
Abstract
Amyloid imaging with [11 C]Pittsburgh Compound-B (PiB) provides in vivo data on plaque deposition in those with, or at risk for, Alzheimer’s disease (AD). We performed a gene-based association analysis of 15 quality-controlled amyloid-pathway associated candidate genes in 103 Alzheimer’s Disease Neuroimaging Initiative participants. The mean normalized PiB uptake value across four brain regions known to have amyloid deposition in AD was used as a quantitative phenotype. The minor allele of an intronic SNP within DHCR24 was identified and associated with a lower average PiB uptake. Further investigation at whole-brain voxel-wise level indicated that non-carriers of the minor allele had higher PiB uptake in frontal regions compared to carriers. DHCR24 has been previously shown to confer resistance against beta-amyloid and oxidative stress-induced apoptosis, thus our findings support a neuroprotective role. Pathway-based genetic analysis of targeted molecular imaging phenotypes appears promising to help elucidate disease pathophysiology and identify potential therapeutic targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD), the most common type of dementia, is characterized by marked decline in memory and other cognitive abilities. An estimated 5.4 million Americans, most over the age of 65, have AD at present and no disease-modifying treatment is available to slow or stop its progression (Alzheimer’s Association, 2011). The hallmark abnormalities of AD are deposits of the protein fragment beta-amyloid (Aβ), which form amyloid plaques, and twisted strands of the protein tau which form neurofibrillary tangles. Amnestic mild cognitive impairment (MCI) is a clinical condition in which an individual has memory complaints and memory impairment that is not normal for his/her age, but does not significantly interfere with daily functioning. Approximately 14–18% of individuals in the population aged 70 years and older have MCI, and approximately 10–15% of these individuals are likely to progress to dementia, particularly AD, each year (Petersen et al., 2009).
Genetic variation has been shown to play a key role in the development of AD. Mutations in APP (amyloid beta (A4) precursor protein), PSEN1 (presenilin 1) and PSEN2 (presenilin 2 (Alzheimer disease 4)) cause early-onset AD (onset <60 years). However, early-onset AD accounts for a small percent (<5%) of AD cases (Bertram, Lill, & Tanzi, 2010). Currently, the most predictive genetic risk factor for the more common late-onset form of AD (onset ≥60 years) is the APOE (apolipoprotein E) ε4 allele. The ε4 allele, a member of a three allele haplotype (composed of ε2, ε3, and ε4 alleles), confers a dose-dependent increase in AD risk of approximately four-fold as compared to non-carriers (Corder et al., 1993; Farrer et al., 1997; Saunders et al., 1993). Recent large-scale case-control genome-wide association studies (GWASs) have identified and replicated additional risk loci including: CLU (clusterin), CR1 (complement component (3b/4b) receptor 1 (Knops blood group)), PICALM (phosphatidylinositol binding clathrin assembly protein), BIN1 (bridging integrator 1), EXOC3L2 (exocyst complex component 3-like 2), MTHFD1L (methylenetetrahydrofolate dehydrogenase (NADP + dependent) 1-like), MS4A4A (membrane-spanning 4-domains, subfamily A, member 4)/MS4A6E (membrane-spanning 4-domains, subfamily A, member 6E), CD2AP (CD2-associated protein), EPHA1 (EPH receptor A1), CD33 (CD33 molecule) and ABCA7 (ATP-binding cassette, sub-family A (ABC1), member 7) (Harold et al., 2009; Hollingworth et al., 2011; Jun et al., 2010; Lambert et al., 2009; Naj et al., 2010; Naj et al., 2011; Seshadri et al., 2010). However, the population attributable fractions, or the proportional reduction in mortality or population disease if a risk factor exposure were reduced to an alternative ideal exposure scenario, for the strongest SNPs at each of CR1, CLU, PICALM, BIN1, EPHA1, MS4A, CD33, CD2AP and ABCA7 loci were estimated to be between 2.72% and 5.97%. Furthermore, the cumulative population-attributable fraction for these non-APOE loci is estimated to be as much as 35% (Naj et al., 2011). Thus, the reported loci do not account for all of the genetic variation associated with the disease. An alternative approach, employing analysis of quantitative phenotypes, may be able to detect other loci associated with AD and MCI.
Biomarkers for AD, such as cerebrospinal fluid (CSF) measurements (Hampel et al., 2010), blood markers, and neuroimaging modalities such as magnetic resonance imaging (MRI) and positron emission tomography (PET), can be used as quantitative phenotypes in genetic association studies to identify additional risk loci. The use of quantitative phenotypes has been shown to require smaller sample sizes for the detection of similar variants with equivalent effect sizes than traditional case-control designs (Potkin et al., 2009). The quantitative phenotype approach has revealed risk loci associated with a number of AD phenotypes including: hippocampal atrophy (Potkin et al., 2009) and rate of decline (Saykin et al., 2010), temporal lobe structure (Stein et al., 2010), brain-wide MRI phenotypes (Shen et al., 2010; Stein et al., 2010) and cerebrospinal fluid measures (Han, Schellenberg, & Wang, 2010; Kim et al., 2011).
[11 C]-Pittsburgh Compound-B (PiB) is a PET ligand that binds to fibrillar Aβ deposits with high affinity (Klunk et al., 2004). The distribution of PiB binding in AD patients closely matches the post-mortem histological distribution of Aβ plaques seen in AD (Rabinovici & Jagust, 2009). Recent data using an alternative [18 F]-labeled amyloid tracer, Florbetapir, demonstrated strong antemortem-postmortem agreement in amyloid burden and distribution (Clark et al., 2011). For PiB, the highest signal is observed in the prefrontal cortex, precuneus and posterior cingulate cortex, followed by the lateral parietal and temporal cortices and striatum, with a lower signal in the occipital cortex, globus pallidus, and thalamus (Rabinovici & Jagust, 2009).
We hypothesized that PiB-PET can serve as a useful endophenotype for genetic analyses to identify new loci associated with AD pathology (Berti, Nacmias, Bagnoli, & Sorbi, 2011). To test this hypothesis, we performed a gene-based association analysis of 15 quality-controlled amyloid-pathway associated candidate genes in 103 Alzheimer’s Disease Neuroimaging Initiative (ADNI) participants who had initial PiB-PET scans as well as genotype data. ADNI is an ongoing multiyear public-private partnership with the goal of testing whether combinations of biomarkers such as MRI, PET, CSF, genetic factors (e.g., single nucleotide polymorphisms (SNPs) and copy number variations), and clinical and neuropsychological assessments can improve early diagnosis and predict progression of MCI and early AD. We used an average of four PiB uptake values, one from each of four brain regions known to have amyloid deposition in AD as the quantitative phenotype. We sought to determine genes and SNPs significantly associated with this phenotype representing a mean regional PiB uptake. Candidate genes and SNPs identified from this analysis were further investigated across the whole brain on a voxel-wise level.
Methods
Alzheimer’s Disease Neuroimaging Initiative
Data used in this study were obtained from the ADNI database (http://adni.loni.ucla.edu/). The ADNI was initiated in 2003 by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration (FDA), private pharmaceutical companies and non-profit organizations. The primary goal of ADNI has been to test whether serial MRI, PET, other biological markers, and clinical and neuropsychological assessments can be combined to measure the progression of MCI and early AD. Determination of sensitive and specific markers of very early AD progression is intended to aid researchers and clinicians to develop new treatments and monitor their effectiveness, as well as lessen the time and cost of clinical trials. Michael W. Weiner, MD, VA Medical Center and University of California-San Francisco is the Principal Investigator of this initiative. This $60 million, multiyear public-private partnership involves many co-investigators from a broad range of academic institutions and private corporations. More than 800 participants, aged 55 to 90, have been recruited from across more than 50 sites in the US and Canada. This includes approximately 200 cognitively normal older individuals (healthy controls or HCs) to be followed for 3 years, 400 patients diagnosed with MCI to be followed for 3 years, and 200 patients diagnosed with early AD to be followed for 2 years. Longitudinal imaging data, including structural 1.5 Tesla MRI scans, were collected on the full sample, and [11 C]PiB- and [18 F]fluorodeoxyglucose (FDG)-PET imaging data were acquired in a subset of participants. Neuropsychological and clinical assessments were collected at baseline, and at follow-up visits of six-to-twelve month intervals. Other available data used in the present analysis included APOE ε4 genotype and a dense set of 489 SNPs in amyloid-pathway associated genes that were genotyped as part of a GWAS in the full ADNI sample. Longitudinal CSF AD biomarkers were also available from a sub-sample. Further information about ADNI can be found in (Weiner et al., 2010) and at http://www.adni-info.org.
The study was conducted after Institutional Review Board approval at each site. Written informed consent was obtained from all study participants, or their authorized representatives.
Participants
One hundred and three participants (25 AD, 56 MCI and 22 HC at time of scan; 95 non-Hispanic Caucasians, three non-Hispanic African Americans, two non-Hispanic Asians, two Hispanic Caucasians and one Caucasian of unknown ethnicity) in the ADNI cohort with initial PiB-PET scans and genotype data were included in the present analyses (Table 1).
PiB-PET data
Detailed methods have been previously reported for acquisition and processing of PiB-PET scans for the ADNI sample (Jagust et al., 2010; Jagust et al., 2009), and are briefly described below.
Acquisition of PiB-PET scans
The PiB-PET study was initiated as an “add-on” project towards the end of the first year of the ADNI study, and 103 participants at 14 participating ADNI PET centers were recruited for PiB-PET imaging. In some participants, PiB-PET scanning was not conducted at the baseline visit, but scans were acquired at month 12 and/or month 24 visits (Table 1). PiB-PET scans were collected on multiple PET instruments and with multiple acquisition sequences. Approximately 10–15 mCi of [11 C]PiB was injected intravenously, followed by in most cases a 50-min uptake period. Data were then acquired from 50 to 70 min in 4–5 min frames. Quality control (QC) was performed on all scans. Raw PiB-PET frames were realigned to the first frame and averaged to create a single image. This image was then set to a standard orientation and voxel size, intensity normalized using a cerebellar grey matter region of interest (ROI), and smoothed to a common resolution of 8 mm full-width at half-maximum. The resulting image was referred to as the “PIB Coreg, Avg, Std Img and Vox Size, Uniform Resolution” and was subsequently made available for download from the LONI ADNI site (http://adni.loni.ucla.edu/). Further information can be found in (Jagust et al., 2009).
Regional assessment of PiB-PET data
Regional normalized PiB uptake values used in this analysis were obtained from a dataset generated by the University of Pittsburgh as described briefly below. PiB-PET data were assessed using an automated ROI template method to sample different brain areas. The MRI template from a 79-year-old MCI participant scanned at the University of Pittsburgh was used to generate the ROIs. Each participant’s PiB-PET scan was co-registered to his/her MRI scan. The individual’s MRI scan was normalized to the MCI template, and the same parameters were applied to normalize his/her PiB-PET scan to the MCI template. The PiB-PET scans were then resliced to the dimensions of the MCI template and mean PiB uptake from each ROI was extracted. For each ROI, the extracted PiB uptake values were renormalized relative to a cerebellar ROI generated from the MCI participant described above. Further information on the post processing steps of the PiB-PET scans by the University of Pittsburgh can be found at http://adni.loni.ucla.edu/research/pet-post-processing/.
Phenotype selection for the gene-based association analysis of amyloid-pathway associated candidate genes
Normalized PiB uptake values from four ROIs (anterior cingulate, frontal cortex, parietal cortex and precuneus) generated by the University of Pittsburgh were obtained for initial PiB-PET scans of all participants from the ADNI database (http://adni.loni.ucla.edu/) (as of November 2010). Only one set of scan data was used per subject. The average PiB uptake from these four brain regions has been previously used to define a cut-off value (average PiB uptake = 1.50), to classify participants as either positive for amyloid deposition (average PiB uptake > 1.50) or negative for amyloid deposition (average PiB uptake < 1.50) (Jagust et al., 2010). We thus decided to use the average PiB uptake value from these four regions as the quantitative phenotype for the genetic association analysis.
Processing of PiB-PET scans for the whole-brain voxel- and cluster-wise analysis
PiB-PET image processing was performed using Statistical Parametric Mapping (Friston et al., 1994) version 5 (SPM5) (http://www.fil.ion.ucl.ac.uk/spm/) implemented using MATLAB v7.1.0.183 (MathWorks, Natick, MA). Initial “PIB Coreg, Avg, Std Img and Vox Siz, Uniform Resolution” PiB-PET images, processed as described above, were obtained in NIfTI format for all available participants from the ADNI database (http://adni.loni.ucla.edu/) (as of October 2010). Additionally, 1.5 Tesla T1-weighted 3D magnetization prepared rapid acquisition gradient echo (MP-RAGE) MRI scans for all participants (Jack et al., 2008) were downloaded from the ADNI database. Each participant’s processed PiB-PET image was co-registered to his/her MRI scan at the same timepoint. One participant who had an initial PiB-PET scan at the month 12 visit did not have an MRI scan at the same timepoint. In this case, the participant’s MRI scan at the month 6 visit was used for the co-registration. The MR images were co-registered to a standard T1-template image, bias corrected, segmented using a T1-weighted MRI template from SPM5, and spatially normalized to Montreal Neurological Institute (MNI) space (Risacher et al., 2009). Normalization parameters obtained from the segmentation of the MP-RAGE scans were then used to normalize each participant’s PiB-PET image to MNI space. All scan processing and normalization steps were subjected to QC on an individual level. The processed PiB-PET images of all participants were then entered into voxel- and cluster-wise statistical analysis.
Genotype data
Acquisition of genotype data
Methods for acquisition and processing of genotype data for the ADNI sample have been previously described (Saykin et al., 2010). The Human610-Quad BeadChip (Illumina, Inc., San Diego, CA) was used to analyze samples with all sources of DNA according to the manufacturer’s protocol (Infinium HD Assay; Super Protocol Guide; rev. A, May 2008). SNP genotypes were generated from bead intensity data using Illumina BeadStudio 3.2 software.
The two APOE SNPs (rs429358, rs7412) that define the ε2, ε3 and ε4 alleles were not available on the Illumina Human610-Quad BeadChip. These SNPs were genotyped separately and were made available in the ADNI database (Potkin et al., 2009; Saykin et al., 2010).
Gene and SNP selection criteria for the gene-based association analysis of amyloid-pathway associated candidate genes
As only a modest size sample (103 participants) had completed the PiB-PET protocol, we explicitly focused our analysis on amyloid-pathway associated genes that were also present in the AlzGene database. The AlzGene database (http://www.alzgene.org/) provides a comprehensive curated list and meta-analysis of genes previously investigated for association to AD (Bertram, McQueen, Mullin, Blacker, & Tanzi, 2007). A search for the Gene Ontology (GO) term or ID “amyloid” was made on the GO website (http://www.geneontology.org/) (Ashburner et al., 2000) (as of November 2010). The search identified 37 genes associated with different GO “amyloid” terms. The APOE gene was excluded from the analysis as the APOE ε4 status was used as a covariate in the analysis (see below). The APP (amyloid beta (A4) precursor protein) gene was not identified in the search results, but given its role as a precursor molecule in the generation of Aβ fragments, this gene was included in the analysis. Twenty of the 37 genes were also present in the AlzGene database (as of November 2010) and these 20 genes were selected for further filtering.
As an initial screening analysis, we selected SNPs that were located in these genes, i.e. SNPs in the untranslated region (UTR), 3′ UTR, 5′ UTR, coding or intronic regions of these genes as defined in the Illumina Human610-Quad Gene Annotation file. 16 genes with 489 SNPs were found to satisfy this criteria and selected for QC performed in PLINK v1.07 (http://pngu.mgh.harvard.edu/~purcell/plink/) (Purcell et al., 2007). Samples were selected if they had a call rate >95%. SNPs were selected if they had a call rate >95% and minor allele frequency >20%. A relatively high minor allele frequency was used to ensure adequate sample size in the three genotype groups to reduce the likelihood of false positives due to small cell sizes. 213 SNPs did not pass this filter. 103 samples and 272 SNPs in 15 genes remained after all QC and filter steps (Table 2) and were entered into the association analysis. The analysis was also repeated restricting the sample to the 95 non-Hispanic Caucasian participants in the present study to confirm that the findings obtained from the analysis with all 103 participants were not biased by subpopulation effects. In addition, a cluster analysis was performed for all 818 ADNI participants using the smartpca program from the EIGENSOFT package (Price et al., 2006). The primary analysis was repeated using the first two principal components as additional covariates.
Statistical analyses
Two approaches were used in this study: (1) a gene-based association approach of amyloid-pathway associated candidate genes, to identify genes and SNPs associated with the average PiB uptake from four brain regions; and (2) a whole brain, voxel- and cluster-wise approach, in which the identified SNPs were tested for associations with PiB uptake across the whole brain. An overview of the steps for both approaches is shown in Fig. 1. Descriptive statistical analysis was carried out in PASW Statistics 18 (SPSS Inc., Chicago, IL).

Overview of steps used for the analyses. a Gene-based association analysis of amyloid-pathway associated candidate genes. b Whole-brain voxel- and cluster-wise analysis. SNP-single nucleotide polymorphism; APOE-apolipoprotein E; APP-amyloid beta (A4) precursor protein; PiB-Pittsburgh Compound B; PET-positron emission tomography; MRI-magnetic resonance imaging; MNI-Montreal Neurological Institute; MP-RAGE-3D magnetization prepared rapid acquisition gradient echo
Gene-based association analysis of amyloid-pathway associated candidate genes
The gene-based association analysis of amyloid-pathway associated candidate genes was performed using the “Set-based tests” option in PLINK v1.07 (Purcell et al., 2007) (http://pngu.mgh.harvard.edu/~purcell/plink/anal.shtml#set). This analysis performs a standard SNP quantitative trait analysis. A mean test statistic for each gene is calculated taking into account the number of significant SNPs in each gene as well as selecting SNPs that are not in linkage disequilibrium (LD) with each other. A permutation is also performed, keeping LD between SNPs constant and permuting the individual labels. An empirical p-value is then reported for each gene with at least one significant SNP. We used the default PLINK parameters in the present analysis: SNPs were considered to be in LD if the correlation coefficient between them was r2 > 0.5; SNPs from the quantitative trait analysis were considered significant with a p-value < 0.05; and a maximum of five significant SNPs from each gene was chosen for calculating the mean test statistic. The analysis was performed using an additive model, i.e., the additive effect of the SNP minor allele on the phenotypic mean was estimated. Factors known to influence amyloid deposition, i.e. age at time of scan (Yamada, Tsukagoshi, Otomo, & Hayakawa, 1988), gender (Hirata-Fukae et al., 2008), diagnosis at time of scan (Jagust et al., 2010) and presence/absence of APOE ε4 allele (Reiman et al., 2009) were included as covariates in the analysis. One hundred thousand permutations were performed.
The empirical p-values obtained for each gene were then analyzed using the Benjamini-Hochberg false discovery rate (FDR) procedure (Benjamini & Hochberg, 1995), with a commonly employed q-value threshold of 0.10 to indicate genes with significant association to the average PiB uptake (van den Oord, 2008). The q-value is the FDR analog of the family-wise error (FWE) adjusted p-value. For a given test, the q-value provides the smallest FDR for which the null can be rejected and a discovery declared. A more powerful approach such as the one used in the whole-brain cluster-wise analysis (Ventura, Paciorek, & Risbey, 2004) (as described below) could not be used because it would require a p-value for each gene. Using the “Set-based tests” procedure in PLINK, a permutation p-value for a gene exists only if there is at least one significant SNP in the gene. The number of significant SNPs identified depends on the parameters specified in the analysis. Comparison graphs were created using SigmaPlot for Windows Version 10.0 (Systat Software, Inc., San Jose, CA).
Power analysis
We performed a power calculation in Quanto (Version 1.2.4, May 2009) (Gauderman & Morrison, 2006) to estimate the power of detecting variation in the mean normalized PiB uptake that can be explained by genetic factors. We also determined the variance in mean normalized PiB uptake explained by APOE ε4 status, as well as a SNP in a novel gene identified in the present study. Age at time of scan, gender, and diagnosis at time of scan were included as covariates in both variance analyses. In addition, APOE ε4 status was included as a covariate in the variance analysis of the identified SNP.
Whole-brain voxel- and cluster-wise analysis
As a follow-up to the findings from the gene-based association analyses, voxel- and cluster-wise differences in normalized PiB uptake among the three genotype groups were assessed in SPM5 using a multiple regression model. Covariates used in the gene-based association analysis (age at time of scan, gender, diagnosis at time of scan and presence/absence of APOE ε4 allele) were also included as covariates in the voxel- and cluster-wise analysis, and an explicit grey matter (GM) mask was used to restrict analysis to GM regions. Voxels were projected onto the three-dimensional rendered brain provided by SPM5 for visualization. The MNI coordinates of the local maximum of each cluster were converted to Talairach coordinates, and were loaded into Talairach Client v2.4.2 (Lancaster et al., 1997; Lancaster et al., 2000) software to determine the associated anatomic labels.
The statistical parametric map of the regression t-statistics was analyzed in SPM5 using a voxel-level significance threshold of p < 0.001 (uncorrected) and minimum cluster size k = 50 contiguous voxels. Voxel- and cluster-level statistics were assessed using both FWE and FDR type I error control. For voxel-level analysis in SPM5, the FWE and FDR corrected p-value statistics were used. For cluster-level analysis, the random field theory corrected p-values were used. In addition, the uncorrected cluster-level p-values were assessed using the FDR algorithm of Ventura et al. (Chumbley, Worsley, Flandin, & Friston, 2010; Chumbley & Friston, 2009; Ventura et al., 2004). The author’s FDR program was accessed through the download fdr.jcli (S/R version) from the software archives at StatLib (http://lib.stat.cmu.edu) and run using R linux-gnu version 2.10.0 (2009-10-26). Clusters were considered significant if their random field theory corrected p-value did not exceed 0.05 or their FDR q-value did not exceed 0.10 (Genovese, Lazar, & Nichols, 2002).
Results
Gene-based association analysis of amyloid-pathway associated candidate genes
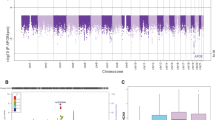
A gene-based association analysis of amyloid-pathway associated candidate genes was performed in 103 ADNI participants to determine genes and SNPs associated with the average normalized PiB uptake from four regions (anterior cingulate, frontal cortex, parietal cortex, and precuneus). Table 3 shows the summary output of the gene-based association analysis obtained from PLINK. The DHCR24 gene was observed to be significantly associated with the average PiB uptake (empirical gene-based p = 0.0049, uncorrected; FDR q = 0.0735). Three SNPs in this gene were included in the quantitative trait analysis, but only one intronic SNP (rs7551288) was found to be significantly associated with the average PiB uptake (p = 0.0028, uncorrected). An additive effect of this SNP was observed (Fig. 2), i.e., 35 participants with no copies of the minor allele (GG genotype) had a higher average PiB uptake compared to 50 participants with one copy of the minor allele (AG genotype) and 18 participants with two copies of the minor allele (AA genotype).

Comparison of average PiB uptake from four brain regions in the three genotype groups (GG, AG and AA) of rs7551288. ‘A’ represents the minor allele and ‘G’ represents the major allele of this SNP
To remove possible effects of age, diagnosis, APOE ε4 status and gender on the average PiB uptake, we included them as covariates in the analyses. The absence/presence of covariates did not significantly alter the pattern of results (data not shown). Further, on performing the analyses without APOE ε4 status as a covariate (but all other covariates included), the DHCR24 gene (empirical gene-based p = 0.0012, uncorrected) and the rs7551288 SNP (p = 0.0008, uncorrected) were observed to be significantly associated with the average PiB uptake. Finally, as there could be an interaction of the rs7551288 SNP with the covariates, we also performed a series of two-way analyses of variance (ANOVA) to determine if there was an interaction effect of the SNP with diagnosis, APOE ε4 status and gender on the unadjusted average PiB uptake. No significant interaction was observed at the 0.05 level (data not shown).
Two analyses were performed to examine the possible influence of population stratification. First, the analysis was performed with only the 95 non-Hispanic Caucasian participants. Similar to the analysis of all 103 participants, the DHCR24 gene was identified, but with a slightly attenuated signal (empirical gene-based p = 0.0167, uncorrected). rs7551288 was again found to be the only SNP within the gene significantly associated with the average PiB uptake (p = 0.0090, uncorrected). Second, we also repeated the analysis using the first two principal components from EIGENSOFT as covariates. Again, the DHCR24 gene (empirical gene-based p = 0.0222) and the rs7551288 SNP (p = 0.0110, uncorrected) remained significantly associated with average PiB uptake.
Power calculations
APOE ε4 status, the strongest AD-associated genetic risk factor, accounts for approximately 12.84% of the variation in average PiB uptake, when accounting for covariates. The identified SNP (rs7551288) in the DCHR24 gene accounts for an additional 6.65% of the variation in average PiB uptake. We calculated that with the present sample size of 103 participants, we had 80% power to detect 18.5% of variation in average PiB uptake explained by genetic factors.
Whole brain voxel- and cluster-wise analysis
Similar to the gene-based association analysis using the ROI approach, a multiple regression was performed to determine the additive effect of the minor allele (A allele) of rs7551288 on a voxel- and cluster-wise level. Age at time of scan, gender, diagnosis at time of scan and presence/absence of APOE ε4 allele were used as covariates in this analysis. Higher mean PiB uptake in participants with the GG genotype group compared to participants with the AG and AA genotype groups (additive model: GG > AG > AA) was found in frontal regions (p < 0.001, uncorrected; k = 50 voxels; Table 4). To illustrate the topography of amyloid deposition associated with this SNP in an unbiased manner, sagittal brain images are presented at p < 0.005 (uncorrected) in Fig. 3. All voxels in the voxel-level analysis were significant after FDR correction (p FDR-corr = 0.044). In the cluster-level analysis, two clusters in the left middle frontal gyrus (MNI peak value coordinates: x = −22, y = −10, z = 64; k = 271 voxels) and the left medial frontal gyrus (MNI peak value coordinates: x = −2, y = 54, z = 22; k = 295 voxels) were observed to be significant after random field theory correction (p corr = 0.038 and 0.028 respectively) and FDR correction (FDR q = 0.067 for each of the two clusters). No regions were observed at the same statistical threshold in the reverse contrast. These results are similar to those obtained in the gene-based association analysis but provide a spatially unbiased illustration of the distribution of PiB uptake differences as a function of DHCR24 genotype.

Visualization of the results of statistical parametric mapping analysis. The regions with significantly (uncorrected p < 0.005, cluster size = 50 voxels) increased PiB uptake in 35 participants with the GG genotype group compared to 50 participants with the AG and 18 participants with the AA genotype groups are shown in color. The red-to-yellow scale indicates increasing statistical significance of differences in PiB uptake
Discussion
A quantitative phenotype approach to genetic association studies offers the advantage of increased power and consequently requires smaller sample sizes to detect significant genetic effects as compared to a traditional case-control design (Potkin et al., 2009). This methodology has been used within ADNI to identify novel loci associated with several AD phenotypes, including: hippocampal atrophy (Potkin et al., 2009), temporal lobe structure (Stein et al., 2010), brain-wide imaging phenotypes (Shen et al., 2010) and cerebrospinal fluid measures (Han et al., 2010; Kim et al., 2011).
To our knowledge, this report represents the first study to use PiB-PET data as a quantitative phenotype to identify genes other than APOE (Drzezga et al., 2009; Morris et al., 2010; Reiman et al., 2009) that are possibly associated with fibrillar amyloid burden, and hence probable AD susceptibility. 103 participants in the ADNI cohort had PiB-PET scans, and an automated ROI template extraction method was applied to sample different brain areas. The average normalized PiB uptake values from anterior cingulate, frontal cortex, parietal and precuneus regions are often used to classify individuals as having either positive or negative amyloid deposition (Jagust et al., 2010). Because these regions are robust indicators of plaque burden as indicated by amyloid deposition, a single overall averaged PiB value of these four regions was hypothesized to represent a suitable quantitative phenotype for genetic analyses.
Given the modest sample size (103 ADNI participants) that had both genotype and phenotype data, we employed a targeted gene-based approach, using an additive model to identify genes associated with the average PiB uptake. We focused our analysis on amyloid-associated genes that were previously studied for association with AD. An intronic SNP (rs7551288) in the DHCR24 gene on chromosome 1 was found to be significantly associated with the average PiB uptake. This SNP appeared to have a protective effect, as the presence of the minor allele (A allele) was associated with a lower average PiB uptake and hence lower amyloid burden (Fig. 2). Despite the modest available sample of 103 participants, the present study had the power to detect genetic effects that explain almost 7% of the variation in average PiB uptake after controlling for covariates used in the analyses. We believe this is quite substantial for an imaging genetics effect.
To determine if the DHCR24 gene and, in particular, the rs7551288 SNP had an effect on other brain regions, a whole brain voxel- and cluster-wise analysis with this SNP was also performed. We investigated whether there were brain regions where there was increased PiB uptake in participants with no copies of the minor allele (GG genotype) compared to those who had one copy (AG genotype) or two copies of the minor allele (AA genotype). The results were similar to those obtained in the gene-based association analysis. Non-carriers of the minor allele (GG genotype) had higher PiB uptake in the frontal regions compared to carriers of the minor allele (AG and AA genotypes) (Fig. 3, Table 4). The frontal cortical areas are regions known to have higher PiB retention in AD patients compared to controls (Jack et al., 2008; Kemppainen et al., 2006; Klunk et al., 2004). Amyloid deposition has been shown to first occur in the cortical areas, followed by accumulation in the entorhinal region (Thal, Rüb, Orantes, & Braak, 2002). We did not observe any differences in PiB uptake between the genotype groups in the hippocampal regions. This is consistent with previous reports (Jack et al., 2008; Kemppainen et al., 2006; Klunk et al., 2004) which did not find differences in PiB uptake in the medial temporal lobe in AD patients compared to controls. The current results, which show that the DHCR24 gene is associated with frontal amyloid burden, suggest that this gene may play a role in the pathogenesis of AD.
The DHCR24 (24-dehydrocholesterol) gene (also known as seladin-1 or SELective AD INdicator-1) encodes the enzyme 3-β-hydroxysterol δ-24-reductase. This enzyme catalyzes the reduction of the δ-24 double bond in desmosterol to produce cholesterol. The gene was initially identified by a differential mRNA display approach (Greeve et al., 2000). Reduced mRNA and protein expression of this gene was found in the inferior temporal lobe compared with the frontal cortex in AD brains. This was in contrast to brains from normal, non-demented control participants, where no differences in mRNA and protein expression were detected among these brain regions. These findings suggest a disease-related alteration in DHCR24 expression.
The DHCR24 gene is thought to interact with cholesterol, estrogen and insulin-like growth factor 1 (IGF1) (Peri et al., 2009). The authors of that study have proposed a “circular” model in which DHCR24 expression is stimulated by estrogen, which, in turn, increases cholesterol synthesis. This leads to increased expression of steroidogenic acute regulatory protein (StAR), which regulates the movement of cholesterol from the outer to the inner mitochondrial membrane, and is thus important for the initiation of steroid synthesis. Genetic variations in the TOMM40 (translocase of outer mitochondrial membrane 40) gene have been associated with AD (Kim et al., 2011; Potkin et al., 2009; Roses et al., 2010; Shen et al., 2010), thus suggesting a possible role for mitochondria in AD pathogenesis. The synthesis of cholesterol induces the synthesis of estradiol, which in turn stimulates the expression of DHCR24. This may happen via IGF1, as estradiol increases IGF1 release, which binds to IGF1 receptors and stimulates the expression of DHCR24.
The DHCR24 gene appears to play a neuroprotective role. It has been shown to confer resistance to Aβ and oxidative stress-induced apoptosis by inhibiting caspase-3 activation, a key mediator of apoptosis (Greeve et al., 2000). Recently, Sarajärvi et al. elucidated the possible molecular mechanisms that could be related to DHCR24 down-regulation in the AD brain (Sarajärvi et al., 2009). The authors found that in SH-SY5Y human neuroblastoma cells under apoptotic conditions, reduced DHCR24 expression resulted in increased caspase-3 activity and enhanced depletion of the BACE1 (beta-site APP-cleaving enzyme-1)-sorting protein GGA3 (golgi-associated, gamma adaptin ear containing, ARF binding protein 3). This resulted in increased BACE1 protein levels, leading to enhanced β-amyloidogenic processing of APP and increased Aβ production. It is important to note that no changes in GGA3 or BACE1 levels were observed with reduced DHCR24 expression under normal growth conditions. These findings may be important, as oxidative stress-induced apoptosis is thought to play a key role in AD pathogenesis (Loh, Huang, De Silva, Tan, & Zhu, 2006). To investigate the genetic association of the DHCR24 gene with AD, Lamsa et al. genotyped four SNPs in 414 Finnish AD cases and 459 controls, and performed allelic and haplotype association analyses (Lämsä et al., 2007). They were able to identify a risk allele, as well as risk and protective haplotypes, and concluded that the gene may be associated with AD. The AlzGene database (Updated 18 April 2011) lists three case-control studies for the DHCR24 gene (http://www.alzgene.org/geneoverview.asp?geneid=515). Four polymorphisms (rs638944, rs600491, rs718265 and rs7374) were investigated in these studies. However, a meta-analyses of these polymorphisms has not been performed to date. Thus, the results in the current study support the hypothesis of a neuroprotective role of DHCR24. Further analysis in independent cohorts is warranted.
It is important to note some limitations of the present study. Only 103 ADNI participants had initial PiB-PET scans as well as genotype data, which is a modest sample size for genetic analysis. In the next phases of ADNI (ADNI-GO and ADNI-2), all participants will receive amyloid PET imaging with the [18 F]-labeled amyloid imaging agent, Florbetapir, providing additional opportunities to examine genetic influence on amyloid deposition in a larger sample. ADNI-GO and ADNI-2 will also recruit a cohort of very mild, early MCI (EMCI) participants. These individuals differ from the late MCI (LMCI) individuals in the ADNI-1 cohort in that they have more mild memory impairments that may represent an earlier prodromal manifestation of AD. Thus, a larger and more diverse sample would be available to perform a similar gene-based analysis, as well as a whole-genome analysis. Even with the limited number of samples, a previously associated AD gene (DHCR24) and an intronic SNP (rs7551288) within this gene were identified in the present analysis, highlighting the potential of using a quantitative imaging phenotype to identify gene regions that may be associated with disease pathology. The present findings support the hypothesis of a neuroprotective role of this gene based on prior independent research, and indicate the potential importance of further investigation of this gene in larger cohorts.
Another limitation is the criteria used to select genes and SNPs for the analyses. Genes associated with the amyloid pathway were selected from a single database (Gene Ontology). It is possible that we may have missed other amyloid-associated genes not represented in this database. In order to reduce the incidence of false positives, we selected SNPs that had a minor allele frequency >20% and thus excluded less common variants that may play a significant role in amyloid deposition. Deep-sequencing of the gene regions analyzed in the present study may help to identify other variants that may mediate amyloid-associated AD pathology. Finally, the inclusion of the presence of APOE ε4 genotype status as a covariate may have attenuated the magnitude of the effect for DHCR24. Nonetheless inclusion of APOE lends support to the hypothesis that DHCR24 contributes new information with regard to AD mechanisms and risk.
Conclusion
In sum, we analyzed the association between variation in selected amyloid-pathway genes and PiB-PET measures of fibrillar amyloid deposition in the ADNI cohort. The analysis identified an intronic SNP (rs7551288) in the DHCR24 gene that was associated with PiB uptake. The SNP appeared to have a protective effect against amyloid deposition in brain regions known to have preferential amyloid plaque burden in AD, consistent with earlier reports of a neuroprotective role of this gene. Replication of this finding in independent cohorts and targeted analysis of this gene are warranted to further evaluate its role in AD susceptibility. These findings encourage further use of pathway-based genetic analysis of targeted molecular imaging phenotypes. This approach appears promising for helping to elucidate disease pathophysiology and may identify potential therapeutic targets.
References
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. Nature Genetics, 25(1), 25–29.
Association, A. (2011). 2011 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia, 7(2), 208–244. doi:10.1016/j.jalz.2011.02.004
Benjamini, Y., & Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological), 57(1), 289–300.
Berti, V., Nacmias, B., Bagnoli, S., & Sorbi, S. (2011). Alzheimer’s disease: genetic basis and amyloid imaging as endophenotype. The Quarterly Journal of Nuclear Medicine and Molecular Imaging, 55(3), 225–236.
Bertram, L., McQueen, M. B., Mullin, K., Blacker, D., & Tanzi, R. E. (2007). Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nature Genetics, 39(1), 17–23. http://www.nature.com/ng/journal/v39/n1/suppinfo/ng1934_S1.html
Bertram, L., Lill, C. M., & Tanzi, R. E. (2010). The genetics of Alzheimer disease: back to the future. Neuron, 68(2), 270–281. doi:10.1016/j.neuron.2010.10.013
Chumbley, J. R., & Friston, K. J. (2009). False discovery rate revisited: FDR and topological inference using Gaussian random fields. NeuroImage, 44(1), 62–70. doi:10.1016/j.neuroimage.2008.05.021
Chumbley, J., Worsley, K., Flandin, G., & Friston, K. (2010). Topological FDR for neuroimaging. NeuroImage, 49(4), 3057–3064. doi:10.1016/j.neuroimage.2009.10.090
Clark, C. M., Schneider, J. A., Bedell, B. J., Beach, T. G., Bilker, W. B., Mintun, M. A., et al. (2011). Use of Florbetapir-PET for imaging β-amyloid pathology. JAMA: The Journal of the American Medical Association, 305(3), 275–283. doi:10.1001/jama.2010.2008
Corder, E., Saunders, A., Strittmatter, W., Schmechel, D., Gaskell, P., Small, G., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science, 261(5123), 921–923. doi:10.1126/science.8346443
Drzezga, A., Grimmer, T., Henriksen, G., Mühlau, M., Perneczky, R., Miederer, I., et al. (2009). Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology, 72(17), 1487–1494. doi:10.1212/WNL.0b013e3181a2e8d0
Farrer, L. A., Cupples, L. A., Haines, J. L., Hyman, B., Kukull, W. A., Mayeux, R., et al. (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. JAMA: The Journal of the American Medical Association, 278(16), 1349–1356. doi:10.1001/jama.1997.03550160069041
Friston, K. J., Holmes, A. P., Worsley, K. J., Poline, J. P., Frith, C. D., & Frackowiak, R. S. J. (1994). Statistical parametric maps in functional imaging: a general linear approach. Human Brain Mapping, 2(4), 189–210. doi:10.1002/hbm.460020402
Gauderman, W., & Morrison, J. (2006). QUANTO 1.1: A computer program for power and sample size calculations for genetic-epidemiology studies, http://hydra.usc.edu/gxe. Retrieved from http://hydra.usc.edu/gxe
Genovese, C. R., Lazar, N. A., & Nichols, T. (2002). Thresholding of statistical maps in functional neuroimaging using the false discovery rate. NeuroImage, 15(4), 870–878. doi:10.1006/nimg.2001.1037
Greeve, I., Hermans-Borgmeyer, I., Brellinger, C., Kasper, D., Gomez-Isla, T., Behl, C., et al. (2000). The human DIMINUTO/DWARF1 homolog seladin-1 confers resistance to Alzheimer’s disease-associated neurodegeneration and oxidative stress. The Journal of Neuroscience, 20(19), 7345–7352.
Hampel, H., Frank, R., Broich, K., Teipel, S. J., Katz, R. G., Hardy, J., et al. (2010). Biomarkers for Alzheimer’s disease: academic, industry and regulatory perspectives. Nature Reviews Drug Discovery, 9(7), 560–574. http://www.nature.com/nrd/journal/v9/n7/suppinfo/nrd3115_S1.html
Han, M.-R., Schellenberg, G., Wang, L.-S., & the Alzheimer’s Disease Neuroimaging Initiative (2010). Genome-wide association reveals genetic effects on human Abeta42 and tau protein levels in cerebrospinal fluids: a case control study. BMC Neurology, 10(1), 90.
Harold, D., Abraham, R., Hollingworth, P., Sims, R., Gerrish, A., Hamshere, M. L., et al. (2009). Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nature Genetics, 41(10), 1088–1093. http://www.nature.com/ng/journal/v41/n10/suppinfo/ng.440_S1.html
Hirata-Fukae, C., Li, H.-F., Hoe, H.-S., Gray, A. J., Minami, S. S., Hamada, K., et al. (2008). Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Research, 1216, 92–103. doi:10.1016/j.brainres.2008.03.079
Hollingworth, P., Harold, D., Sims, R., Gerrish, A., Lambert, J.-C., Carrasquillo, M. M., et al. (2011). Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nature Genetics, 43(5), 429–435. http://www.nature.com/ng/journal/v43/n5/abs/ng.803.html#supplementary-information
Jack, C. R., Bernstein, M. A., Fox, N. C., Thompson, P., Alexander, G., Harvey, D., et al. (2008). The Alzheimer’s disease neuroimaging initiative (ADNI): MRI methods. Journal of Magnetic Resonance Imaging, 27(4), 685–691. doi:10.1002/jmri.21049
Jack, C. R., Lowe, V. J., Senjem, M. L., Weigand, S. D., Kemp, B. J., Shiung, M. M., et al. (2008). 11 C PiB and structural MRI provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain, 131(3), 665–680. doi:10.1093/brain/awm336
Jagust, W. J., Landau, S. M., Shaw, L. M., Trojanowski, J. Q., Koeppe, R. A., Reiman, E. M., et al. (2009). Relationships between biomarkers in aging and dementia. Neurology, 73(15), 1193–1199. doi:10.1212/WNL.0b013e3181bc010c
Jagust, W. J., Bandy, D., Chen, K., Foster, N. L., Landau, S. M., Mathis, C. A., et al. (2010). The Alzheimer’s disease Neuroimaging initiative positron emission tomography core. Alzheimer’s & Dementia, 6(3), 221–229. doi:10.1016/j.jalz.2010.03.003
Jun, G., Naj, A. C., Beecham, G. W., Wang, L.-S., Buros, J., Gallins, P. J., et al. (2010). Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Archives of Neurology, 67(12), 1473–1484. doi:10.1001/archneurol.2010.201
Kemppainen, N. M., Aalto, S., Wilson, I. A., Någren, K., Helin, S., Brück, A., et al. (2006). Voxel-based analysis of PET amyloid ligand [11 C]PIB uptake in Alzheimer disease. Neurology, 67(9), 1575–1580. doi:10.1212/01.wnl.0000240117.55680.0a
Kim, S., Swaminathan, S., Shen, L., Risacher, S. L., Nho, K., Foroud, T., et al. (2011). Genome-wide association study of CSF biomarkers Aβ1-42, t-tau, and p-tau181p in the ADNI cohort. Neurology, 76(1), 69–79. doi:10.1212/WNL.0b013e318204a397
Klunk, W. E., Engler, H., Nordberg, A., Wang, Y., Blomqvist, G., Holt, D. P., et al. (2004). Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Annals of Neurology, 55(3), 306–319. doi:10.1002/ana.20009
Lambert, J.-C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nature Genetics, 41(10), 1094–1099. http://www.nature.com/ng/journal/v41/n10/suppinfo/ng.439_S1.html
Lämsä, R., Helisalmi, S., Hiltunen, M., Herukka, S. K., Tapiola, T., Pirttilä, T., et al. (2007). The association study between DHCR24 polymorphisms and Alzheimer’s disease. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics, 144B(7), 906–910. doi:10.1002/ajmg.b.30532
Lancaster, J. L., Rainey, L. H., Summerlin, J. L., Freitas, C. S., Fox, P. T., Evans, A. C., et al. (1997). Automated labeling of the human brain: a preliminary report on the development and evaluation of a forward-transform method. Human Brain Mapping, 5(4), 238–242. doi:10.1002/(sici)1097-0193(1997)5:4<238::aid-hbm6>3.0.co;2-4
Lancaster, J. L., Woldorff, M. G., Parsons, L. M., Liotti, M., Freitas, C. S., Rainey, L., et al. (2000). Automated Talairach Atlas labels for functional brain mapping. Human Brain Mapping, 10(3), 120–131. doi:10.1002/1097-0193(200007)10:3<120::aid-hbm30>3.0.co;2-8
Loh, K. P., Huang, S. H., De Silva, R., Tan, B. K., & Zhu, Y. Z. (2006). Oxidative stress: apoptosis in neuronal injury. Current Alzheimer Research, 3(4), 327–337.
Morris, J. C., Roe, C. M., Xiong, C., Fagan, A. M., Goate, A. M., Holtzman, D. M., et al. (2010). APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Annals of Neurology, 67(1), 122–131. doi:10.1002/ana.21843
Naj, A. C., Beecham, G. W., Martin, E. R., Gallins, P. J., Powell, E. H., Konidari, I., et al. (2010). Dementia revealed: novel chromosome 6 locus for late-onset Alzheimer disease provides genetic evidence for folate-pathway abnormalities. PLoS Genetics, 6(9), e1001130.
Naj, A. C., Jun, G., Beecham, G. W., Wang, L.-S., Vardarajan, B. N., Buros, J., et al. (2011). Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nature Genetics, 43(5), 436–441. http://www.nature.com/ng/journal/v43/n5/abs/ng.801.html#supplementary-information
Peri, A., Danza, G., Benvenuti, S., Luciani, P., Deledda, C., Rosati, F., et al. (2009). New insights on the neuroprotective role of sterols and sex steroids: the seladin-1/DHCR24 paradigm. Frontiers in Neuroendocrinology, 30(2), 119–129. doi:10.1016/j.yfrne.2009.03.006
Petersen, R. C., Roberts, R. O., Knopman, D. S., Boeve, B. F., Geda, Y. E., Ivnik, R. J., et al. (2009). Mild cognitive impairment: ten years later. Archives of Neurology, 66(12), 1447–1455. doi:10.1001/archneurol.2009.266
Potkin, S. G., Guffanti, G., Lakatos, A., Turner, J. A., Kruggel, F., Fallon, J. H., et al. (2009). Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for Alzheimer’s disease. PloS One, 4(8), e6501.
Potkin, S. G., Turner, J. A., Guffanti, G., Lakatos, A., Torri, F., Keator, D. B., et al. (2009). Genome-wide strategies for discovering genetic influences on cognition and cognitive disorders: methodological considerations. Cognitive Neuropsychiatry, 14(4), 391–418.
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., & Reich, D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics, 38(8), 904–909. doi:10.1038/ng1847
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics, 81(3), 559–575. doi:10.1086/519795
Rabinovici, G. D., & Jagust, W. J. (2009). Amyloid imaging in aging and dementia: testing the amyloid hypothesis in vivo. Behavioural Neurology, 21(1), 117–128.
Reiman, E. M., Chen, K., Liu, X., Bandy, D., Yu, M., Lee, W., et al. (2009). Fibrillar amyloid-β burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proceedings of the National Academy of Sciences, 106(16), 6820–6825. doi:10.1073/pnas.0900345106
Risacher, S. L., Saykin, A. J., West, J. D., Shen, L., Firpi, H. A., & McDonald, B. C. (2009). Baseline MRI predictors of conversion from MCI to probable AD in the ADNI cohort. Current Alzheimer Research, 6(4), 347–361.
Roses, A. D., Lutz, M. W., Amrine-Madsen, H., Saunders, A. M., Crenshaw, D. G., Sundseth, S. S., et al. (2010). A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. The Pharmacogenomics Journal, 10(5), 375–384. http://www.nature.com/tpj/journal/v10/n5/suppinfo/tpj200969s1.html
Sarajärvi, T., Haapasalo, A., Viswanathan, J., Mäkinen, P., Laitinen, M., Soininen, H., et al. (2009). Down-regulation of seladin-1 increases BACE1 levels and activity through enhanced GGA3 depletion during apoptosis. The Journal of Biological Chemistry, 284(49), 34433–34443. doi:10.1074/jbc.M109.036202
Saunders, A. M., Strittmatter, W. J., Schmechel, D., St. George-Hyslop, P. H., Pericak-Vance, M. A., Joo, S. H., et al. (1993). Association of apolipoprotein E allele ϵ4 with late–onset familial and sporadic Alzheimer’s disease. Neurology, 43(8), 1467.
Saykin, A. J., Shen, L., Foroud, T. M., Potkin, S. G., Swaminathan, S., Kim, S., et al. (2010). Alzheimer’s disease neuroimaging initiative biomarkers as quantitative phenotypes: genetics core aims, progress, and plans. Alzheimer’s & Dementia, 6(3), 265–273. doi:10.1016/j.jalz.2010.03.013
Seshadri, S., Fitzpatrick, A. L., Ikram, M. A., DeStefano, A. L., Gudnason, V., Boada, M., et al. (2010). Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA: The Journal of the American Medical Association, 303(18), 1832–1840. doi:10.1001/jama.2010.574
Shen, L., Kim, S., Risacher, S. L., Nho, K., Swaminathan, S., West, J. D., et al. (2010). Whole genome association study of brain-wide imaging phenotypes for identifying quantitative trait loci in MCI and AD: a study of the ADNI cohort. NeuroImage, 53(3), 1051–1063. doi:10.1016/j.neuroimage.2010.01.042
Stein, J. L., Hua, X., Lee, S., Ho, A. J., Leow, A. D., Toga, A. W., et al. (2010). Voxelwise genome-wide association study (vGWAS). NeuroImage, 53(3), 1160–1174. doi:10.1016/j.neuroimage.2010.02.032
Stein, J. L., Hua, X., Morra, J. H., Lee, S., Hibar, D. P., Ho, A. J., et al. (2010). Genome-wide analysis reveals novel genes influencing temporal lobe structure with relevance to neurodegeneration in Alzheimer’s disease. NeuroImage, 51(2), 542–554. doi:10.1016/j.neuroimage.2010.02.068
Thal, D. R., Rüb, U., Orantes, M., & Braak, H. (2002). Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology, 58(12), 1791–1800.
van den Oord, E. J. C. G. (2008). Controlling false discoveries in genetic studies. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics, 147B(5), 637–644. doi:10.1002/ajmg.b.30650
Ventura, V., Paciorek, C. J., & Risbey, J. S. (2004). Controlling the proportion of falsely rejected hypotheses when conducting multiple tests with climatological data. Journal of Climate, 17(22), 4343–4356. doi:10.1175/3199.1
Weiner, M. W., Aisen, P. S., Jack Jr, C. R., Jagust, W. J., Trojanowski, J. Q., Shaw, L., et al. (2010). The Alzheimer’s Disease Neuroimaging Initiative: Progress report and future plans. Alzheimer’s and Dementia, 6(3), 202–211.e207. doi: 10.1016/j.jalz.2010.03.007
Yamada, M., Tsukagoshi, H., Otomo, E., & Hayakawa, M. (1988). Systemic amyloid deposition in old age and dementia of Alzheimer type: the relationship of brain amyloid to other amyloid. Acta Neuropathologica, 77(2), 136–141. doi:10.1007/bf00687423
Acknowledgments
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health (NIH) Grant U01 AG024904). ADNI is funded by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), and through generous contributions from the following: Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson and Johnson, Eli Lilly and Co., Medpace, Inc., Merck and Co., Inc., Novartis AG, Pfizer Inc, F. Hoffman-La Roche, Schering-Plough, Synarc, Inc., and Wyeth, as well as non-profit partners the Alzheimer’s Association and Alzheimer’s Drug Discovery Foundation, with participation from the U.S. Food and Drug Administration. Private sector contributions to ADNI are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129, K01 AG030514, the Dana Foundation and U01 AG032984 Alzheimer’s Disease Genetics Consortium (ADGC) grant (PI: Schellenberg).
Samples from the National Cell Repository for AD (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the NIA, were used in this study. Additional support for data analysis was provided by NIA R01 AG19771 and P30 AG10133. The authors thank contributors, including the ADNI sites that collected samples used in this study, as well as patients and their families, whose help and participation made this work possible.
We thank the following people for their contributions to the ADNI genotyping project: (1) genotyping at the Translational Genomics Institute, Phoenix AZ: Jennifer Webster, Jill D. Gerber, April N. Allen, and Jason J. Corneveaux; and (2) sample processing, storage and distribution at the NIA-sponsored NCRAD: Kelley Faber and Colleen Mitchell.
Disclosures
Dr. Shen receives support from the NIH (NIBIB R03 EB008674 [PI], NIBIB R03 EB008674S1 [PI], UL1RR025761 [PI of an Indiana CTSI pilot project]), and the NSF (IIS-1117335 [PI]).
Ms. Risacher receives support from the NIH (CTSI Pre-doctoral training grant, TL1 RR025759).
Dr. Yoder is supported by NIAAA R21AA016901 [PI], NIDA R03DA024774 [PI], NIAAA R01AA017661 [coinvestigator], NIA P30AG010133-20 [coinvestigator, Neuroimaging Core], The Indiana State Department of Health [PI], the IUPUI Research Support Funds Grant [PI] and the Indiana CTSI Clinical Research Center, UL RR025761.
Dr. Nho receives support from the NIH (NLM T15LM07117 [post-doctoral trainee]).
Dr. Foroud serves on the editorial boards of Psychiatric Genetics, Journal of Bone and Mineral Research, Behavior Genetics, Parkisonism and Related Disorders, and Journal of Studies on Alcohol and Drugs; has served as a consultant for the Institute for Behavioral Genetics at the University of Colorado; and receives research support from the NIH (NIA 5U24AG021886 [PI], R01 AR047822 [PI], R01 NS037167 [PI], N01NS32357 [PI], U01AA014809 [PI], R01 CA141668 [PI], and P01 AG018397 [PI]), Families of Spinal Muscular Atrophy, the Muscular Dystrophy Association, HP Therapeutics Foundation, and CHDI Foundation (previously High Q Foundation).
Dr. Potkin serves on scientific advisory boards and speakers’ bureaus for and has received funding for travel and speaker honoraria from AstraZeneca, Bioline, Bristol Myers-Squibb, Cortex Pharmaceuticals, Inc., Dainippon Sumitomo Pharma, Janssen, Novartis, Merck Serono, Pfizer Inc., Otsuka Pharmaceutical Co., Ltd., Roche, and Minister Pharmaceuticals plc; serves on the editorial board of Brain Imaging and Behavior; conducts clinical procedures/imaging studies at the UCI Brain Imaging Center and Neuropsychiatric Clinical Research Division; and receives research support from the Transdisciplinary Imaging Genetics Center (TIGC) P20 RR020837, and the NIH (NCRR U24 RR021992 [PI], U01 AG032984 [PI], NCIRE AG036535 [coinvestigator]).
Dr. Huentelman receives research support from the NIH/NINDS (R01 NS059873 [PI]) and the Arizona Alzheimer’s Consortium.
Dr. Craig has received research support from the Arizona Science Foundation, Arizona Alzheimer’s Research Center, and the NIH (U01 HL086528 [PI] and R01 MH078151 [coinvestigator]).
Dr. Jagust has served on a scientific advisory board for Genentech, Inc., Ceregene, Schering-Plough Corp., and Merck & Co; and receives research support from the NIH (AG027859 [PI], AG027984 [PI], and AG024904 [coinvestigator]) and from the Alzheimer’s Association.
Dr. Koeppe receives research support from Elan Corporation and from the NIH (U01 AG024904 [coinvestigator], P01 NS15655 [coinvestigator], R01 HL079540 [coinvestigator], R01 DA022520 [coinvestigator], and R01 DA016423 [coinvestigator]).
Dr. Mathis serves on a scientific advisory board for Neuroptix Corporation; has received funding for travel and speaker honoraria from Elan Corporation, GE Healthcare, Bayer-Schering Pharma, IBA, and Takeda Pharmaceutical Company Limited; serves on the editorial board of Nuclear Medicine and Biology; may accrue revenue on over 20 active US and international patents, 1996-present re: amyloid imaging agents; serves as a consultant for GE Healthcare, Elan Corporation, Wyeth, and Novartis; estimates that 30% of his academic effort is spent on PiB imaging, which might benefit the commercial license holder of the technology, GE Healthcare, and the University of Pittsburgh as the licensor of the technology; has received/receives research support from GE Healthcare, Neuroptix Corporation, the NIH (AG018402 [PI]), the US Department of Energy, the Dana Foundation, and the Anonymous Foundation; holds stock options in Neuroptix Corporation; and has received license fees and will receive future royalties from GE Healthcare (amyloid imaging agents for brain applications) and Neuroptix Corporation (amyloid imaging agents for eye applications).
Dr. Jack serves a consultant for Elan Corporation and GE Healthcare; receives research support from Pfizer, Inc., the NIH (NIA R01-AG11378 [PI] and U01 AG024904-01 [coinvestigator]), and the Alexander Family Alzheimer’s Disease Research Professorship of the Mayo Foundation; and holds stock in GE Healthcare and Johnson & Johnson.
Dr. Weiner serves on scientific advisory boards for Bayer Schering Pharma, Eli Lilly and Company, Nestlé, CoMentis, Inc., Neurochem Inc., Eisai Inc., Avid Radiopharmaceuticals Inc., Aegis Therapies, Genentech, Inc., Allergan, Inc., Lippincott Williams and Wilkins, Bristol-Myers Squibb, Forest Laboratories, Inc., Pfizer Inc., McKinsey & Company, Mitsubishi Tanabe Pharma Corporation, and Novartis; has received funding for travel from Nestlé and Kenes International and to attend conferences not funded by industry; serves on the editorial board of Alzheimer’s & Dementia; has received honoraria from the Rotman Research Institute and BOLT International; serves as a consultant for Elan Corporation; receives research support from Merck and Co., Avid Radiopharmaceuticals Inc., the NIH (U01 AG024904 [PI], P41 RR023953 [PI], R01 AG10897 [PI], P01 AG19724 [coinvestigator], P50 AG23501 [coinvestigator], R24 RR021992 [coinvestigator], R01 NS031966 [coinvestigator], and P01 AG012435 [coinvestigator]), the US Department of Defense, the Veterans Administration, and the State of California; and holds stock in Synarc and Elan Corporation.
Dr. Saykin serves as Editor-in-Chief of Brain Imaging and Behavior; has served as a consultant to Baxter International Inc., Bristol-Myers Squibb, and Pfizer Inc.; and has received research support from Pfizer Inc., Eli Lilly and Company, Siemens AG, Welch Allyn Inc., the NIH (R01 CA101318 [PI], R01 AG19771 [PI], RC2 AG036535 [Genetics Core Leader], P30 AG10133-18S1 [Imaging Core Leader], and U01 AG032984 [Site PI and Chair, Genetics Working Group]), the Indiana Economic Development Corporation (IEDC #87884), and the Foundation for the NIH.
All other authors report no disclosures.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.ucla.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.ucla.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Swaminathan, S., Shen, L., Risacher, S.L. et al. Amyloid pathway-based candidate gene analysis of [11C]PiB-PET in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort. Brain Imaging and Behavior 6, 1–15 (2012). https://doi.org/10.1007/s11682-011-9136-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11682-011-9136-1