A., Beaumont, M. A., Burger, J., & Thomas, M. G. (2009). The origins of lactase persistence in Europe. PLoS Computional Biology, 5(8), e1000491.
Kimura, M., & Weiss, G. H. (1964). The stepping-stone model of population structure and the decrease of genetic correlation with distance. Genetics, 49, 561–576.
Kruskal, J. B. (1964). Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika, 29, 1–27.
Kyriacou, C. P., Peixoto, A. A., Sandrelli, F., Costa, R., & Tauber, E. (2008). Clines in clock genes: Fine-tuning circadian rhythms to the environment. Trends in Genetics, 24, 124–132.
Lazaridis, I., Patterson, N., Mittnik, A., Renaud, G., Mallick, S., Kirsanow, K., Sudmant, P. H., Schraiber, J. G., Castellano, S., Lipson, M., & Berger, B. (2014). Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature, 513, 409–413.
Legendre, P. (1993). Spatial autocorrelation: Trouble or new paradigm? Ecology, 74, 1659–1673.
Maes, G. E., & Volckaert, F. A. M. (2002). Clinal genetic variation and isolation by distance in the European eel Anguilla anguilla (L.). Biological Journal of the Linnean Society, 77, 509–521.
Malécot, G. (1973). Isolation by distance. In N. E. Morton (Ed.), Genetic structure of populations. University of Hawaii Press.
Mallet, J., Barton, N., Lamas, G., Santisteban, J., Muedas, M., & Eeley, H. (1990). Estimates of selection and gene flow from measures of cline width and linkage disequilibrium in Heliconius hybrid zones. Genetics, 124, 921–936.
Moran, P. A. P. (1950). Notes on continuous stochastic phenomena. Biometrika, 37, 17–23.
Moreira, A. S., Horgan, F. G., Murray, T. E., & Kakouli-Duarte, T. (2015). Population genetic structure of Bombus terrestris in Europe: Isolation and genetic differentiation of Irish and British populations. Molecular Ecology, 24, 3257–3268.
Pérez-Losada, J., & Fort, J. (2018). A serial founder effect model of phonemic diversity based on phonemic loss in low-density populations. PLoS ONE, 13, e0198346.
Razgour, O. (2015). Beyond species distribution modeling: A landscape genetics approach to investigating range shifts under future climate change. Ecological Informatics, 30, 250–256.
Reynolds, J., Weir, B. S., & Cockerman, C. C. (1983). Estimation of the coancestry coefficient: Bases for a short-term genetic distance. Genetics, 105, 767–779.
Rousset, F. (1997). Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics, 145, 1219–1228.
Roy, J., Blanckenhorn, W. U., & Rohner, P. T. (2018). Largely flat latitudinal life history clines in the dung fly Sepsis fulgens across Europe (Diptera: Sepsidae). Oecologia, 187, 851–862.
Sammon, J. W. (1969). A nonlinear mapping for data structure analysis. IEEE Transactions on Computers, 18, 401–409.
Slatkin, M. (1973). Gene flow and selection in a cline. Genetics, 75, 733–756.
Sokal, R. R., & Oden, N. L. (1978). Spatial autocorrelation in biology: 2. Some biological implications and four applications of evolutionary and ecological interest. Biological Journal of the Linnean Society, 10, 229–249.
Sotka, E. E., & Palumbi, S. R. (2006). The use of genetic clines to estimate dispersal distances of marine larvae. Ecology, 87, 1094–1103.
Weir, B. S., & Goudet, J. (2017). A unified characterization of population structure and relatedness. Genetics, 206, 2085–2103.
Wright, S. (1943). Isolation by distance. Genetics, 28, 114–138.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest in this paper.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peña, J.A., Gómez-Pérez, L. & Alfonso-Sánchez, M.A. On the Trail of Spatial Patterns of Genetic Variation. Evol Biol 49, 84–91 (2022). https://doi.org/10.1007/s11692-021-09552-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11692-021-09552-y