
Abstract
Wnt signaling is a key pathway that helps organize development of the nervous system. It influences cell proliferation, cell fate, and cell migration in the developing nervous system, as well as axon guidance, dendrite development, and synapse formation. Given this wide range of roles, dysregulation of Wnt signaling could have any number of deleterious effects on neural development and thereby contribute in many different ways to the pathogenesis of neurodevelopmental disorders. Some major psychiatric disorders, including schizophrenia, bipolar disorder, and autism spectrum disorders, are coming to be understood as subtle dysregulations of nervous system development, particularly of synapse formation and maintenance. This review will therefore touch on the importance of Wnt signaling to neurodevelopment generally, while focusing on accumulating evidence for a synaptic role of Wnt signaling. These observations will be discussed in the context of current understanding of the neurodevelopmental bases of major psychiatric diseases, spotlighting schizophrenia, bipolar disorder, and autism spectrum disorder. In short, this review will focus on the potential role of synapse formation and maintenance in major psychiatric disorders and summarize evidence that defective Wnt signaling could contribute to their pathogenesis via effects on these late neural differentiation processes.
Similar content being viewed by others


Wnt signaling pathways
The Wnt family of signaling molecules are small secreted glycoproteins best known for their role as critical regulators of cell fate specification, cell proliferation, and cell migration during development. There are 19 Wnt ligands made from different genetic loci in humans and a similar number in other mammals. Wnt signaling is mediated by Wnt ligands binding to the Frizzled (Fzd) family of receptors, the co-receptors low density lipoprotein receptor-related protein 5 and 6 (LRP5/6), or alternatively the Ryk and Ror receptor tyrosine kinases (Komiya and Habas 2008). Wnts can initiate several signaling pathways through activation of these different receptor types and complexes (Fig. 1).

Wnts signal through a “canonical” pathway involving β-catenin mediated transcriptional regulation (at left) and through “non-canonical” pathways involving calcium and Rac/JNK-mediated signaling (at right). In the absence of Wnt, the β-catenin destruction complex, whose core members are the scaffold proteins axin and adenomatous polyposis coli (APC) and the protein kinase glycogen synthase kinase 3β (GSK3β), binds and phosphorylates β-catenin. Phospho-β-catenin is then ubiquitinated and destroyed in the proteosome. Wnt binding to Fzd and LRP5/6 activates Disheveled, which breaks up the β-catenin destruction complex. This leads to accumulation of β-catenin in the cytosol and translocation of β-catenin into the nucleus, where it forms complexes with LEF/TCF transcription factors to promote transcription of selected gene targets. Wnt binding to Fzd can also trigger the Wnt/calcium signaling pathway involving calcium-dependent activation of CamK and PKC, or to the Wnt/PCP signaling pathway which leads to regulation of small GTPases and JNK. Wnt signaling is inhibited extracellularly by Dkk proteins, which specifically bind to LRP5/6 and thereby antagonize only Wnt/β-catenin signaling, or by secreted Frizzled-related proteins (SFRPs) and Wnt inhibitory factor (WIF), which bind Wnts extracellularly to antagonize all Wnt-activated signaling pathways
The so-called canonical Wnt/β-catenin signaling pathway involves Wnt binding to Fzd and LRP5/6, followed by Fzd binding to the scaffold protein Disheveled (dsh in fly, Dvl in vertebrates). Dvl then binds and destabilizes the β-catenin destruction complex, a group of proteins including glycogen synthase kinase 3 (GSK3) and the scaffold proteins axin and adenomatous polyposis coli (APC), among others. The constitutively functioning destruction complex binds β-catenin and targets it for poly-ubiquitination and degradation in the proteosome. When the β-catenin destruction complex is destabilized by Wnt activation and Dvl binding, β-catenin accumulates in the cytoplasm and in the nucleus, where it binds members of the lymphoid enhancer-binding factor/T cell-specific transcription factor (LEF/TCF) transcription factor family, preventing their repression of gene transcription and functioning as a transcription co-activator (Komiya and Habas 2008; Gao and Chen 2010). The Wnt/β-catenin signaling pathway has been intensely studied as a key regulator of cell proliferation and cell fate during development, including neural development (Machon et al. 2007; Zhou et al. 2004, 2006; Galercan et al. 2000).
There is also “non-canonical” β-catenin-independent signal transduction that occurs downstream of Wnts binding to Fzds and other receptors. In the Wnt/Ca++ signaling pathway, Wnt signaling through Fzd and Dvl initiates release of intracellular stores of calcium, driving calcium-dependent signaling in the cell through such targets as atypical protein kinase C and calcium/calmodulin-dependent protein kinase II (Kohn and Moon 2005). Wnt signaling also contributes to the planar cell polarity (PCP) signaling pathway. Like previously mentioned pathways, Wnt/PCP signaling depends on Fzd and Dvl, but with Rho GTPases and the Jun N-terminal kinase (JNK) as downstream targets rather than the β-catenin destruction complex (Gao and Chen 2010; Kohn and Moon 2005; Habas et al. 2003; Habas and He 2006). PCP signaling, like Wnt/β-catenin signaling, is a key pathway in organizing the developing organism, and many genes in both the Wnt/β-catenin and PCP signaling pathways were first discovered in Drosophila because mutations of these genes led to disorganization of body structures such as the eye and wing hairs (Vladar et al. 2009; Simons and Mlodzik 2008). PCP signaling in vertebrates is associated with organization of cilia in the inner ear, hair growth, and, most critically, with elongation of the body axis and formation of the neural tube through the process of convergent extension (Simons and Mlodzik 2008; Mlodzik 2002).
Wnt ligands are often subgrouped based on the downstream pathway they activate in vivo, which is presumably a function of their affinity for certain receptor complexes combined with the signaling competence of available target tissues. For example, Wnt1, Wnt2, Wnt3a, Wnt7a, and Wnt7b are typically considered activators of the β-catenin pathway, whereas Wnt4 and Wnt5a are typically considered activators of β-catenin-independent forms of signaling (Moon et al. 1997).
Wnts and nervous system development
In vertebrates, Wnts have well-established roles in brain development, dating back to the discovery that knockout of the Wnt1 gene in mice produces dramatic hypotrophy of the cerebellum and midbrain (Thomas and Capecchi 1990). This has since been discovered to reflect a general principle of Wnt signaling during brain development, in which an anteroposterior gradient (low Wnt signaling for anterior structures, high for posterior structures) is important for proper regional specification (Machon et al. 2007; Hoch et al. 2009; Patapoutian and Reichardt 2000). Wnt/β-catenin signaling is critical for specification of the hippocampus (Galercan et al. 2000; Lee et al. 2000) and for proliferation of neuronal precursors (Zhou et al. 2006), as well as for directing the radial migration of neurons into the cortex (Zhou et al. 2004). The roles that Wnt/β-catenin signaling plays in these neurodevelopmental processes and their potential and real implications for neurodevelopmental and/or psychiatric disorders are the subject of much investigation and have already been widely discussed in the scientific literature (Freese et al. 2010; De Ferrari and Moon 2006; Toro and Deakin 2006; Li et al. 2005; Mao et al. 2009) and will therefore not be a further focus of this review.
Wnt/PCP signaling is also tremendously important for nervous system development in vertebrates (Tissir and Goffinet 2010). The importance of PCP signaling is most notable in the process of convergent extension that produces the notochord and, eventually, the spine. During morphogenesis of these structures, cells must converge from the sides of the organism to the midline, and disruption of PCP signaling leads to a failure in this process. One common and clinically relevant consequence of disruptions in PCP signaling is failure of neural tube closure. In mild cases, the neural tube fails to close only at the lower end of the spinal cord producing spina bifida; in severe cases, the whole spine and even the skull may fail to close leaving the spinal cord and brain exposed, a lethal condition known as craniorachischisis. The family of Dvl proteins and the four-pass transmembrane protein Van Gogh-like 2 (Vangl2) are core components of the PCP signaling pathway; knockout of the Dvl genes causes craniorachischisis (Etheridge et al. 2008), as does the Looptail (Lp) allele of Vangl2 in mice homozygous for the mutation (Strong and Hollander 1947; Kibar et al. 2001). The molecular basis for these phenotypes downstream of the Vangl and Dvl proteins is complex but involves the family of Rho small GTPases and their regulation of actin dynamics and cytoskeletal organization. For example, Dvl1 has been shown to bind Rac directly and promote its activity (Rosso et al. 2005). Vangl2 can bind and redistribute Rac to adherens junctions, while disruption of Vangl2 signaling results in loss of Rho activity in the mouse Lp mutant (Phillips et al. 2009).
Wnts and axon guidance
Wnt signaling is important for axon development and guidance (Bovolenta et al. 2006; Zou 2004). Generally, Wnt–Fzd signaling has been found to mediate attractive cues (Lyuksyutova et al. 2003; Wang et al. 2006a) whereas Wnt–Ryk signaling mediates repulsive cues (Keeble et al. 2006; Liu et al. 2005) to extending axons. Specifically, Wnt4–Fzd3 signaling directs axonal projection of spinal cord neurons after crossing the midline (Lyuksyutova et al. 2003), while a Wnt5a–Ryk interaction helps direct axons routed through the corpus callosum (Keeble et al. 2006) and corticospinal tract (Liu et al. 2005). Interestingly, the relevant Wnt–Fzd pathway seems to involve β-catenin, but not the entire canonical Wnt pathway in which β-catenin drives gene transcription (Zou 2004).
In addition to providing guidance cues, Wnt signaling regulates cytoskeletal dynamics in the growth cone and during axon branching. Wnt7a has been shown to control growth cone spreading and axon branching in the cerebellum (Lucas and Salinas 1997; Hall et al. 2000), as does Wnt3a in spinal cord neurons (Krylova et al. 2002; Purro et al. 2008). It was also found that the effect of Wnt7a on axons could be mimicked by inhibition of GSK3β, suggesting that a downstream microtubule-regulating pathway is mediated by GSK3β activity (Lucas and Salinas 1997; Hall et al. 2000). Dvl1 is involved in this process, as shown by deficits in microtubule dynamics in Dvl1 knockout animals (Purro et al. 2008). Concordantly, a recent study identified complementary cell biological roles for Dvl1 and Vangl2 in regulating Fzd3 signaling downstream of Wnt5a during commissural growth cone guidance (Shafer et al. 2011).
Wnts and dendrite formation
Less is presently known about the role of Wnt signaling in dendrite development. Studies in cultured hippocampal neurons (HCNs) have shown that an increase in Wnt7b (Rosso et al. 2005) or in Wnt2 are both associated with increases in complexity of dendritic arbors and that Wnt2 transcription is upregulated by neuronal activity in this system (Wayman et al. 2006). The Ror tyrosine kinases promote dendrite branching (Paganoni et al. 2010), as does the scaffold protein Dvl1, both through Rac and JNK (Rosso et al. 2005), as well as through a β-catenin-dependent (but non-canonical) pathway (Yu and Malenka 2003). It should be noted that, as with the β-catenin pathway involved in axon remodeling, the β-catenin-mediated response reported in this study is not due to LEF/TCF transcription and seems to rely instead on regulation of cell adhesion through β-catenin’s interaction with adhesion molecules such as the cadherins.
Wnt signaling and the synapse
Wnt signaling is also implicated in synapse formation and maintenance. The first evidence that Wnt signaling contributes to synapse development came from a study implicating Wnt7a in axon extension and in clustering of presynaptic proteins in cerebellar granule neurons (Lucas and Salinas 1997). Wnt signaling has also been implicated in synapse formation at the neuromuscular junction (NMJ) in fruit flies (Wu et al. 2010) and in Caenorhabditis elegans (Davis and Ghosh 2007) and in the hippocampus and cerebellum of the mammalian CNS (Farias et al. 2010; Speese and Budnik 2007; Tang 2007).
Wnt signaling at the NMJ
Wg (the Drosophila homolog of Wnt1) has both pre- and postsynaptic effects at the Drosophila NMJ, where Wg is released in conjunction with neuronal activity. During anterograde signaling, Wg made in the presynaptic motor neuron travels across the synaptic cleft in association with lipid and a transmembrane protein called evenness interrupted (Korkut et al. 2009). It then binds to the Fzd2 receptor expressed by postsynaptic muscle cells, leading to internalization and cleavage of Fzd2, with the intracellular domain of the receptor transported into the muscle nucleus where it may directly regulate transcription (Mathew et al. 2005). Wg release also causes changes in the motor neuron itself in an autocrine manner by binding to presynaptic Fzd2. The machinery downstream of this includes Dvl and GSK3-mediated phosphorylation of microtubule associated proteins, with the net effect being stabilization of microtubules and increases in local axonal complexity (Ciani and Salinas 2007). A molecularly similar signal transduction mechanism may contribute to increases in presynaptic transmitter release and to synaptic potentiation upon repeated depolarization (Ataman et al. 2008).
In contrast, the Wnt lin-44 in C. elegans is a non-permissive signal for synapse formation in the posterior of the worm; null mutation of lin-44 or its receptor lin-17, a Fzd receptor, causes expansion of synapse formation into a region where that neuron’s synapses do not normally form, as does removal of dsh-1, the C. elegans Disheveled gene (Klassen and Shen 2007). Evidence therefore suggests that Wnts can provide either positive or negative signals for synapse formation at the NMJ, depending on the organism and cellular context.
Wnts and synapse formation in the mammalian CNS
The hippocampus has been intensely studied as a model for synapse formation in mammalian forebrain neurons because of its stereotyped anatomy and the relative ease of obtaining adequately pure populations of hippocampal pyramidal neurons for culture, in addition to its behavioral relevance in memory formation (Banker and Goslin 1998). Wnt3, Wnt5a, Wnt7a, and Wnt7b are all present in the mammalian hippocampus, although Wnt3 is not widely expressed (Rosso et al. 2005; Davis et al. 2008). The Wnt receptors Fzd3, Fzd5, Fzd8, and Fzd9 are also all expressed in the postnatal hippocampus (Davis et al. 2008; Zhou et al. 2010). These Wnts and Fzds are therefore natural candidates for considering the role of Wnt signaling at mammalian CNS synapses.
Current understanding of the role of the “canonical” Wnts—those associated with Wnt/β-catenin signaling—in synapse formation is straightforward. Available evidence concurs that Wnt3a, Wnt7a, and Wnt7b raise nuclear levels of β-catenin and promote organization of presynaptic sites in the cerebellum (Lucas and Salinas 1997; Ahmad-Annuar et al. 2006) as well as in HCNs (Davis et al. 2008).
Wnt signaling through Wnt7a/7b in mice has also been shown to be increased by exposure to a stimuli-enriched environment, with a corresponding increase in synaptic terminal complexity in CA3 hippocampal neurons receiving input from mossy fiber axons from the dentate gyrus (Gogolla et al. 2009). These changes, however, seem to be through some mechanism other than the full canonical Wnt/β-catenin signaling pathway because their signaling outcome, as far as is presently known, is not β-catenin-mediated transcriptional regulation. They have instead been linked to changes in microtubule organization (Hall et al. 2000), APC-mediated protein clustering (Farias et al. 2007), and β-catenin mediated cell–cell adhesion (Bamji et al. 2003), the last of which has not been shown to be triggered by Wnt/Fzd/Dvl signaling at all.
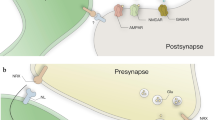
In contrast, there is contradictory evidence for the role of Wnt/PCP signaling mediated by Wnt5a in synapse formation. One study found that Wnt3, Wnt7a, or Wnt7b application all increased the number of presynaptic protein clusters in Wnt-treated neurons, whereas application of Wnt5a did not increase nuclear β-catenin and decreased the number of presynaptic protein clusters. Based on this and other evidence, the authors concluded that Wnt/β-catenin signaling is positive and Wnt/PCP signaling negative for synapse formation (Davis et al. 2008). In striking contrast, a set of studies from another group has found that application of Wnt5a increased clustering of both excitatory and inhibitory postsynaptic proteins through calcium and JNK-mediated pathways while not affecting presynaptic sites; the authors therefore concluded that Wnt/Ca2+ and Wnt/PCP signaling promote synapse formation (Farias et al. 2009; Varela-Nallar et al. 2010; Cuitino et al. 2010). Another study found that transfection of astrocytes with Wnt5a led to an increase in presynaptic puncta in co-cultured HCNs and that this increase was dependent on the non-canonical Wnt receptor Ror (Paganoni et al. 2010). The three sets of observations are not necessarily irreconcilable, as the experimental methods of each study vary at several points. If all published observations are correct, Wnt signaling through Wnt5a can either promote or inhibit synapse formation in vivo, depending on local conditions, and does so through both the Wnt/Ca2+ and Wnt/PCP pathways. Known connections between Wnt signaling and synapse formation in vertebrates are summarized in Fig. 2.

Wnts exert both pre- and post-synaptic effects. Presynaptic Wnt signaling through Wnt3 and Wnt7 acts through a Dvl pathway to stabilize microtubules and also influences presynaptic clustering of acetylcholine receptors (AChRs) through an APC-mediated mechanism (upper left). Presynaptic β-catenin is also involved in synapse formation and maintenance through cell–cell adhesion mechanisms, though a relationship between this and Wnt signaling remains speculative. Wnt5a-activated signaling through the Ror kinase is also thought to contribute to an increase in presynaptic sites through a mechanism that has not yet been determined (upper right). Postsynaptic Wnt signaling driven by Wnt5a probably acts through a Dvl/Rac/JNK pathway to regulate the actin cytoskeleton (lower right). Application of Wnt3a, Wnt 7a, or Wnt7b is sufficient to induce β-catenin translocation to the nucleus and may well also influence synapses (not shown). As in the presynaptic terminal, β-catenin is present in the postsynaptic density of excitatory synapses where it is involved in cell–cell adhesion. Also not shown is a highly divergent postsynaptic pathway established at the Drosophila neuromuscular junction involving cleavage and translocation of Fzd receptors to the nucleus; there is evidence that a mechanistically similar pathway operates at some vertebrate synapses
Wnt signaling transducers and synapse formation
Some key downstream components of Wnt signaling pathways have also been strongly implicated in synapse formation. For example, Fzd proteins have been linked to synapse formation in two quite disparate ways. First, Fzd2 has been shown to regulate postsynaptic transcription at the NMJ through a mechanism involving cleavage of Fzd2 and transport of a fraction of the cleaved protein to the nucleus (Mathew et al. 2005; Mosca and Schwarz 2010). Second, Fzd1 has been found to mediate Wnt3a signaling (Varela-Nallar et al. 2009) and Fzd5 to mediate Wnt7a signaling (Sahores et al. 2010) leading to increased synapse formation, in particular the organization of new presynaptic terminals, in cultured hippocampal neurons.
In Drosophila, the arrow Wg receptor that is homologous to mammalian LRP5/6 has also been implicated in synapse formation at the NMJ through a presynaptic pathway involving Wg, dsh, and shaggy/GSK3, but which is linked to regulation of microtubule dynamics rather than to β-catenin-dependent transcriptional regulation (Miech et al. 2008).
Several scaffold proteins associated with Wnt signaling, including APC, axin, Dvl1, and the Dvl-binding Wnt signaling modulator Dact1, have all been shown to function at the synapse as well. APC participates in Wnt-induced presynaptic clustering of nAChR receptors (Farias et al. 2007); axin localizes to the synapse and binds the synaptic scaffold molecule S-SCAM (Hirabayashi et al. 2004). Loss of the key Wnt signaling transductor Dvl1 has been shown to produce a loss of presynaptic sites in the cerebellum (Ahmad-Annuar et al. 2006) and a reduction in dendrite complexity in hippocampal neurons (Rosso et al. 2005). Loss of Dact1 has also been shown to result in a loss of dendrite complexity, as well as a loss of dendritic spines and excitatory (but not inhibitory) synapses in hippocampal neuron culture, a deficit traced to decreases in active Rac (Okerlund et al. 2010).
β-Catenin has been linked to synapse formation through a role in the clustering of presynaptic proteins (Bamji et al. 2003; Lee et al. 2008; Sun et al. 2009) and through regulation of cadherin-mediated adhesion (Arikkath and Reichardt 2008; Uchida et al. 1996; Benson and Tanaka 1998). It has also been shown to mediate transcription postsynaptically after NMDA receptor activation (Abe and Takeichi 2007). However, reduction of β-catenin levels in postnatal forebrain leads to only subtle behavioral changes in mice (Gould et al. 2008).
Wnts and the neurodevelopmental hypothesis of psychiatric disorders
Since Wnt signaling is important in organizing the developing brain, it is plausible that defects in Wnt signaling contribute to illnesses such as autism, bipolar disorder, and schizophrenia. Although our understanding of the biological basis for these psychiatric disorders remains limited, contemporary models for their pathophysiology generally posit neurodevelopmental origins (Harrison and Weinberger 2005; Bourgeron 2009; Guilmatre et al. 2009; Sutcliffe 2008). Accumulating evidence suggests that improper formation or regulation of synapses is an important contributor to these neurodevelopmental diseases, especially since they are associated with only mild gross anatomical defects (Guilmatre et al. 2009; Newey et al. 2004; von Bohlen und Halbach 2009; Penzes et al. 2011). In addition, numerous proteins contributing to Wnt signaling have been implicated in the etiology of these disorders through genetic, biochemical, and pharmacological analyses (De Ferrari and Moon 2006; Hur and Zhou 2010; Gould and Manji 2002; Kozlovsky et al. 2002).
Wnt signaling and schizophrenia
Schizophrenia is a psychiatric disorder characterized by “positive” symptoms such as delusions, hallucinations, and disorganized speech and “negative” symptoms such as lack of emotional affect and motivation (American Psychiatric Association 2000). Neuroanatomical defects associated with schizophrenia are subtle but include enlargement of the lateral ventricles (Steen et al. 2006), disorganization of forebrain structures (Harrison 1999), loss of neuropil, and fewer synapses in pyramidal neurons (Harrison and Weinberger 2005; Hill et al. 2006; Ross et al. 2006; Jarskog et al. 2007). The neurodevelopmental hypothesis of schizophrenia posits that such defects are caused by dysregulation of brain development (Harrison and Weinberger 2005; Guilmatre et al. 2009; Ross et al. 2006; Jarskog et al. 2007; Cantor and Geschwind 2008), and the Wnt signaling pathway is a candidate for such a dysregulated system. Since, as discussed above, Wnt signaling is important for brain regionalization, development of the hippocampus, neuronal proliferation, neuronal migration, and synapse formation, dysregulation of Wnt signaling could simultaneously produce a reduction in hippocampal volume, disorganization of forebrain structures (through incorrect specification, proliferation, and/or migration), and synaptic defects such as those seen in schizophrenia.
Schizophrenia has a strong genetic component (Sullivan et al. 2003). Although no Wnt ligands have been identified as genes of interest in genome-wide association studies (GWAS) or other linkage studies, Wnt1 has been reported to be upregulated in schizophrenic brains (Miyaoka et al. 1999), and some genes associated with susceptibility to the disease are core components of the Wnt signaling pathway. Perhaps most striking is a recent finding that the Wnt/β-catenin-activated transcription factor TCF4 is one of only three genes with a single nucleotide polymorphism (SNP) significantly correlated with schizophrenia at the level of the whole genome (Stefansson et al. 2009). The other genes identified in the study were in the major histocompatibility complex and neuregulin, a protein involved in neuronal migration and synaptic function which had previously been strongly linked to the genetics of schizophrenia (Buonanno 2010). TCF4 itself has been shown to have a role in brain development (Broźozka et al. 2010; Kunz et al. 2004; Cho and Dressler 1998), and TCF4 haploinsufficiency causes Pitt–Hopkins syndrome, which includes severe mental retardation as one of its features (Brockschmidt et al. 2007; de Pontual et al. 2009). TCF4 overexpression in the brain has also been reported to cause memory deficits and, importantly, deficits in prepulse inhibition, a neurophysiological correlate of schizophrenia and other psychiatric disorders (Broźozka et al. 2010).
Dkk proteins—extracellular proteins that compete with Wnts for binding to LRP5/6 receptors and thereby disrupt the Fzd-LRP5/6 complex that initiates Wnt/β-catenin signaling—have also been implicated in schizophrenia via several genetic screens. Dkk1, Dkk3, and Dkk4 have all been identified as genes of interest in genome analyses of mutations associated with schizophrenia (Aleksic et al. 2010; Proitsi et al. 2008; Ftouh et al. 2005), as has the Dkk1 co-receptor KREMEN1 (Aleksic et al. 2010). Intriguingly, the human Dkk1 homolog was first isolated via interaction with neuregulin (Fedi et al. 1999).
The role of the Dkk family members in brain development is still relatively undefined. Dkk1 is expressed only at very low levels in the healthy brain but is upregulated in response to neurodegeneration (Caraci et al. 2008). Dkk3 is the most strongly and widely expressed Dkk family member in the brain during development (Diep et al. 2004) and is upregulated in Alzheimer’s patients, possibly also as a response to neurodegeneration (Zenzmaier et al. 2009); female Dkk3 knockout mice were also observed to be slightly hyperactive (Barrantes et al. 2006). Dkk4 is not known to be strongly expressed in the brain at any time, but it is upregulated in some brain regions in response to elevated β-catenin levels during development (Diep et al. 2004). No specifically synaptic role for any of these three Dkk proteins has been identified, although application of exogenous Dkk1 has been found to reduce clustering of presynaptic proteins in hippocampal neuronal culture (Davis et al. 2008).
GSK3 is another key regulator of Wnt signaling with genetic and pharmacological ties to schizophrenia. Polymorphisms in the GSK3β locus have been linked to susceptibility for schizophrenia (Scassellati et al. 2004; Souza et al. 2008), and as described in the first section, regulation of GSK3β activity is central to the Wnt/β-catenin signaling pathway (Wu and Pan 2010). However, it is worth emphasizing that GSK3β, along with many other components of the Wnt signal transduction machinery, also plays prominent roles in several biochemical and signaling pathways independent of Wnts (Hur and Zhou 2010; Cohen and Frame 2001; Jope and Roh 2006). Therefore, an important general caveat in considering such evidence is that some of these non-Wnt related biochemical roles—not only for GSK3β but also for the other Wnt signaling components mentioned—may in the end be more relevant to psychiatric pathogenesis.
Other genes linked to schizophrenia that can also modulate Wnt signaling include Disrupted In SChizophrenia 1 (DISC1), a scaffold protein whose genetic locus is mutated in affected members of a family with a high incidence of schizophrenia (Millar et al. 2000). DISC1 helps regulate neuronal migration and synapse formation (Hayashi-Takagi et al. 2010; Singh et al. 2010) and has been linked to Wnt/β-catenin signaling both through interaction with GSK3β (Mao et al. 2009) as well as with the scaffold protein DIX Domain Containing 1 (DIXDC1) (Singh et al. 2010).
Also suggestive is that human chromosomal region 8p, genetically implicated in neuropsychiatric and other diseases (Harrison and Weinberger 2005; Tabares-Seisdedos and Rubenstein 2009), contains at least three Wnt-signaling-related genes (FZD3, DKK4, and SFRP1). FZD3 in particular has been put forward as a potential schizophrenia susceptibility locus (Katsu et al. 2003; Yang et al. 2003; Zhang et al. 2004), although successive genetic linkage studies have cast doubt on initial findings linking mutations in the gene to schizophrenia incidence in the general population (Ide et al. 2004; Wei and Hemmings 2004). Importantly, Fzd3 has roles in axon guidance (Lyuksyutova et al. 2003; Wang et al. 2006a; Shafer et al. 2011) and PCP signaling (Wang et al. 2006b; Shafer et al. 2011). The roles of Dkk4 (discussed above) and Sfrp1 in brain development have received relatively little analysis, but deletion of Sfrp1 has been shown to have no obvious effect on gross anatomy of the nervous system in mice (Trevant et al. 2008).
In addition to these genetic and other connections, antipsychotics such as haloperidol and clozapine have been reported to increase protein levels of β-catenin, GSK3β, and Dvl3 in the brains of adult rats, whereas amphetamine, which can induce psychosis, has the opposite effect (Alimohamad et al. 2005). Haloperidol has also been reported to reduce phospho-GSK3β (Kozlovsky et al. 2006), providing another biochemical link between GSK3 activity and schizophrenia pharmacotherapeutics. Interestingly, changes in Wnt signaling induced by antipsychotic administration in a cellular assay can be mimicked by overexpression of Dvl3 (Sutton et al. 2007). Taken together, the genetic and pharmacological data suggest a potential connection between Wnt signaling and the pathogenesis and therapeutics of schizophrenia.
Wnt signaling and autism
Autism is a childhood-onset psychiatric disorder characterized by impairment of social interaction, difficulty in communication, and perseverative stereotyped behaviors (American Psychiatric Association 2000). As with schizophrenia, a defect in synapse formation is a leading current hypothesis for its pathogenesis (Bourgeron 2009; Guilmatre et al. 2009; Sutcliffe 2008). The genes currently most tightly linked to autism causality, although only accounting in themselves for a small percentage of cases, include the neuroligin family of adhesion proteins and associated molecules which are important for synapse formation and maintenance (Bourgeron 2009; Guilmatre et al. 2009; Sutcliffe 2008; Südhof 2008; Chubykin et al. 2005). Autism is also associated with macrocephaly in early stages of development (McCaffery and Deutsch 2005), an increased number of neurons in the brain (Casanova et al. 2006), and misplaced or misoriented neurons due to incorrect neuronal migration, especially for Purkinje neurons (Wegiel et al. 2010; Palmen et al. 2004). Since Wnt signaling is an important factor in proliferation, cell fate specification, migration, and synaptogenesis, a defect in Wnt signaling could produce all of these defects, and it is reasonable to look for connections between deficits in Wnt signaling and autism.
There is some evidence for a direct genetic link between Wnt2 and autism spectrum disorders in rare cases. Two studies have found a correlation between mutation of the WNT2 locus and the incidence of autism in different populations (Marui et al. 2010; Wassink et al. 2001), although this finding has not been replicated by other studies (McCoy et al. 2002; Li et al. 2004). Wnt2 has also been found to be expressed at lower levels in a mouse model of fragile X syndrome than in wild-type mice (Zhang et al. 2009), and while typified by mental retardation, a significant fraction of patients with fragile X syndrome also have autistic symptoms.
No similar genetic links have yet been found with Wnt receptors, but Wnt signaling has also been related to autism through downstream signaling components and transcriptional targets. For example, Dvl1 knockout mice show behavioral impairments of potential relevance to autism, including loss of sociability (decreased huddling) and abnormal social interactions (reduced barbering of facial whiskers; Lijam et al. 1997); this is especially notable in light of the effects of Dvl1 knockout on synapses (Rosso et al. 2005; Ahmad-Annuar et al. 2006). Wnt signaling is also linked to autism through TSC1 and 2, scaffold proteins that are mutated in tuberous sclerosis (a developmental disorder that, like fragile X syndrome, often includes features of autism; Wiznitzer 2004). TSC1 and 2 regulate Wnt/β-catenin signaling through interaction with GSK3 and axin (Mak et al. 2003). The transcription factor Engrailed 2 (En2), a Wnt/β-catenin transcriptional target (Kuemerle et al. 2007), has been genetically linked to autism both through human genetics showing association of SNPs with the disease (Benayed et al. 2009; Yang et al. 2010; Sen et al. 2010) and through a mouse model showing that En2 knockout produces cerebellar patterning deficits similar to those seen in autism, as well as social behavioral deficits (Kuemerle et al. 2007; Cheh et al. 2006). Finally, fragile X mental retardation protein (FMRP), an RNA-binding protein strongly associated with autistic-like symptoms and mental retardation when mutated (Pfeiffer and Huber 2009; Bassell and Warren 2008), has recently been shown to play a role in Wnt signaling in a FMRP knockout mouse (Luo et al. 2010), offering another potential connection between Wnt signaling and autism. Interestingly, some, but not all, of the autism-relevant behavioral phenotypes observed in a FMR1 knockout mouse are also observed in a mouse model in which GSK3 is constitutively active (Mines et al. 2010).
Wnt signaling and bipolar disorder
Bipolar disorder, another psychiatric disorder potentially linked to changes in neural plasticity (Schloesser et al. 2008) and synapse formation (Eastwood and Harrison 2009), is characterized by phases of mania, in which the subject feels an abnormal surge of energy and heightened mood, sometimes leading to delusions and even psychosis, alternating with phases of depression, with accompanying loss of energy and negative mood (American Psychiatric Association 2000). Based on GWAS and linkage studies, there is no demonstrated genetic association between the genetics of bipolar disorder and genes for either the Wnt ligands themselves or any of their known receptors. However, as with schizophrenia, Wnt signaling is implicated in the pathogenesis and therapeutics of bipolar disorder, with the pharmacological link between Wnt/β-catenin signaling, GSK3 activity, and lithium being fairly provocative (Gould and Manji 2002).
Lithium has been used to treat bipolar disorder for decades and is still the most widely used treatment for the disorder (Hirschowitz et al. 2010). Among its other effects, including inhibition of inositol phosphatases (Beaulieu and Caron 2008; Quiroz et al. 2004), lithium inhibits GSK3 and promotes β-catenin-mediated transcriptional activation at therapeutic doses (Hedgepeth et al. 1997; Klein and Melton 1996). Intriguingly, mice missing one copy of GSK3β show reduced immobility in the forced swim test, a behavioral measure of antidepressant effects, as do mice dosed with lithium at therapeutic concentrations (O’Brien et al. 2004). GSK3β and Wnt/β-catenin signaling have therefore been of interest in bipolar disorder, and human genetics has revealed that copy number variation in the 3′ non-coding region of the GSK3β locus is more frequent in bipolar patients than in controls (Lachman et al. 2007). In a meta-analysis of all candidates, the GSK3β locus was found to be implicated in bipolar disorder by the widest array of studies, including association analysis, gene expression, pharmacogenomics, structural variants, and mouse models (Luykx et al. 2009).
The simplest pharmaceutical model based on these findings would be one in which administration of lithium corrects an underlying GSK3 hyperactivity associated with bipolar pathogenesis. Since in the absence of a Wnt ligand GSK3 activity promotes β-catenin degradation and inhibits the pathway, one might predict that there should be abnormal downregulation of β-catenin-mediated transcriptional targets in untreated bipolar patients. Paradoxically, available evidence suggests instead that there is an upregulation of β-catenin transcriptional targets in bipolar patients (Matigian et al. 2007; Zandi et al. 2008). In this regard, it is important that GSK3 operates as a switch; while inhibiting the pathway in the absence of a signal, it also participates in Wnt/β-catenin signal pathway activation by phosphorylating the LRP5/6 receptor (Zeng et al. 2005) and transcriptional co-activators (Scassellati et al. 2004) in the presence of a signal. These signal-promoting activities of GSK3 might therefore be more relevant to the pathophysiology of bipolar disorder.
Wnt signaling in psychiatry: “to β or not to β”
Although β-catenin-dependent and β-catenin-independent Wnt signaling are both implicated in neural development, studies of Wnt signaling in disease have historically centered mostly on the canonical Wnt/β-catenin signaling pathway, the first described and molecularly best-understood Wnt pathway, whereas other types of Wnt signaling have generally been less thoroughly interrogated for connections to disease in general and to mental illness in particular. Given structural alterations found in major psychiatric disorders including modest changes in the sizes, shapes, cellular architecture, and differential growth characteristics of some brain regions, it remains reasonable to explore potential contributions of Wnt/β-catenin signaling to psychiatric pathogenesis. Simultaneously, given the importance of cytoskeletal regulation and calcium release in synapse formation and function (Luo 2002; Koh 2006; Nakayama et al. 2000), potential connections between Rho GTPases and mental illness (Newey et al. 2004), the sensitivity of Wnt/PCP signaling to dosage effects including those caused by genetic perturbations in mammalian animal models (Etheridge et al. 2008; Wang et al. 2006c; Suriben et al. 2009), and evidence that signal transduction mechanisms at synapses downstream of Wnts are highly divergent from previously described pathways, it may be that Wnt/PCP, Wnt/Ca++, and other “non-canonical” forms of Wnt signaling are as important or more important than Wnt/β-catenin signaling in the pathogenesis of psychiatric disorders. These pathways therefore deserve further scrutiny in studies of neural differentiation, synapse maintenance, synaptic plasticity, and their consequences for complex behavior and the etiology of major mental illnesses.
References
Abe K, Takeichi M. NMDA-receptor activation induces calpain-mediated b-catenin cleavages for triggering gene expression. Neuron. 2007;53:387–97.
Ahmad-Annuar A, Ciani L, Simeonidis I, Herreros J, Fredj NB, Rosso SB, et al. Signaling across the synapse: a role for Wnt and Dishevelled in presynaptic assembly and neurotransmitter release. J Cell Biol. 2006;174(1):127–39.
Aleksic B, Kushima I, Ito Y, Nakamura Y, Ujike H, Suzuki M, et al. Genetic association study of KREMEN1 and DKK1 and schizophrenia in a Japanese population. Schizophr Res. 2010;118:113–7.
Alimohamad H, Sutton LP, Mouyal J, Rajakumar N, Rushlow WJ. The effects of antipsychotics on β-catenin, glycogen synthase kinase-3, and Dishevelled in the ventral midbrain of rats. J Neurochem. 2005;95:513–25.
Arikkath J, Reichardt LF. Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends Neurosci. 2008;31(9):487–94.
Ataman B, Ashley J, Gorczyca M, Ramachandran P, Fouquet W, Sigrist SJ, et al. Rapid activity-dependent modifications in synaptic structure and function require bidirectional Wnt signaling. Neuron. 2008;57:705–18.
Bamji SX, Shimazu K, Kimes N, Huelsken J, Birchmeier W, Lu B, et al. Role of β-catenin in synaptic vesicle localization and presynaptic assembly. Neuron. 2003;40:719–31.
Banker G, Goslin K, editors. Culturing nerve cells. 2nd ed. Cambridge: MIT; 1998.
Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–14.
Beaulieu J-M, Caron MG. Looking at lithium: molecular moods and complex behavior. Mol Interv. 2008;8(5):230–41.
Benayed R, Choi J, Matteson PG, Gharani N, Kamdar S, Brzustowicz LM, et al. Autism-associated haplotype affects the regulation of the homeobox gene, Engrailed 2. Biol Psychiatry. 2009;66:911–7.
Benson DL, Tanaka H. N-cadherin redistribution during synaptogenesis in hippocampal neurons. J Neurosci. 1998;18(17):6892–904.
Bourgeron T. A synaptic trek to autism. Curr Opin Neurobiol. 2009;19:231–4.
Bovolenta P, Rodriguez J, Esteve P. Frizzled/Ryk mediated signaling in axon guidance. Development. 2006;133:4399–408.
Brockschmidt A, Todt U, Ryu S, Hoischen A, Landwehr C, Birnbaum S, et al. Severe mental retardation with breathing abnormalities (Pitt–Hopkins syndrome) is caused by haploinsufficiency of the neuronal bHLH transcription factor TCF4. Hum Mol Genet. 2007;16(12):1488–94.
Broźozka MM, Radyushkin K, Wichert SP, Ehrenreich H, Rossner MJ. Cognitive and sensorimotor gating impairments in transgenic mice overexpressing the schizophrenia susceptibility gene Tcf4 in the brain. Biol Psychiatry. 2010;68:33–40.
Buonanno A. The neuregulin signaling pathway and schizophrenia: from genes to synapses and neural circuits. Brain Res Bull. 2010;83:122–31.
Cantor RM, Geschwind DH. Schizophrenia: genome, interrupted. Neuron. 2008;58:165–7.
Caraci F, Busceti C, Biagioni F, Aronica E, Mastroiacovo F, Cappucio I, et al. The Wnt antagonist, Dickkopf-1, as a target for the treatment of neurodegenerative disorders. Neurochem Research. 2008;33:2401–6.
Casanova MF, Van Kooten IAJ, Switala AE, Van Engeland H, Heinsen H, Steinbusch HWM, et al. Minicolumnar abnormalities in autism. Acta Neuropathol. 2006;112:287–303.
Cheh MA, Millonig JH, Roselli LM, Ming X, Jacobsen E, Kamdar S, et al. En2 knockout mice display neurobehavioral and neurochemical alterations relevant to autism spectrum disorder. Brain Res. 2006;1116:166–76.
Cho EA, Dressler GR. TCF-4 binds β-catenin and is expressed in distinct regions of the embryonic brain and limbs. Mech Dev. 1998;77:9–18.
Chubykin AA, Liu X, Comoletti D, Tsigelny I, Taylor P, Südhof TC. Dissection of synapse induction by neuroligins: effect of a neuroligin mutation associated with autism. J Biol Chem. 2005;280(23):22365–74.
Ciani L, Salinas PC. c-Jun N-terminal kinase (JNK) cooperates with GSK3b to regulate Dishevelled-mediated microtubule stability. BMC Cell Biol. 2007;8:27.
Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–76.
Cuitino L, Godoy JA, Farias GG, Couve A, Bonansco C, Fuenzalida M, et al. Wnt-5a modulates recycling of functional GABAA receptors on hippocampal neurons. J Neurosci. 2010;30:8411–20.
Davis E, Ghosh A. Should I stay or should I go: Wnt signals at the synapse. Cell. 2007;130:593–6.
Davis E, Zou Y, Ghosh A. Wnts acting through canonical and non-canonical signaling pathways exert opposite effects on hippocampal synapse formation. Neural Dev. 2008;3:17.
De Ferrari GV, Moon RT. The ups and downs of Wnt signaling in prevalent neurological disorders. Oncogene. 2006;25:7545–53.
de Pontual L, Mathieu Y, Glozio C, Rio M, Malan V, Boddaert N, et al. Mutational, functional, and expression studies of the TCF4 gene in Pitt–Hopkins syndrome. Hum Mutat. 2009;30(4):669–76.
Barrantes Idel B, Montero-Pedrazuela A, Guadaño-Ferraz A, Obregon M-J, de Mena RM, Gailus-Durner V, et al. Generation and characterization of dickkopf3 mutant mice. Mol Cell Biol. 2006;26(6):2317–26.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th ed. Washington, DC: American Psychiatric Association; 2000.
Diep DB, Hoen N, Backman M, Machon O, Krauss S. Characterization of the Wnt antagonists and their response to conditionally activate Wnt signaling in the developing mouse forebrain. Dev Brain Research. 2004;153:261–70.
Eastwood SL, Harrison PJ. Markers of glutamate synaptic transmission and plasticity are increased in the anterior cingulate cortex in bipolar disorder. Biol Psychiatry. 2009;67:1010–6.
Etheridge SL, Ray S, Li S, Hamblet NS, Lijam N, Tsang M, et al. Murine Dishevelled 3 functions in redundant pathways with Dishevelled 1 and 2 in normal cardiac outflow tract, cochlea, and neural tube development. PLoS Gen. 2008;4(11):e1000259.
Farias GG, Valles AS, Colombres M, Godoy JA, Toledo EM, Lukas RJ, et al. Wnt-7a induces presynaptic colocalization of α7-nicotinic acetylcholine receptors and adenomatous polyposis coli in hippocampal neurons. J Neurosci. 2007;27(20):5313–25.
Farias GG, Alfaro I, Cerpa W, Grabowski CP, Godoy JA, Bonansco C, et al. Wnt-5a/JNK signaling promotes the clustering of PSD-95 in hippocampal neurons. J Biol Chem. 2009;284:15857–66.
Farias GG, Godoy JA, Cerpa W, Varela-Nallar L, Inestrosa NC. Wnt signaling modulates pre- and postsynaptic maturation: therapeutic considerations. Dev Dyn. 2010;239:94–101.
Fedi P, Bafico A, Soria AN, Burgess WH, Miki T, Bottaro DP, et al. Isolation and biochemical characterization of the human Dkk-1 homologue, a novel inhibitor of the mammalian Wnt signaling. J Biol Chem. 1999;274(27):19465–72.
Freese JL, Pino D, Pleasure SJ. Wnt signaling in development and disease. Neurobiol Dis. 2010;38:148–53.
Ftouh S, Akbar MT, Hirsch SR, de Belleroche JS. Down-regulation of Dickkopf 3, a regulator of the Wnt signalling pathway, in elderly schizophrenic subjects. J Neurochem. 2005;94:520–30.
Galercan J, Miyashita-Lin EM, Devaney E, Rubenstein JL, Grosschedl R. Hippocampus development and generation of dentate gyrus granule cells is regulated by Lef1. Development. 2000;127:469–82.
Gao C, Chen Y-G. Dishevelled: the hub of Wnt signaling. Cell Signal. 2010;22:717–27.
Gogolla N, Galimberti I, Deguchi Y, Caroni P. Wnt signaling mediates experience-related regulation of synapse numbers and mossy fiber connectivities in the adult hippocampus. Neuron. 2009;62:510–25.
Gould TD, Manji HK. The Wnt signaling pathway in bipolar disorder. Neuroscientist. 2002;8:497–511.
Gould TD, O’Donnell KC, Picchini AM, Dow ER, Chen G, Manji HK. Generation and behavioral characterization of β-catenin mice forebrain-specific conditional knock-out mice. Behav Brain Research. 2008;189:117–25.
Guilmatre A, Dubourg C, Mosca A-L, Legallic S, Goldenberg A, Drouin-Garraud V, et al. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry. 2009;66(9):947–56.
Habas R, He X. Activation of Rho and Rac by Wnt/Frizzled signaling. Meth Enzymol. 2006;406(38):500–11.
Habas R, Dawid IB, He X. Co-activatin of Rac and Rho by Wnt/Frizzled signaling is required for vertebrate gastrulation. Genes Dev. 2003;17:295–309.
Hall AC, Lucas FR, Salinas PC. Axonal remodeling and synaptic differentiation in the cerebellum is regulated by Wnt-7a. Cell. 2000;100:525–35.
Harrison PJ. The neuropathology of schizophrenia: a critical review of the data and their interpretation. Brain. 1999;122:593–624.
Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68.
Hayashi-Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H, Dunlop AJ, et al. Disrupted-in-schizophrenia 1 (DISC1) regulates spines of the glutamate synapse through Rac1. Nat Neurosci. 2010;13:327–32.
Hedgepeth CM, Conrad LJ, Zhang J, Huang H-C, Lee VMY, Klein PS. Activation of the Wnt signaling pathway: a molecular mechanism for lithium action. Dev Biol. 1997;185:82–91.
Hill JJ, Hashimoto T, Lewis DA. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2006;11:557–66.
Hirabayashi S, Nishmura W, Iida J, Kansaku A, Kishida S, Kikuchi A, et al. Synaptic scaffolding molecule interacts with axin. J Neurochem. 2004;90:332–9.
Hirschowitz J, Kolevzon A, Garakani A. The pharmacological treatment of bipolar disorder: the question of modern advances. Harv Rev Psychiatry. 2010;18(5):266–78.
Hoch RV, Rubenstein JL, Pleasure S. Genes and signaling events that establish regional pattering of the mammalian forebrain. Semin Cell Dev Biol. 2009;20:378–86.
Hur E-M, Zhou F-Q. GSK3 signalling in neural development. Nat Rev Neurosci. 2010;11:539–51.
Ide M, Muratake T, Yamada K, Iwayama-Shigeno Y, Iwamoto K, Takao H, et al. Genetic and expression analyses of FZD3 in schizophrenia. Biol Psychiatry. 2004;56(6):462–5.
Jarskog LF, Miyamoto S, Lieberman JA. Schizophrenia: new pathological insights and therapies. Annu Rev Med. 2007;58:49–61.
Jope RS, Roh M-S. Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr Drug Targets. 2006;7(11):1421–34.
Katsu T, Ujike H, Nakano T, Tanaka Y, Nomura A, Nakata K, et al. The human frizzled-3 (FZD3) gene on chromosome 8p21, a receptor gene for Wnt ligands, is associated with the susceptibility to schizophrenia. Neurosci Lett. 2003;353(1):53–6.
Keeble TR, Halford MM, Seaman C, Kee N, Macheda M, Anderson RB, et al. The Wnt receptor Ryk is required for Wnt5a-mediated axon guidance on the contralateral side of the corpus callosum. J Neurosci. 2006;26(21):5840–8.
Kibar Z, Vogan KJ, Groulx N, Justice MJ, Underhill DA, Gros P. Ltap, a mammalian homolog of Drosophila strabismus/Van Gogh, is altered in the mouse neural tube mutant loop-tail. Nat Genet. 2001;28(3):251–5.
Klassen MP, Shen K. Wnt signaling positions neuromuscular connectivity by inhibiting synapse formation in C. elegans. Cell. 2007;130:704–16.
Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci. 1996;93:8455–9.
Koh C-G. Rho GTPases and their regulators in neuronal functions and development. Neurosignals. 2006;15:228–37.
Kohn AD, Moon RT. Wnt and calcium signaling: b-catenin independent pathways. Cell Calcium. 2005;38:439–46.
Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. 2008;4(2):68–75.
Korkut C, Ataman B, Ramachandran P, Ashley J, Barria R, Gherbesi N, et al. Trans-synaptic transmission of vesicular Wnt signals through Evi/Wntless. Cell. 2009;139:393–404.
Kozlovsky N, Belmaker RH, Agam G. GSK-3 and the neurodevelopmental hypothesis of schizophrenia. Eur Neuropsychopharmacol. 2002;12:13–25.
Kozlovsky N, Amar S, Belmaker RH, Agam G. Psychotropic drugs affect Ser9-phosphorylated GSK-3 beta protein levels in rodent frontal cortex. Int J Neuropsychopharmacol. 2006;9(3):337–42.
Krylova O, Herreros J, Cleverley KE, Ehler E, Henriquez JP, Hughes SM, et al. Wnt-3, expressed by motoneurons, regulates terminal arborization of neurotrophin-3 responsive spinal sensory neurons. Neuron. 2002;35:1043–56.
Kuemerle B, Gulden F, Cherosky N, Williams E, Herrup K. The mouse Engrailed genes: a window into autism. Behavioral Brain Research. 2007;176(1):121–32.
Kunz M, Herrmann M, Wedlich D, Gradl D. Autoregulation of canonical Wnt signals controls midbrain development. Dev Biol. 2004;273:390–401.
Lachman HM, Pedrosa E, Petruolo OA, Cockerham M, Papolos A, Novak T, et al. Increase in GSK3β gene copy number variation in bipolar disorder. Am J Med Genet. 2007;144B:259–65.
Lee SMK, Tole S, Grove E, McMahon AP. A local Wnt-3a signal is required for development of the mammalian hippocampus. Development. 2000;127:457–67.
Lee S-H, Peng I-F, Ng YG, Yanagisawa M, Bamji SX, Elia LP, et al. Synapses are regulated by the cytoplasmic tyrosine kinase Fer in a pathway mediated by p120 catenin, Fer, Shp-2, and β-catenin. J Cell Biol. 2008;183(5):893–908.
Li J, Nguyen L, Gleason C, Lotspeich L, Spiker D, Risch N, et al. Lack of evidence for an association between WNT2 and RELN polymorphism and autism. Am J Med Genet. 2004;126B:51–7.
Li F, Chong ZZ, Maiese K. Vital elements of the Wnt–Frizzled signaling pathway in the nervous system. Curr Neurovasc Res. 2005;2(4):331–40.
Lijam N, Paylor R, McDonald MP, Crawley JN, Deng C-X, Herrup K, et al. Social interaction and sensorimotor gating abnormalities in mice lacking Dvl1. Cell. 1997;20:895–905.
Liu Y, Shi J, Lu C-C, Wang Z-B, Lyuksyutova AI, Song X-J, et al. Ryk-mediated Wnt repulsion regulates posterior-directed growth of corticospinal tract. Nat Neurosci. 2005;8(9):1151–9.
Lucas FR, Salinas PC. WNT-7a induces axonal remodeling and increases synapsin I levels in cerebellar neurons. Dev Biol. 1997;192:31–44.
Luo L. Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu Rev Cell Dev Biol. 2002;18:601–35.
Luo Y, Shan G, Guo W, Smrt RD, Johnson EB, Li X, et al. Fragile X mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 2010;6:e1000898.
Luykx JJ, Boks MPM, Terwindt APR, Bakker S, Kahn RS, Ophoff RA. The involvement of GSK3b in bipolar disorder: integrating evidence from multiple types of genetic studies. Eur Neuropsychopharmacol. 2009;20:357–68.
Lyuksyutova AI, Lu C-C, Milanesio N, King LA, Guo N, Wang Y, et al. Anterior-posterior guidance of commissural axons by Wnt–Frizzled signaling. Science. 2003;302:1984–8.
Machon O, Backman M, Machonova O, Kozmik Z, Vacik T, Andersen L, et al. A dynamic gradient of Wnt signaling controls initiation of neurogenesis in the mammalian cortex and cellular specification in the hippocampus. Dev Biol. 2007;311:223–37.
Mak BC, Takemaru K-I, Kenerson HL, Moon RT, Young RS. The tuberin–hamartin complex negatively regulates b-catenin signaling activity. J Biol Chem. 2003;278(8):5947–51.
Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, et al. DISC1 regulates neural progenitor proliferation via modulation of GSK3β/β-catenin signaling. Cell. 2009;136(6):1017–31.
Marui T, Funatogawa I, Koishi S, Yamamoto K, Matsumoto H, Hashimoto O, et al. Association between autism and variants in the wingless-type MMTV integration site family member 2 (WNT2) gene. Int J Neuropsychopharmacol. 2010;13:443–9.
Mathew D, Ataman B, Chen J, Zhang Y, Cumberledge S, Budnik V. Wingless signaling at synapses is through cleavage and nuclear import of receptor DFrizzled2. Science. 2005;310:1344–7.
Matigian N, Windus L, Smith H, Filippich C, Pantelis C, McGrath J, et al. Expression profiling in monozygotic twins discordant for bipolar disorder reveals dysregulation of the WNT signalling pathway. Mol Psychiatry. 2007;12:815–25.
McCaffery P, Deutsch CK. Macrocephaly and the control of brain growth in autistic disorders. Prog Neurobiol. 2005;77:38–56.
McCoy PA, Shao Y, Wolpert CM, Donnelly SL, Ashley-Koch A, Abel HL, et al. No association between the WNT2 gene and autistic disorder. Am J Med Genet. 2002;114:106–9.
Miech C, Pauer H-U, He X, Schwarz TL. Presynaptic local signaling by a canonical wingless pathway regulates development of the Drosophila neuromuscular junction. J Neurosci. 2008;28:10875–84.
Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CAM, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9(9):1415–23.
Mines MA, Yuskaitis CJ, King MK, Beurel E, Jope RS. GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS One. 2010;5(3):e9706.
Miyaoka T, Seno H, Ishino H. Increased expression of Wnt-1 in schizophrenic brains. Schizophr Res. 1999;38:1–6.
Mlodzik M. Planar cell polarization: do the same mechanisms govern Drosophila tissue polarity and vertebrate gastrulation? Trends Genet. 2002;18(11):564–71.
Moon RT, Brown JD, Torres M. WNTs modulate cell fate and behavior during vertebrate development. Trends Genet. 1997;13(4):157–62.
Mosca TJ, Schwarz TL. The nuclear import of Frizzled2-C by importins-β11 and α2 promotes postsynaptic development. Nat Neurosci. 2010;13:935–45.
Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20(14):5329–38.
Newey SE, Velamoor V, Govek E-E, Van Aelst L. Rho GTPases, dendritic structure, and mental retardation. J Neurobiol. 2004;64:58–74.
O’Brien WT, Harper AD, Jove F, Woodgett JR, Maretto S, Piccolo S, et al. Glycogen synthase kinase-3β haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci. 2004;24(30):6791–8.
Okerlund ND, Kivimae S, Tong CK, Peng I-F, Ullian EM, Cheyette BNR. Dact1 is a postsynaptic protein required for dendrite, spine, and excitatory synapse development in the mouse forebrain. J Neurosci. 2010;30(12):4362–8.
Paganoni S, Bernstein J, Ferreira A. Ror1–Ror2 complexes modulate synapse formation in hippocampal neurons. Neuroscience. 2010;165:1261–74.
Palmen SJMC, Van Engeland H, Hof PR, Schmitz C. Neuropathological findings in autism. Brain. 2004;127:2572–83.
Patapoutian A, Reichardt LF. Roles of Wnt proteins in neural development and maintenance. Curr Opin Neurobiol. 2000;10:392–9.
Penzes P, Cahill ME, Jones KA, Van Leeuwen J-E, Woolfrey KM. Dendrite spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14(3):285–93.
Pfeiffer BE, Huber KM. The state of synapses in fragile X syndrome. Neuroscientist. 2009;15:549–67.
Phillips HM, Murdoch JN, Chaudhry B, Copp AJ, Henderson DJ. Vangl2 acts via RhoA signaling to regulate polarized cell movement during development of the proximal outflow tract. Circ Res. 2009;96:292–9.
Proitsi P, Li T, Hamilton G, Di Forti M, Collier D, Killick R, et al. Positional pathway screen of Wnt signaling genes in schizophrenia: association with Dkk4. Biol Psychiatry. 2008;63:13–6.
Purro SA, Ciani L, Hoyos-Flight M, Stamatakou E, Siomou E, Salinas PC. Wnt regulates axon behavior through changes in microtubule growth directionality: a new role for adenomatous polyposis coli. J Neurosci. 2008;28(34):8644–54.
Quiroz JA, Gould TD, Manji HK. Molecular effects of lithium. Mol Interv. 2004;4(5):259–72.
Ross CA, Margolis RL, Reading SAJ, Pletnikov M, Coyle JT. Neurobiology of schizophrenia. Neuron. 2006;52:139–53.
Rosso SB, Sussman DJ, Wynshaw-Boris A, Salinas PC. Wnt signalling through Dishevelled, Rac and JNK regulates dendritic development. Nat Neurosci. 2005;8(1):34–42.
Sahores M, Gibb A, Salinas PC. Frizzled-5, a receptor for the synaptic organizer Wnt7a, regulates activity-mediated synaptogenesis. Development. 2010;137:2215–25.
Scassellati C, Bonvicini C, Perez J, Bocchio-Chiavetto L, Tura GB, Rossi G, et al. Association study of −1727 A/T, −50 C/T and (CAA)n repeat GSK-3β gene polymorphisms with schizophrenia. Neuropsychobiology. 2004;50:16–20.
Schloesser RJ, Huang J, Klein PS, Manji HK. Cellular plasticity cascades in the pathophysiology and treatment of bipolar disorder. Neuropsychopharmacology. 2008;33:110–33.
Sen B, Singh AS, Sinha S, Chatterjee A, Ahmed S, Ghosh S, et al. Family-based studies indicate association of Engrailed 2 with autism in an Indian population. Genes Brain Behav. 2010;9:248–55.
Shafer B, Onishi K, Lo C, Colakoglu G, Zou Y. Vangl2 promotes Wnt/planar cell polarity-like signaling by antagonizing Dvl1-mediated feedback inhibition in growth cone guidance. Dev Cell. 2011;20(2):177–91.
Simons M, Mlodzik M. Planar cell polarity signaling: from fly development to human disease. Annu Rev Genet. 2008;42:517–40.
Singh KK, Ge X, Mao Y, Drane L, Meletis K, Samuels BA, et al. Dixdc1 is a critical regulator of DISC1 and embryonic cortical development. Neuron. 2010;67:33–48.
Souza RP, Romano-Silva MA, Lieberman JA, Meltzer HY, Wong AHC, Kennedy JL. Association study of GSK3 gene polymorphisms with schizophrenia and clozapine response. Psychopharmacology. 2008;200:177–86.
Speese SD, Budnik V. Wnts: up-and-coming at the synapse. Trends Neurosci. 2007;30(6):268–75.
Steen RG, Mull C, McClure R, Hamer RM, Lieberman JA. Brain volume in first-episode schizophrenia: systematic review and meta-analysis of magnetic resonance imaging studies. Br J Psychiatry. 2006;188:510–8.
Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–8.
Strong LC, Hollander WF. Hereditary loop-tail in the house mouse. J Hered. 1947;40:329–34.
Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455(7215):903–11.
Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait. Arch Gen Psychiatry. 2003;60:1187–92.
Sun Y, Aiga M, Yosida E, Humbert PO, Bamji SX. Scribble interacts with beta-catenin to localize synaptic vesicles to synapses. Mol Biol Cell. 2009;20:3390–400.
Suriben R, Kivimae S, Fisher DA, Moon RT, Cheyette BNR. Posterior malformations in Dact1 mutant mice arise through misregulated Vangl2 at the primitive streak. Nat Genet. 2009;41(9):977–85.
Sutcliffe JS. Insights into the pathogenesis of autism. Science. 2008;321:208–9.
Sutton LP, Honardoust D, Mouyal J, Rajakumar N, Rushlow WJ. Activation of the canonical Wnt pathway by the antipsychotics haloperidol and clozapine involves Dishevelled-3. J Neurochem. 2007;102:153–69.
Tabares-Seisdedos R, Rubenstein JL. Chromosome 8p as a potential hub for developmental neuropsychiatric disorders: implications for schizophrenia, autism and cancer. Mol Psychiatry. 2009;14(6):563–89.
Tang S-J. The synaptic Wnt signaling hypothesis. Synapse. 2007;61:866–8.
Thomas KR, Capecchi MR. Targeted disruption of the murine int-1 proto-oncogene resulting in severe abnormalities in midbrain and cerebellar development. Nature. 1990;346:847–50.
Tissir F, Goffinet AM. Planar cell polarity signaling in neural development. Curr Opin Neurobiol. 2010;20:1–6.
Toro CT, Deakin JFW. Adult neurogenesis and schizophrenia: a window on abnormal early brain development? Schizophr Res. 2006;90:1–14.
Trevant B, Gaur T, Hussain S, Symons J, Komm BS, Bodine PVN, et al. Expression of secreted Frizzled related protein 1 a Wnt antagonist, in brain, kidney, and skeleton is dispensable for normal embryonic development. J Cell Physiol. 2008;217:113–1226.
Uchida N, Honjo Y, Johnson KR, Wheolock MJ, Masatoshi T. The catenin/cadherin adhesion system is localized in synaptic junctions bordering transmitter release zones. J Cell Biol. 1996;135:767–79.
Varela-Nallar L, Grabowski CP, Alfaro IE, Alvarez AR, Inestrosa NC. Role of the Wnt receptor Frizzled-1 in presynaptic differentiation and function. Neural Dev. 2009;4:41–55.
Varela-Nallar L, Alfaro IE, Serrano FG, Parodi J, Inestrosa NC. Wingless-type family member 5a (Wnt-5a) stimulates synaptic differentiation and function of glutamatergic synapses. Proc Natl Acad Sci. 2010;107(49):21164–9.
Vladar EK, Antic D, Axelrod JD. Planar cell polarity signaling: the developing cell’s compass. Cold Spring Harb Perspect Biology. 2009;1:a002964.
von Bohlen und Halbach O. Structure and function of dendritic spines within the hippocampus. Ann Anat. 2009;191:518–31.
Wang Y, Zhang J, Mori S, Nathans J. Axonal growth and guidance defects in Frizzled3 knock-out mice: a comparison of diffusion tensor magnetic resonance imaging, neurofilament staining, and genetically directed cell labeling. J Neurosci. 2006a;26(2):355–64.
Wang Y, Guo N, Nathans J. The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J Neurosci. 2006b;26:2147–56.
Wang J, Hamblet NS, Mark S, Dickinson ME, Brinkman BC, Segil N, et al. Dishevelled genes mediate a conserved mammalian PCP pathway to regulate convergent extension during neurulation. Development. 2006c;133:1767–78.
Wassink TH, Piven J, Vieland VJ, Huang J, Swiderski RE, Pietila J, et al. Evidence supporting WNT2 as an autism susceptibility gene. Am J Med Genet. 2001;105:406–13.
Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, et al. Activity-dependent dendritic arborization mediated by CaM-kinase 1 activation and enhanced CREB-dependent transcription of Wnt2. Neuron. 2006;50:897–909.
Wegiel J, Kuchna I, Nowicki K, Imaki H, Wegiel J, Marchi E, et al. The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010;119:755–70.
Wei J, Hemmings G. Lack of a genetic association between the frizzled-3 gene and schizophrenia in a British population. Neurosci Lett. 2004;366(3):336–8.
Wiznitzer M. Autism and tuberous sclerosis. J Child Neurol. 2004;19:675–9.
Wu D, Pan W. GSK3: a multifacted kinase in Wnt signaling. Trends Biochem Sci. 2010;35(3):161–8.
Wu H, Xiong WC, Lin M. To build a synapse: signaling pathways in neuromuscular junction assembly. Development. 2010;127137:1017–33.
Yang J, Si T, Ling Y, Ruan Y, Han Y, Wang X, et al. Association study of the human FZD3 gene with schizophrenia. Biol Psychiatry. 2003;54(11):1298–301.
Yang P, Shu B-C, Hallmayer JF, Lung F-W. Intronic single nucleotide polymorphisms of Engrailed homeobox 2 modulate the disease vulnerability of autism in a Han Chinese population. Neuropsychobiology. 2010;62:104–15.
Yu X, Malenka RC. β-Catenin is critical for dendritic morphogenesis. Nat Neurosci. 2003;6(11):1169–77.
Zandi PP, Belmonte PL, Willour VL, Goes FS, Badner JA, Simpson SG, et al. Association study of Wnt signaling pathway genes in bipolar disorder. Arch Gen Psychiatry. 2008;65(7):785–93.
Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, et al. A dual-kinase mechanism for Wnt coreceptor phosphorylation and activation. Nature. 2005;438(7069):873–7.
Zenzmaier C, Marksteiner J, Kiefer A, Berger P, Humpel C. Dkk-3 is elevated in CSF and plasma of Alzheimer’s disease patients. J Neurochem. 2009;110:653–61.
Zhang Y, Yu X, Yuan Y, Ling Y, Ruan Y, Si T, et al. Positive association of the human frizzled 3 (FZD3) gene haplotype with schizophrenia in Chinese Han population. Am J Med Genet B Neuropsychiatr Genet. 2004;129B:16–9.
Zhang A, Shen C-H, Ma SY, Ke Y, El Idrissi A. Altered expression of autism-associated genes in the brain of fragile X mouse model. Biochem Biophys Res Commun. 2009;379:920–3.
Zhou C-J, Zhao C, Pleasure SJ. Wnt signaling mutants have decreased granule cell production and radial glial scaffolding abnormalities. J Neurosci. 2004;24(1):121–6.
Zhou CJ, Borello U, Rubenstein JL, Pleasure S. Neuronal production and precursor proliferation defects in the neocortex of mice with loss of function in the canonical Wnt signaling pathway. Neuroscience. 2006;142:1119–31.
Zhou W, Zhang Y, Li Y, Wei YS, Liu G, Liu DP, et al. A transgenic Cre mouse line for the study of cortical and hippocampal development. Genesis. 2010;48(5):343–50.
Zou Y. Wnt signaling in axon guidance. Trends Neurosci. 2004;27(9):528–32.
Acknowledgments
This work was supported by the US National Institutes of Health R21MH085995 (B.N.R.C), Autism Speaks (N.D.O.: Predoctoral Fellowship 4654), and Simons Foundation Autism Research Initiative (B.N.R.C.).
Conflict of interest
The authors declare they have no competing financial or other conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Okerlund, N.D., Cheyette, B.N.R. Synaptic Wnt signaling—a contributor to major psychiatric disorders?. J Neurodevelop Disord 3, 162–174 (2011). https://doi.org/10.1007/s11689-011-9083-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11689-011-9083-6