
Abstract
Microdeletions and microduplications encompassing a ~593-kb region of 16p11.2 have been implicated as one of the most common genetic causes of susceptibility to autism/autism spectrum disorder (ASD). We report 45 microdeletions and 32 microduplications of 16p11.2, representing 0.78% of 9,773 individuals referred to our laboratory for microarray-based comparative genomic hybridization (aCGH) testing for neurodevelopmental and congenital anomalies. The microdeletion was de novo in 17 individuals and maternally inherited in five individuals for whom parental testing was available. Detailed histories of 18 individuals with 16p11.2 microdeletions were reviewed; all had developmental delays with below-average intelligence, and a majority had speech or language problems or delays and various behavioral problems. Of the 16 individuals old enough to be evaluated for autism, the speech/behavior profiles of seven did not suggest the need for ASD evaluation. Of the remaining nine individuals who had speech/behavior profiles that aroused clinical suspicion of ASD, five had formal evaluations, and three had PDD-NOS. Of the 19 microduplications with parental testing, five were de novo, nine were maternally inherited, and five were paternally inherited. A majority with the microduplication had delayed development and/or specific deficits in speech or language, though these features were not as consistent as seen with the microdeletions. This study, which is the largest cohort of individuals with 16p11.2 alterations reported to date, suggests that 16p11.2 microdeletions and microduplications are associated with a high frequency of cognitive, developmental, and speech delay and behavior abnormalities. Furthermore, although features associated with these alterations can be found in individuals with ASD, additional factors are likely required to lead to the development of ASD.
Similar content being viewed by others

Introduction
Only 10–20% of individuals with ASD have a known etiology, which includes single-gene disorders and cytogenetic abnormalities (Abrahams and Geschwind 2008). This has prompted a search for additional genetic susceptibility loci. Several such studies discovered a recurrent ~593-kb microdeletion flanked by segmental duplications at 16p11.2 in multiple affected individuals (AUTS14, OMIM 611913), although three of these studies identified the microdeletion in the same four families from the Autism Genetic Resource Exchange (AGRE) (Sebat et al. 2007; Kumar et al. 2008; Weiss et al. 2008; Christian et al. 2008; Marshall et al. 2008). Screening studies have estimated the prevalence of either the microdeletion or microduplication to be approximately 1% of individuals with autism (15/1740, 4/397, and 4/427 individuals with ASD) (Weiss et al. 2008; Christian et al. 2008; Marshall et al. 2008). Numerous studies suggest variability in expressivity and penetrance. The microdeletion has been reported in individuals with cognitive impairment but without autism (Ghebranious et al. 2007; Rosenberg et al. 2006; Shiow et al. 2008; Bijlsma et al. 2009); in 1.76% (9/512) of children (including two monozygotic twins) with mental retardation, developmental delay or ASD (Weiss et al. 2008); in 0.33% (14/4284) of patients with mental retardation or multiple congenital anomalies (Bijlsma et al. 2009); and in 1.23% (1/81) of another smaller sample of individuals with mental retardation, dysmorphic features and a normal karyotype (Rosenberg et al. 2006). Moreover, both events have been found in control populations, including a group that contained individuals with bipolar disorder and a population of 19,000 unscreened individuals (Table 1) (Weiss et al. 2008). The detection of this microdeletion and microduplication in parents, in control populations (Kumar et al. 2008; Weiss et al. 2008; Bijlsma et al. 2009; Fernandez et al. 2009; Glessner et al. 2009) and in non-autistic individuals (Ghebranious et al. 2007; Rosenberg et al. 2006; Shiow et al. 2008; Bijlsma et al. 2009; Walsh et al. 2008), as well as the lack of clinical details in individuals classified with autism poses a challenge for diagnosis and research. Furthermore, only two studies have explored the broader phenotypes of these individuals in-depth (Bijlsma et al. 2009; Fernandez et al. 2009). Therefore, in the largest study to date, we characterized the clinical features associated with microdeletions and microduplications of 16p11.2 to provide a genotype-phenotype correlation of individuals with these events.
Results
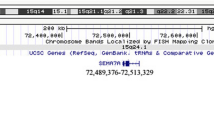
Between November 2007 and October 2008, we analyzed 9,773 individuals, 820 of whom had an ASD as the indication for study, using whole-genome bacterial artificial chromosome or oligonucleotide microarrays with expanded coverage of 16p11.2. Other indications for study among these individuals were developmental delays, dysmorphic features, congenital anomalies, and/or seizures. We identified 45 microdeletions (0.46%) encompassing 16p11.2 (Table 2). Six (0.73%) of the 820 individuals with an ASD as the indication for study had the microdeletion. Three of these 45 individuals had another clinically significant finding on aCGH and were excluded from further study (Table 3). Analysis of the microdeletions using an ultra high-density NimbleGen array with 120,146 probes within 16p11.2 confirmed a 548-kb common interval spanning chr16:29559100-30107210 (Hg18) (Fig. 1). This common interval encompasses about 29 genes (see Supplemental Note and Table S1). High-resolution aCGH also showed that the proximal and distal breakpoints of the 16p11.2 rearrangement map within highly identical clusters of segmental duplications (Fig. 1). Within these segmental duplication clusters, two highly homologous directly oriented duplication blocks were identified. These duplication blocks are 147 kb and 72 kb in size with a sequence identity of 99.5% and 98.7%, respectively, and map to both the proximal and distal breakpoints according to the UCSC Genome Browser, Hg18, Build 36 (Table S2). Misalignment of these directly oriented homologous segmental duplication blocks flanking the disease-associated 16p11.2 genomic region, during meiosis potentially predisposes to non-allelic homologous recombination (NAHR) events resulting in microdeletions or microduplications (Lupski 1998).

High-resolution microarray analysis of 16p11.2 rearrangements. Refinement of 16p11.2 microdeletion breakpoints by high-density microarray analysis, for a representative set of cases, is shown. Note that probes with log2 ratios above or below a threshold of 1.5 standard deviations from the normalized mean log2 ratio are colored green (duplication) or red (deletion), respectively. Dotted lines represent breakpoint regions (BP). Segmental duplications flanking the 16p11.2 rearrangements are also shown. The orange and blue boxes represent homologous segmental-duplication blocks, 147 kb and 72 kb respectively, participating in the NAHR event for this particular rearrangement
For the 45 individuals with the 16p11.2 microdeletion, 29 maternal samples and 19 paternal samples were tested, and inheritance was determined in 22 individuals. The microdeletion was de novo in 17 individuals (77%) and was maternally inherited in five individuals (23%). One of these mothers was reported to be normal; another mother had mental retardation, schizophrenia, and Tourette syndrome; a third had intellectual impairment; and a fourth had short-term memory loss secondary to accidental trauma but no history of delays. Information was unavailable on the fifth mother.
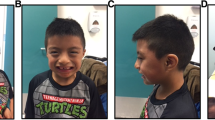
Detailed clinical information was available for 18 individuals with microdeletions in our cohort (Table 4, Table S3). The majority of individuals had minimal dysmorphic features, and these features were variable (Fig. 2). Ear abnormalities were the most common, with eight subjects (44%) with fleshy or prominent ears and four subjects (22%) with hypoplastic or simple ears. Major organ defects were uncommon in our study population, although three had aortic abnormalities: either abnormal aortic valves, hypoplastic aortic arch, or enlarged aortas. One individual reported here has a history of seizures, a second has staring spells, and a third has an abnormal EEG without seizure activity. All 18 individuals had developmental delays with below-average intelligence. A majority of the individuals (94%) had speech problems or delays, including speech delays greater than other delays, poor articulation, lower verbal intelligence and difficulty in reading skills. Fourteen of the individuals in our population (78%) were reported to have some type of behavioral problem, most commonly attention deficit-hyperactivity disorder (ADHD) or poor attention span (44%) and aggression or outbursts (39%).

Physical features in individuals with 16p11.2 microdeletions. Individuals are aged <1 year a, 2 years b, 3 years c, d, 4 years e, f, 8 years g, 9 years h, 10 years i, j, 18 years k and 20 years l. m Foot of individual shown in (k). Note 2–3 syndactyly and small toenails. n Profile of individual shown in (g). Note hypoplastic ear with Darwinian tubercle and fleshy lobe. Ears are also fleshy and/or prominent in individuals (b, e, i, j). Individual (g) also has bilateral iris coloboma. Other features noted include frontal bossing (f, j, k), flattened midface (h, j, k), broad nose (b, e), retrognathia (a, j, k), short, thick, or webbed neck (e, g, j), downturned mouth (e, g, h) and synophrys (k, j)
Of the 16 individuals with the microdeletion old enough to be evaluated for autism, seven were not described as autistic and had not had ASD testing. Although a majority of these individuals had speech delay, their behavioral problems, when present, were different than those needed for ASD diagnosis, including poor attention span and aggression, and several were specifically reported to be social and not have any stereotypic behaviors. Mild ASD features were reported for the remaining nine individuals, five of whom had a formal ASD evaluation. Of these five, three received a diagnosis of PDD-NOS, and the other two did not meet ASD criteria. PDD-NOS diagnoses were given to one female (based on an Autism Diagnostic Observation Schedule, ADOS) and two males (based on a Childhood Autism Rating Scale, CARS, one with a score of 30.5, with the diagnostic cutoff being 30). One female who did not meet ASD criteria had a CARS score of 24. The ADI-R for this same female showed mild impairment in social interactions (score 22) and communication skills (score 15), but she did not have restrictive, repetitive, or stereotypic patterns of behavior. The other female who did not meet ASD criteria had an extensive interdisciplinary developmental evaluation, including observations in multiple settings, and she showed multiple behaviors inconsistent with ASD, including social interactions, varied interests, and maintenance of conversations. Formal evaluations were not performed on the remaining three males and one female with features suggestive of ASD, although one male and one female had been given a PDD-NOS label.
During the study period, we also identified 32 microduplications (0.33% of all cases tested) encompassing 16p11.2 (Table 2). Like the microdeletions, analysis of the microduplications on ultra-dense tiling arrays confirmed a 548-kb common interval spanning chr16:29559100-30107210 (Hg18) with breakpoints that were all within the flanking segmental duplications (Fig. 1). Three (0.37%) of the 820 individuals with an ASD as the indication for study had the microduplication. Four of these 32 individuals had another clinically significant finding on aCGH and were excluded from further study (Table 5).
Among the 32 individuals with the 16p11.2 microduplication, 24 maternal samples and 15 paternal samples were tested, and inheritance was determined in 19 individuals. The microduplication was de novo in five individuals (26%), maternally inherited in nine (47%) and paternally inherited in five (26%). Information was available on four mothers and two fathers. One mother had childhood seizures and intermittent hypoglycemia; another had suspected multiple sclerosis; a third mother had speech articulation problems, mood swings, depression, anxiety, pain, and bouts of head banging when upset; a fourth mother had learning disabilities. One father had childhood seizures, delays, and ADHD-like features, and the other father was described as autistic-like.
Detailed clinical information was available for 10 individuals with microduplications in our cohort (Table 6, Table S4). Three of these individuals had clinical diagnoses—Beckwith-Wiedemann syndrome, PKU and methylmalonic aciduria—likely independent of the microduplication. Two individuals had a history of abuse, causing physical or psychological trauma that could have resulted in subsequent behavioral issues, complicating conclusions about the phenotype associated with the microduplication. Minimal dysmorphic features were reported in these patients, with epicanthal folds (70%) and a broad or prominent nose (40%) the most common features (Fig. 3). Only one individual had a heart abnormality, a patent ductus arteriosus (PDA), although a sister of one of these individuals had a ventricular septal defect (VSD) and was found to have the microduplication. Another individual had congenital diaphragmatic hernia (CDH), horseshoe kidneys and malrotation, which was interpreted as a field defect. A majority of individuals had delayed development (80%), with the same number reporting specific deficits in speech or language (80%). Behavior problems were present in half, with four out of five individuals with behavior problems showing aggression or outbursts. Of the eight individuals with microduplications old enough to be evaluated for autism, none had formal ASD evaluation, although one male had a suspected diagnosis of PDD-NOS or autism.

Physical features in individuals with 16p11.2 microduplications. a, b 7-year-old male with epicanthal folds, broad nose, frontal hair whorl, wide mouth and low-set ears. c, d, e Siblings aged 6, 2, and their mother (age 38), all of whom have a 16p11.2 microduplication. Note thin upper lip and prominent forehead
Discussion
The 16p11.2 microdeletion (n = 45) and microduplication (n = 32) represent 3.1% of all abnormalities reported by our laboratory, second only to the 22q11.2 velocardiofacial/DiGeorge syndrome region (n = 60 deletions and 22 duplications; 3.3% of all abnormalities). During this same period, we identified 13 Smith-Magenis syndrome (SMS) deletions (population frequency of ~1/15,000) (Elsea and Girirajan 2008) and 28 Williams syndrome deletions (population frequency of ~1/7,500) (Stromme et al. 2002), suggesting the population frequency of 16p11.2 microdeletions may be greater than 1/5,000 and the microduplication greater than 1/7,500. These may be overestimates of frequency because some cases of SMS and Williams syndrome are diagnosed by other methods, and therefore not all individuals with these syndromes will have aCGH, while 16p11.2 abnormalities would not be expected to be diagnosed by other methods. Conducting Fisher’s exact test to compare our population to our group of 2493 controls (Itsara et al. 2009), in which no microdeletions were found and one microduplication was found (Table 1), yields a two-tailed p of 5.2 × 10−5 for the microdeletion and an odds ratio of 8.19 and two-tailed p of 0.0084 for the microduplication. Therefore, both the microdeletion and microduplication have a significantly higher frequency in our patient population when compared to our control group. We conducted a review of these individuals’ phenotypes to better understand the features associated with these recurrent genomic alterations.
Observation of our cohort suggests that language delays and behavior problems are common among individuals with 16p11.2 microdeletions, which is consistent with previous reports (Kumar et al. 2008; Weiss et al. 2008; Marshall et al. 2008; Ghebranious et al. 2007; Rosenberg et al. 2006; Shiow et al. 2008; Bijlsma et al. 2009; Fernandez et al. 2009), although some reports have suggested that cognitive or behavioral impairment may be present without speech delay (Sebat et al. 2007; Weiss et al. 2008; Ghebranious et al. 2007; Bijlsma et al. 2009). While some patients have dysmorphic features, others are specifically nondysmorphic (Kumar et al. 2008; Weiss et al. 2008; Marshall et al. 2008; Ghebranious et al. 2007; Rosenberg et al. 2006; Bijlsma et al. 2009; Fernandez et al. 2009), and there is no recognizable facial gestalt. There may be an association with aortic or aortic valve abnormalities, seen in three individuals in our report and a set of twins in the literature (Ghebranious et al. 2007). Pyloric stenosis may also be infrequently associated with this microdeletion, seen in one individual in our report and two others in the literature (Bijlsma et al. 2009). Seizures were present in two individuals in our cohort, which is less frequent than previously reported, particularly in ASD cohorts (Kumar et al. 2008; Weiss et al. 2008; Ghebranious et al. 2007; Fernandez et al. 2009).
It is difficult to ascertain a common phenotype among individuals with 16p11.2 microduplications, and it is further complicated by the presence of other diagnoses and complicated social histories in patients in our cohort. Furthermore, we have an ascertainment bias, as we are only testing individuals whose phenotype is suggestive of a genetic condition. The literature has reports of this microduplication in a variety of individuals. While most microduplications have been found from screening cohorts of autistic individuals, they have also been found in parents, in individuals with bipolar disorder, in individuals with schizophrenia and in controls (Table 1) (Kumar et al. 2008; Weiss et al. 2008; Fernandez et al. 2009; Walsh et al. 2008). In previous studies, microduplications were usually, but not always, found in a higher frequency in abnormal cohorts than in normal controls (Table 1); our cohort does show a significant increase in this frequency. Speech and/or motor delay is seen in a majority of individuals in our cohort and has been reported in other individuals (Weiss et al. 2008; Fernandez et al. 2009). Behavior problems may be associated with this microduplication, observed in half of the individuals in our cohort and in those in the literature from ASD cohorts (Kumar et al. 2008; Weiss et al. 2008; Marshall et al. 2008; Fernandez et al. 2009). Similar to the microdeletion, individuals with the microduplication may not have dysmorphic features (Weiss et al. 2008; Marshall et al. 2008; Fernandez et al. 2009), and dysmorphic features are not consistent among individuals. The only major malformation seen in more than one individual is a congenital diaphragmatic hernia, one in our cohort and one in a previous report (Marshall et al. 2008; Fernandez et al. 2009).
Inheritance of 16p11.2 microdeletions and microdupliations is notably different, with a majority of microdeletions being de novo and the majority of microduplications being inherited. A combination of our report with those in the literature shows a de novo microdeletion in 36/50 (72%) of families with known inheritance (de novo microdeletions have been found in two sets of monozygotic twins and in two sibs due to paternal germline mosaicism). Including our cases, this microdeletion has been described in eight parents—three fathers and five mothers—with various developmental problems and in five apparently normal parents, one father and four mothers (Sebat et al. 2007; Weiss et al. 2008; Marshall et al. 2008; Ghebranious et al. 2007; Shiow et al. 2008; Bijlsma et al. 2009; Fernandez et al. 2009). In contrast, a combination of our report with those in the literature shows a de novo microduplication in 7/28 (25%) of families with known inheritance. Inheritance is mixed—maternal in twelve, paternal in seven and unspecified in two (Kumar et al. 2008; Weiss et al. 2008; Marshall et al. 2008; Fernandez et al. 2009; Walsh et al. 2008). Phenotypic information is unavailable for a majority of parents in our cohort, and the three parents with the microduplication in the literature who have been described include a mother with learning disabilities, a father with bipolar disorder, and a mother with depression, anxiety, learning disabilities and behavior problems who also carried a 1-Mb deletion on 7q31.2 (Kumar et al. 2008; Fernandez et al. 2009). An apparently healthy 8-year-old sister of a proband who also carried the microduplication has been described (Fernandez et al. 2009). Compared to microdeletions, the greater number of inherited microduplications suggests this alteration is more likely to be passed on to subsequent generations, which could be due to a less-severe phenotype caused by the microduplication. While our report and the literature show a majority of the inherited microdeletions and microduplications are from mothers, in our cohort, this is likely due to the greater availability of maternal samples. Therefore, there is no current evidence for a parent-of-origin bias or imprinting of this region.
Because individuals with 16p11.2 alterations commonly have speech delays and behavior problems, their features may be more likely to arouse clinical suspicion of ASD than individuals without similar neurodevelopmental problems. However, some of the individuals in our cohort were not considered to have autistic features. Additionally, while we found 16p11.2 abnormalities more commonly among individuals referred for ASD, this difference was not significant (Table 2). These groupings are not precise, however, as they are based on indications for study, which are commonly inaccurate or incomplete. Several cases without an ASD in the indication for study were determined to have an ASD, and one with an indication for study of autism specifically did not meet criteria for an ASD diagnosis. Our study is also limited by the lack of rigorous phenotypic analysis for all patients, especially for ASD. However, it is likely that other factors are required in these individuals to cause the development of ASD, a hypothesis supported by reports in the literature of families with 16p11.2 microdeletions and microduplications (Fernandez et al. 2009; Glessner et al. 2009). This microdeletion has been reported in single autistic individuals and not in their affected siblings in four multiplex families (Kumar et al. 2008; Marshall et al. 2008; Fernandez et al. 2009). The microduplication has been found in multiple affected siblings in two families and in unaffected siblings in two families, but it has not been found in affected siblings in one family (Weiss et al. 2008; Marshall et al. 2008; Fernandez et al. 2009). In our report, there is a 2-year-old male with a microduplication and suspected ASD diagnosis with a sister with mild PDD-NOS, a brother with anger issues and emotional lability and a mother with behavioral abnormalities (head banging), depression and anxiety. The mother and sister were found to have the microduplication, whereas the brother did not (Fig. 3). In some of these previously reported individuals, the siblings without the microdeletion are more severely affected in some aspect of their behavior or development (Fernandez et al. 2009). Of the three AGRE multiplex families with affected children discordant for the 16p11.2 microdeletion, the sibling without the microdeletion has later speech development in two families, and in the third family the sibling without the microdeletion has a higher Autism Diagnostic Interview-Revised (ADI-R) communication score than his brother, indicating greater impairment (Kumar et al. 2008). Two families also show a higher ADI-R social score in the sibling without the microdeletion. In addition, in one pair of siblings who both have the 16p11.2 microdeletion, the sister has higher ADI-R and ADOS scores than her brother in most categories (Kumar et al. 2008; Weiss et al. 2008); because females are the less-affected sex, it would be predicted that in a male and female with the same genetic factors for autism, the male would be more severely affected. Consistent with males being the more susceptible gender, two families that are not part of the AGRE cohort have been reported, both of which have a son and daughter affected with ASD, with the son having a more severe phenotype. However, in one case, the daughter has the 16p11.2 microdeletion, and in the other the son has the microdeletion (Fernandez et al. 2009). These multiplex families suggest factors, genetic and/or environmental, other than the 16p11.2 alteration play a role in the children’s autistic features.
Glessner et al. have also suggested that 16p11.2 alterations may not be sufficient to cause ASD, citing the discordance in these multiplex families in the literature as well as a non-significant difference between ASD cases and controls in their cohort (Glessner et al. 2009). Our study differs from this in finding a significant increase of the 16p11.2 microdeletion and microduplication among individuals undergoing clinical aCGH testing as compared to controls. This difference may be due to the fact that their report included smaller deletions and duplications within the 16p11.2 region and/or to their use of children as controls. Of note, a subsequent publication of their control group does not contain any abnormalities in this region (Shaikh et al. 2009).
Microdeletions and microduplications of the 16p11.2 region are emerging as common findings in individuals with developmental delay, although the phenotypic consequences are only beginning to be understood. In our study population, the microdeletions and microduplications are associated with a high frequency of cognitive, developmental and speech delay and behavior issues, with the microdeletions showing a higher incidence of these features than the microduplications. Although 16p11.2 abnormalities have mostly been identified in ASD cohorts, our cohort demonstrates a phenotypic range from behavioral problems that were not considered autistic to formal diagnoses of PDD-NOS in three individuals, suggestive of a spectrum of developmental and speech delays and behavioral issues that overlaps with ASD. The disparity in numbers of individuals with ASD among cohorts may be the result of differences in study inclusion criteria. Because the microdeletion and microduplication have been reported in normal individuals and controls and demonstrate phenotypic variability in families, additional genetic or environmental factors may be required for manifestation of clinical features. Further studies of individuals with 16p11.2 microdeletions and microduplications from control populations and populations with a variety of abnormal phenotypes are necessary to increase the understanding of these genomic alterations and their associated phenotypes.
Subjects and methods
Ethics statement
Consent for publication of photographs was obtained for the individuals with 16p11.2 microdeletions and microduplications shown here.
Subjects and controls
From November 2007 through October 2008, we performed microarray analysis on 9,773 consecutive individuals whose specimens were submitted to Signature Genomic Laboratories, with indications for study including developmental delay, dysmorphic features, congenital anomalies, and/or seizures. The indication for study was autism, autistic features, pervasive developmental delay (PDD), or Asperger syndrome in 820 of these samples. Referring physicians of individuals found to have 16p11.2 microdeletions or microduplications were contacted to help coordinate release of clinical information for publication.
The control set consisted of 2493 individuals whose copy-number variations had been identified using Illumina SNP microarrays in the author’s (EE’s) laboratory as described in Itsara et al. (2009) These included 936 middle-age (40–70 years) individuals living in the United States being tested as part of statin trials and cholesterol; 671 National Institute of Neurological Disorders and Stroke (NINDS) samples of European descent with no family history of or any first-degree relative with amyotrophic lateral sclerosis, ataxia, autism, brain aneurysm, dystonia, Parkinson disease or schizophrenia and 886 individuals sampled from 51 different world populations as part of the HGDP collection.
BAC microarray analysis
Microarray-based comparative genomic hybridization (aCGH) was performed on some individuals with a bacterial artificial chromosome (BAC) microarray (the SignatureChipWG®; Signature Genomic Laboratories, Spokane, WA) as previously described (Ballif et al. 2008a; Bejjani et al. 2005).
Oligonucleotide aCGH
Oligonucleotide-based microarray analysis was performed on some individuals using a 105 K-feature whole-genome microarray (SignatureChip Oligo Solution™, made for Signature Genomic Laboratories by Agilent Technologies, Santa Clara, CA) as previously described (Ballif et al. 2008b).
CNV validation by high-resolution aCGH
Further refinement of 16p11.2 CNV breakpoints were performed by custom high-density oligonucleotide arrays (NimbleGen Systems). The high-density array consisted of a total 135,000 isothermal probes (45–75 bp) including 120,146 probes covering a 4-Mb 16p11.2 region (chr16: 27,500,000 to 31,500,000) with a mean spacing 1 probe every 19 bp. As a control for probe hybridizations and for normalizing the array data, we also included 15,118 probes to a 7.5-Mb 16p12 region with a mean probe spacing of 451 bp. All microarray hybridization experiments were performed as described previously (Selzer et al. 2005), using a single unaffected male (GM15724, [Corriell]) as reference.
Fluorescence in situ hybridization
All 16p11.2 microdeletions detected by array CGH were confirmed and visualized by metaphase fluorescence in situ hybridization (FISH) BAC clones CTD-3159M2 or CTD-2515O10 (Shaffer et al. 1994). Parental samples for individuals with microdeletions were assayed by FISH.
References
Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–55.
Ballif BC, Theisen A, Coppinger J, Gowans GC, Hersh JH, et al. Expanding the clinical phenotype of the 3q29 microdeletion syndrome and characterization of the reciprocal microduplication. Mol Cytogenet. 2008a;1:8.
Ballif B, Theisen A, McDonald-McGinn D, Zackai E, Hersh J, et al. Identification of a previously unrecognized microdeletion syndrome of 16q11.2q12.2. Clin Genet. 2008b.
Bejjani BA, Saleki R, Ballif BC, Rorem EA, Sundin K, et al. Use of targeted array-based CGH for the clinical diagnosis of chromosomal imbalance: is less more? Am J Med Genet A. 2005;134:259–67.
Bijlsma EK, Gijsbers AC, Schuurs-Hoeijmakers JH, van Haeringen A, Fransen van de Putte DE, et al. Extending the phenotype of recurrent rearrangements of 16p11.2: Deletions in mentally retarded patients without autism and in normal individuals. Eur J Med Genet. 2009.
Christian SL, Brune CW, Sudi J, Kumar RA, Liu S, et al. Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol Psychiatry. 2008;63:1111–7.
Elsea SH, Girirajan S. Smith-Magenis syndrome. Eur J Hum Genet. 2008;16:412–21.
Fernandez BA, Roberts W, Chung B, Weksberg R, Meyn S, et al. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J Med Genet. 2009.
Ghebranious N, Giampietro PF, Wesbrook FP, Rezkalla SH. A novel microdeletion at 16p11.2 harbors candidate genes for aortic valve development, seizure disorder, and mild mental retardation. Am J Med Genet A. 2007;143A:1462–71.
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–73.
Itsara A, Cooper GM, Baker C, Girirajan S, Li J, et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009;84:148–61.
Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17:628–38.
Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–22.
Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–88.
Rosenberg C, Knijnenburg J, Bakker E, Vianna-Morgante AM, Sloos W, et al. Array-CGH detection of micro rearrangements in mentally retarded individuals: clinical significance of imbalances present both in affected children and normal parents. J Med Genet. 2006;43:180–6.
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–9.
Selzer RR, Richmond TA, Pofahl NJ, Green RD, Eis PS, et al. Analysis of chromosome breakpoints in neuroblastoma at sub-kilobase resolution using fine-tiling oligonucleotide array CGH. Gene Chromosome Canc. 2005;44:305–19.
Shaffer LG, McCaskill C, Han JY, Choo KH, Cutillo DM, et al. Molecular characterization of de novo secondary trisomy 13. Am J Hum Genet. 1994;55:968–74.
Shaikh TH, Gai X, Perin JC, Glessner JT, Xie H, et al. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009;19:1682–90.
Shiow LR, Paris K, Akana MC, Cyster JG, Sorensen RU, et al. Severe combined immunodeficiency (SCID) and attention deficit hyperactivity disorder (ADHD) associated with a coronin-1A mutation and a chromosome 16p11.2 deletion. Clin Immunol. 2008.
Stromme P, Bjornstad PG, Ramstad K. Prevalence estimation of Williams syndrome. J Child Neurol. 2002;17:269–71.
Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–43.
Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–75.
Acknowledgements
We thank the families, physicians, and genetic counselors who agreed to contribute clinical information to this report. We also thank Aaron Theisen for his editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Jill A. Rosenfeld and Justine Coppinger have contributed equally to this work
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 61 kb)
Table S1
Genes within 16p11.2 microdeletion/microduplication common interval (DOC 64.5 kb)
Table S2
pairwise sequence similarity of segmental duplications between the proximal and the distal 16p11.2 breakpoints (DOC 25 kb)
Table S3
Clinical features of individuals in this cohort with 16p11.2 microdeletions. (DOC 47 kb)
Table S4
Clinical features of individuals in this cohort with 16p11.2 microduplications. (DOC 42 kb)
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Rosenfeld, J.A., Coppinger, J., Bejjani, B.A. et al. Speech delays and behavioral problems are the predominant features in individuals with developmental delays and 16p11.2 microdeletions and microduplications. J Neurodevelop Disord 2, 26–38 (2010). https://doi.org/10.1007/s11689-009-9037-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11689-009-9037-4