
Abstract
Chinese peony (Paeonia lactiflora Pall.) is both medicinally and aesthetically beneficial. Powdery mildew is a common fungal disease that seriously jeopardizes the value of numerous species, including peonies as a crop. In order to provide a basis for the prevention and treatment of peony powdery mildew, we examined the microbial diversity, the malondialdehyde (MDA) concentrations and antioxidant enzyme activities of peony leaves infected with three levels of powdery mildew to determine any modifications to the leaf's antioxidant enzyme systems and microbial community structure following the onset of disease. The results show that the MDA content rose as the degree of infection became worse. Antioxidant enzyme activity rose and then declined. Following the initiation of powdery mildew, fungal community diversity decreased, whereas there was not any appreciable change in bacterial communities according to microbial diversity sequencing. The relative abundance of more than half of fungal species decreased, with the bacterial genera displaying both abundant and diminished communities with less pronounced alterations in their community structure after the disease spread. Significant different taxa that were critical to the organization of each microbiome were found. Correlation analysis showed that the relative abundance of powdery mildew pathogenic fungal genus Erysiphe was correlated with those of 11 fungal genera and one bacterial genus. Among them, Aureobasidium, Neosetophoma and Sclerostagonospora showed significant positive correlations with Erysiphe and MDA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The perennial herbaceous Paeonia lactiflora is a member of the Paeoniaceae family and a traditional Chinese flower with both ornamental and therapeutic uses. Several studies have shown that the leaf extract, paeoniflorin, has antidepressant and anticancer effects (Zhang et al. 2018; Wang et al. 2021a). However, powdery mildew infests peonies during periods of heavy rain and drastically reduces the flowers’ beauty and market value. Both the globally distributed powdery mildew fungus, Erysiphe paeoniae, and the native Chinese powdery mildew fungus, Podosphaera paeoniae, are found in Europe, Asia, North America, and Australia. Both species are common to peonies (Takamatsu et al. 2006). Using microbial diversity sequencing, only Erysiphe paeoniae was discovered in this study, a specific parasitic fungus primarily affecting leaves and growing only in living plant tissues (Li et al. 2022).
At present, the main control of powdery mildew is the use of fungicides, however, current fungicides have reduced the susceptibility of powdery mildew to chemicals, and increased the resistance of powdery mildew pathogens (Vielba-Fernández et al. 2020). Fungicides used in excessive quantities is also hazardous to the environment as well as to human and animal health. As a result, effective disease control approaches that are feasible and effective are required. Microbial diversity analysis has demonstrated its significance as science and technology has progressed. For example, the process can indicate changes in microbial populations in soils, intestines, plants, and other ecosystems (Steinrucken et al. 2016; Niu et al. 2017; O’Dea et al. 2021; Wang et al. 2021b; Xi et al. 2021).
Several studies have shown that plant diseases and environmental factors alter plant microbial community structure. With the disease stress on plants, pathogens compete with the microbiome for available nutrients to infect tissues, according to high-throughput sequencing of sick inter-root microorganisms (Chapelle et al. 2016). Using this sequencing, chestnut trees infected with the Chinese chestnut yellow crinkle disease, researchers have found that phytopathogens have a significant influence on microbial community structure (Ren et al. 2020). Microbial diversity sequencing has been applied to the study of the effect of plant diseases on inter-root microorganisms. Based on variance in the 16S rRNA genes of V3-V4 and the fungal ITS2 region, illumina amplification sequencing was used to examine microbial community structure and diversity in the rhizosphere of powdery mildew-infected and uninfected strawberry plants in the greenhouse. Powdery mildew infection reduced the number of operational taxonomic units (OTUs) and prokaryotic and fungal community richness/diversity indices in the rhizosphere compared to the healthy rhizosphere (Yang et al. 2020). Some researchers used high-throughput sequencing technology to study the effect of root microbiota on healthy and root-rot peas and found that the bacterial and fungal diversities in diseased and rhizosphere samples were higher than that in healthy roots (Zakir et al. 2021). Microbial diversity sequencing has also been applied to the study of the effect of plant diseases on microbial communities in different parts of plants. Powdery mildew (Golovinomyces orontii) showed many of the resident fungi associated with Arabidopsis thaliana leaves, but the fungal community structure in the roots remained constant according to Durán et al. (2021). Further research revealed considerable alterations in bacterial leaf population while root bacteria remained constant. With environmental stress, nickel concentrations were added to agricultural fields growing alfalfa in Tunisia by Helaoui (Helaoui et al. 2020). There was a decline in soil enzyme activity. With substantial nickel pollution however, a considerable increase in soil microbial biomass was observed. Environmental stress has also been analyzed jointly with disease stresses, low nitrogen fertilizer, and high levels of southern leaf blight disease (Cochliobolus heterostrophus) infection to treat maize inbred B73. When the disease was severe, lower species richness alpha diversity was linked to increased severity of southern leaf blight disease. The fall in bacterial alpha diversity was accelerated by nitrogen application (Manching et al. 2014).
Several researchers have also combined metabolite analysis with an analysis of microbial diversity (Sha et al. 2017; Liu et al. 2021a; Wang et al. 2021b; Xu et al. 2021; Saghaï et al. 2022). Physiological markers such as plant antioxidant enzymes can represent the degree of stress imposed on plants by adverse conditions, and microbial diversity analysis is used to explain changes in the microbial community structure of leaves following the emergence of disease (Manching et al. 2014; Wu et al. 2021). As a result, combining microbial diversity analysis with the assessment of leaf antioxidant enzymes and other physiological markers can reveal the extent of leaf damage and changes in their internal environment, laying the groundwork for the screening of powdery mildew biocontrol strains. It has been shown that antioxidant enzyme systems and malondialdehyde content of plants can respond to the degree of stress caused by adversity (Wang et al. 2018a, b).
Therefore, we measured physiological indicators and analyzed the microbial diversity of peony leaves at different disease levels to understand how powdery mildew affects leaf physiological and biochemical levels as well as how different fungal and bacterial populations behave at different disease levels. Correlation analysis was used to predict the biocontrol strains of powdery mildew, and to provide a reference for biological control.
Materials and methods
Collection of leaf samples of Paeonia lactiflora
Paeonia lactiflora leaves were collected from a flower garden on the Northeast Forestry University campus in Harbin, China (45.72 N, 126.68 E) on August 9, 2021. The samples were divided into three categories: healthy leaves HL (no lesions, with a powdery layer on the surface), mildly diseased leaves MDL (white powdery layer on 10% of the surface), and severely diseased leaves SDL (white powdery layer on more than 50% of the surface). Healthy leaves were taken from healthy plants surrounded by diseased plants. Each 1.0 g sample was frozen in liquid nitrogen in centrifuge tubes and sequenced for microbial diversity and physiological leaf characteristics. The leaf images are shown in Supplementary Fig. S1. Three replicates were used for each set of experiments.
Leaf physiological index
Three groups of peony leaves with different disease levels were tested for some physiological indexes of the antioxidant enzyme system. Malondialdehyde content (MDA) was determined using the Peng method (Zeng et al. 2018). Spectrophotometric measurements examined antioxidant enzyme systems in leaves and included superoxide dismutase (SOD), catalase (CAT), and peroxidase (POD). Analysis of SOD activity used a modified xanthine oxidase method. Peroxidase and catalase activities were determined according to Tauqeer et al. (2016). Malondialdehyde, superoxide dismutase, catalase and peroxidase kits were all provided by the Beijing Science Solarbio Technology Co.
PCR amplification and sequencing
The structure of the leaf microbial community was analyzed by high-throughput sequencing of ITS rDNA and 16 s rDNA. The Biomarker Technologies Corporation, Beijing, China, performed all operations, including complete interleaf DNA extraction, amplification, library creation, sequencing, and data analysis. The full length of the fungal ITS rDNA gene was amplified using fungal-specific primers for ITS1 (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) (Shi et al. 2021). Using bacterial-specific primer pairs 27F (5′-AGAGTTTGATCTGGCTCAG-3′) and 1492R (5′-GGTTACTTGTTACGACTT-3′), the entire length of the bacterial 16S rDNA gene was amplified (Shi et al. 2021). The PacBio SMRT RS II DNA sequencing platform was used to sequence amplified libraries (Pacific Biosciences (PacBio), Menlo Park, CA, USA). Low-quality or off-target sequences were screened by the PacBio cycle-consistent sequencing technology (Mosher et al. 2014).
Sequence processing
After extracting the total DNA of the samples, primers were designed according to the conserved regions. The sequencing junction was added at the end of the primers to perform PCR amplification and to purify, quantify and homogenize the products to form a sequencing library, and the built library was first subjected to quality control. The qualified library was sequenced by Illumina Novaseq 6000 and the raw image data files from high-throughput sequencing (such as Illumina Novaseq and other sequencing platforms) were transformed into sequenced reads by base calling analysis. The results were stored in a FASTQ (abbreviated as fq) file format which contained the sequence information of sequenced reads and their corresponding quality information.
The raw reads from the sequencing were filtered using Trimmomatic v0.33 software, and the primer sequences were identified and removed using cutadapt 1.9.1 software to obtain clean reads. Clean reads for each sample were spliced by overlap using Usearch v10 software. The stitched data was filtered for length according to the range of different regions. Using UCHIME v4.2 software, the chimeric sequences were identified and removed to obtain the final effective reads. The resulting high-quality sequences were clustered into operational classification units (OTUs) based on a 97% similarity and uploaded to NCBI. The BioProject was PRJNA842361.
Statistical methods
On BMK Cloud (www.biocloud.net), relevant metrics such as operational taxonomic unit (OTU) richness, alpha diversity index (Ace, Chao1, Shannon, and Simpson), and beta diversity were analyzed using software such as QIIME2 (White et al. 2009; Chen and Boutros 2011; Wang et al. 2012; Edgar 2013). The classification level of PCOA analysis was OUT and the distance algorithm was Bray–Curtis (Ricotta and Podani 2017). Microbial diversity and physiological indices were subjected to a one-way ANOVA using IBM SPSS 25.0 software (SPSS Inc., Chicago, IL, USA) with Duncan’s post hoc test. Significant differences in leaf physiological indices and microbial diversity indices between treatments were determined at a p < 0.05 level. The correlation analysis method was based on Spearman. The 30 fungi and bacteria with the highest relative abundance and leaf physiological indicators were selected for correlation analysis, performed on the Tutools platform (https://www.cloudtutu.com), a free online data analysis website.
Results
Effect of powdery mildew on α-diversity of interleaf microorganisms
The structure and composition of fungal and bacterial communities in leaves at three disease levels were examined using Illumina sequencing technology. The fungal rRNA internal transcribed spacer region (ITS) and bacterial 16S rRNA gene were used as targets for sequencing nine leaf samples. After double-ended reads quality checking and splicing, 4,319,173 fungal reads pairs were recovered sequenced from the three disease levels for a total of 4,138,404 clean reads. Each sample yielded at least 440,420 clean reads, with an average of 459,823. After double-ended reads quality screening and splicing, 719,385 pairs were sequenced, yielding 717,486 clean reads. Each sample yielded at least 79,394 clean reads, with an average of 79,721.
Sequencing showed the differences in fungal and bacterial alpha diversity among the three disease levels (Table 1). Bacterial diversity and richness indices were higher than fungal ones at all disease levels. The Shannon and Simpson fungal diversity indices were greater in healthy leaves (HL) than in mildly diseased (MDL) and severely diseased leaves (SDL); Shannon index was significantly higher in HL than in SDL (p < 0.05). This is consistent with healthy leaves having the largest number of unique fungal OTUs (Fig. 1a). Fungal richness indices (Chao1) and (ACE) were likewise considerably higher in HL than in the other disease levels and the ACE index was significantly higher in HL than in SDL (p < 0.05). The Shannon and Simpson bacterial diversity indices were not significantly different; HL had higher indices (Chao 1 and ACE) than MDL and SDL, and a significantly higher ACE index than SDL (p < 0.05). This is consistent with HL having the highest total number of OTUs (Fig. 1b).

Venn diagram of shared or unique OTUs of (a) fungi and (b) bacteria under different degrees of powdery mildew disease of peony; OTUs clustered together with 97% similarity. HL: healthy leaves; MDL: mildly diseased leaves; SDL: severely diseased leaves
The onset of powdery mildew changed the alpha diversity of the microbial communities, reduced their abundance, and decreased their diversity but had less effect on the diversity of bacterial communities.
Effect of powdery mildew on the structure of interleaf microbial communities
A community structure analysis examined the impact of powdery mildew infestation on interleaf fungal and bacterial populations. In the community structure analysis (Fig. 2a and b), it was found that the β-NTI values of some fungi were > 2, indicating that the community structure of some fungi among groups were mainly affected by the decisive factor. That was the level of powdery mildew. On the other hand, the bacterial β-NTI values were concentrated in the range of –2 to + 2, indicating that the bacterial community structure was mainly affected by random factors. Combined with the Bray–Curtis based PCoA analysis (Fig. 2c), it was found that MDL and SDL were closer in the fungal samples and further from HL. This indicates that the fungal microbial community structure of healthy leaves is different from those of diseased leaves. In the PCoA bacterial analysis (Fig. 2d), the distribution was different from that of fungi. Fungi levels in healthy leaves were closer to those in mildly diseased leaves. This indicates that the bacterial community structure of severely diseased leaves was quite different. In the ternary phase diagram (Fig. 3a), Ascomycota had the most diverse fungal genera. And the majority of each fungal phylum was closer to healthy leaves, indicating that healthy leaves had more fungal species. Most of the bacterial genera were distributed in the center of the ternary phase diagram, i.e., most were not far from all three disease levels. Only some of the bacterial genera in Actinobacteria and Proteobacteria were clustered in the severely diseased leaves (Fig. 3b). These findings suggest that powdery mildew affects the structure of fungal communities more than bacterial communities. It affected the fungal community structure at the initiation of the disease, while affecting bacterial community structure only when the degree of powdery mildew was severe.

PCOA analysis of (a) fungal and (b) bacterial samples with different disease degrees, and Bray–Curtis-based distance algorithm and classification levels of OTUs. Community structure analysis of (c) fungi and (d) bacteria under different disease degrees, showed the Beta-NTI values of leaf microbial communities under different disease degrees. HL: healthy leaves; MDL: mildly diseased leaves; SDL: severely diseased leaves

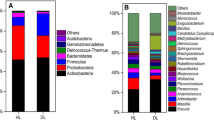
Ternary phase diagram of the horizontal distribution of (a) fungal and (b) bacterial genera under different disease degrees, dotted with the same color and shaped represent genera under the same phylum, fungi select the top four phyla in relative abundance, and bacteria selected relative abundance top five phyla. Abundance histograms for the top ten phyla of relative abundance of (c) fungi and (d) bacteria at different levels of disease, and for the top twenty genera of relative abundance of (e) fungi and (f) bacteria. HL: healthy leaves; MDL: mildly diseased leaves; SDL: severely diseased leaves
By using fungal diversity analysis, 11 phyla, 34 classes, 80 orders, 153 families, 278 genera, and 307 species were identified (Fig. 3c and e). The top 10 phyla with the largest species relative abundance were chosen for investigation at the phylum level, and the top 20 genera with the highest species relative abundance were analyzed at the genus level. Among the fungi, Ascomycota and Basidiomycota were the dominant phyla of peony interleaf fungi. After the disease onset, the relative abundance of Ascomycota increased, 54.9% in healthy leaves, 82.5% in mildly diseased leaves and 83.5% in severely diseased leaves. While the relative abundance of Basidiomycota decreased, 36.0% in healthy leaves, 15.1% in mildly diseased leaves and 10.3% in severely diseased leaves. Among the fungal genera, Erysiphe was dominant and increased after disease onset, 6.3% in healthy leaves, 63.6% in mildly diseased and 52.1% in severely diseased leaves. Respectively, while the relative abundance of most other fungal genera decreased, e.g., Aspergillus 5.7% in healthy leaves, 1.6% in mildly diseased leaves, and 1.1% in severely diseased leaves. Cladosporium 5.6% in healthy leaves, 1.3% in mildly diseased leaves, and 0.7% in severely diseased leaves. Colletotrichum 3.8% in healthy leaves, 0.0% in mildly diseased leaves, and 0.0% in severely diseased leaves.
The structural changes of bacterial communities were different from those of fungal communities; 31 phyla, 75 classes, 185 families, 325 orders, 666 genera, and 744 species were identified based on bacterial diversity analysis. The top ten bacterial phyla in relative abundance and the top twenty genera in relative abundance were studied using distribution histograms (Fig. 3d and f). The bacterial phyla Firmicutes, Proteobacteria, Bacteroidetes, and Actinobacteria were dominant. At the onset of the disease, most bacterial levels did not change significantly. The relative abundance of Firmicutes decreased 47.6% in healthy leaves, 46.9% in mildly diseased leaves, and 28.6% in severely diseased leaves. Proteobacteria 25.7% in healthy leaves, 23.1% in mildly diseased leaves, and 36.2% in severely diseased leaves. Actinobacteria 3.3% in healthy leaves, 3.0% in mildly diseased leaves, and 11.7% in severely diseased leaves. Among the bacterial genera, uncultured__bacterium__f__Lachnospiraceae and Lachnospiraceae_NK4A136 were dominant. The relative abundance of most bacteria decreased when the disease increased, e.g., uncultured_bacterium_f_Lachnospiraceae 11.0% in healthy leaves, 11.2% in mildly diseased leaves, and 5.6% in severely diseased leaves. A small proportion of bacteria increased in relative abundance, e.g., Sphingomonas by 1.2% in healthy leaves, and 1.3% and 5.4% in mildly and severely diseased leaves, respectively.
With regards to the changes in fungal and bacterial genera (Supplementary Fig. S2), more than half of the fungal genera decreased in relative abundance after the onset of the disease. However, bacterial genera have both increased and reduced communities, and community structure changes were more limited. This show that powdery mildew infestation had different effects on structural changes of fungal and bacterial species. The structure of the fungal community was affected immediately after the onset of the disease while the structure of the bacterial species was not affected until after the heavy onset of powdery mildew.
Taxonomic biomarkers of the microbial community
Taxonomic biomarkers were analyzed using LEfSe; 51 and 50 differentially rich fungal and bacterial taxa, respectively, were identified (Fig. 4). For fungal communities, all different taxa of healthy, mild and severely diseased leaves were concentrated between Ascomycota and Basidiomycota. In healthy leaves, Agaricomycetes, Cladosporiaceae, Cladosporium, Cladosporiales, and Fusarium asiaticum contributed most to differences in fungal community structure, in mildly diseased leaves, it was Podosphaera astericola and in severely diseased leaves, it was Filobasidiales. For bacterial communities, it was Gracilibacteria in healthy leaves, Desulfovibrio in mildly diseased leaves, and in severely diseased leaves, it was Hymenobacter. Cytophagales, and Hymenobacteraceae contributed the most to the differences in bacterial community structure. Among the fungi, the most different taxa were found in healthy leaves, while among the bacteria, the most variable were found in severely diseased leaves. This shows that there were differences in the community structures of fungi and bacteria during the infestation of peony powdery mildew disease.

LEfSe analysis indicated the relative abundance of microorganisms at different taxonomic levels. Fungi: a branching plots of different taxa from horizontal phylum to species, with different color nodded representing significantly abundant and important taxa in the sample, and (c) LDA scores, with higher LDA scores for taxa having a greater impact on the taxa. Bacteria: b branching plots of different taxa from horizontal phylum to genus, with different colored nodded representing significantly abundant and important taxa in the sample, (d) LDA scores, with higher LDA scores for taxa having a greater influence. HL: healthy leaves; MDL: mildly diseased leaves; SDL: severely diseased leaves
Effect of powdery mildew on physiological indicators
To understand the effect of powdery mildew on antioxidant enzyme systems of peony leaves, the physiological indicators of peroxidase (POD), malondialdehyde (MDA), superoxide dismutase (SOD), and catalase (CAT) were evaluated from microorganisms catalase from healthy and diseased peony leaves to see how powdery mildew affected physiology and biochemistry. Peroxidase, superoxide dismutase, catalase and malondialdehyde contents after disease onset gradually increased (Table 2). Superoxide dismutase, catalase, and peroxidase activities also significantly increased (p < 0.05). However, after further increase of the disease, these activities were slightly reduced but still higher than in healthy leaves. This is related to the ability of plants to repair themselves in the face of disease stress.
Correlation of physiological indicators and interleaf microbial communities with powdery mildew biocontrol strains
The relationship between physiological and biochemical properties, Erysiphe and microbial communities, was investigated to predict the strains of peony powdery mildew for biological control. Correlation coefficients using the Spearman rank method, correlation heat maps and network diagrams were constructed for leaf physiological indicators and the top thirty genera of fungi and bacteria (Fig. 5). In the genus Erysiphe only the specie Erysiphe paeoniae exists in poney leaves (Table S1). The correlation between the relative abundance of microorganisms in this experiment is represented by the correlation coefficient (R), where there is a significant correlation when |R|> 0.95; |R|≥ 0.8 is highly correlated; 0.5 ≤|R|< 0.8 is moderately correlated; 0.3 ≤|R|< 0.5 means a low degree of correlation; |R|< 0.3 means a very weak relationship and is considered irrelevant. The selection condition of correlation analysis was p < 0.05. The leaf physiological indicators related to the relative abundance of Erysiphe were peroxidase (0.73), malondialdehyde (0.70), superoxide dismutase (0.82) and catalase (0.63). Fungi related to Erysiphe were Fusarium (− 0.83), Aspergillus (− 0.92), Mortierella (− 0.78), Aureobasidium (0.78), Penicillium (− 0.73), Neosetophoma (0.67), Botryotrichum (− 0.68), Sclerostagonospora (0.88), Zygosaccharomyce (− 0.68), Kazachstania (− 0.57). Bacteria related to Erysiphe was Ruminococcus_1 (− 0.77) (Table 3). Among them, Aspergillus and Penicillium were significantly correlated with various physiological indicators of leaves, including malondialdehyde, peroxidase, superoxide dismutase and catalase.

Correlation heat map between microbial communities at the genus level and leaf physiological indicators (a), where p > 0.05 was empty. The red font on the abscissa was physiological indicators, black was fungi, green was bacteria, and asterisks represent significant correlations with Erysiphe unit. Correlation network analysis diagram (b), the green dotted represent physiological indicators, and the red dotted represent microorganisms. In the correlation network analysis diagram (c), the green dotted represent fungi, and the red dotted represent bacteria. Spearman method was used for correlation heat map, and the Pearson method for all correlation network analyses
Using the heat map and network diagram in the correlation analysis (Fig. 5a and c), it was found that there were more significantly correlated genera in the fungal communities, and more significantly correlated genera in the bacterial communities. However, there were fewer significantly correlated genera between the fungal and bacterial communities. This suggests that in peony leaves in various stages of the disease, the link between fungal and bacterial networks was limited. Using the correlation network diagram (Fig. 5b), it is seen that superoxide dismutase and catalase activities were most closely associated with microbial communities in physiological indicators.
The majority of physiological indicators revealed significant positive correlations between fungi and fungi, as well as bacteria and bacteria, indicating that bacterial and fungal networks in peony leaves had more symbiotic or parasitic relationships, and they were fighting changes in survival environment and resources together. Some of the strains of the genera that exhibited strong associations with Erysiphe are used as research paths for powdery mildew ecological management or for the screening of Erysiphe antagonists. Because each physiological indicator could indicate leaf health and internal environment, strains with a high correlation with the physiological index may also be advantageous for peony leaf strains. Some of the strains from the genera that exhibited strong associations with Erysiphe was used as research paths for powdery mildew ecological management or the screening of Erysiphe antagonists. Because each physiological indicator can indicate leaf health, strains with a high correlation with the physiological index may also be advantageous for peony leaf strains. Peroxidase, malondialdehyde, superoxide dismutase, and catalase were most close linked to Aspergillus and Penicillium, and Aureobasidium, Neosetophoma and Sclerostagonospora showed a significant positive correlation with Erysiphe and MDA.
Discussion
Plant pathogenic fungi would reduce their sensitivity due to their adaptation to fungicides, so the use of mechanisms such as antagonism and competition among microorganisms would become the main means of long-term effective biological control research (Kim et al. 2017; Claassen et al. 2021; Martínez-Diz et al. 2021; Mao et al. 2022). Therefore, we analyzed the microbial diversity of peony leaves at different disease levels in order to contribute to the biological control of powdery mildew.
This study showed that the onset of peony powdery mildew reduced the alpha diversity of fungi (Table 1). This is similar to the findings of Manching (Manching et al. 2014). However, there was no significant change in the bacteria diversity after disease onset, while the abundance of communities decreased (Table 1). In the analysis of community structure, fungal community structure began to change after the onset of the disease, while the structure of the bacterial communities began to change when the disease was more severe (Fig. 2). Among the top twenty genera with the highest relative abundance, more than half of the fungal communities showed a decrease in the relative abundance of species after the onset of disease (Fig. 3e, Fig. S2a). In contrast, the structure of the interleaf bacterial community changed significantly only at severe disease levels. The variation in leaf-associated bacterial communities was more limited, with both abundant and reduced taxa (Fig. 3f, Fig. S2b). This is similar to the findings of Durán et al. (2021). In the fungal PCoA analysis, SDL2 had a unique community structure (Fig. 2a) and in the genera community structure analysis, the relative abundance of Erysiphe was significantly lower than in other samples in the same group and all samples in leaves that were mildly diseased (Figs. S2a and S3a). The relative abundance of Botryotrichum, Mortierella, Plectosphaerella, Chaetomium, Pseudaleuria, Talaromyces, and Fusarium in SDL2 was higher than in severely diseased leaves and in all mildly diseased samples. Most were phytopathogenic and saprophytic fungi (Mychele et al. 2019; Han et al. 2020; Liu et al. 2021b). It has been shown that when pathogenic fungi invade plant tissues, the defense mechanisms of the plant are disrupted, the balance of microbial community structure in plants are broken, and other pathogenic microorganisms as well as trophozoites enter tissues more easily (Steinrucken et al. 2016). Therefore, it is speculated that this situation in SDL2 may be the result of the uneven mixing of samples with the more severely diseased leaves in SDL2. The bacterial community structure of SDL2 was also different from other samples in SDL, and similar to that of samples in HL and MDL (Fig. S2b, Fig. S3b). This indicates that the bacterial community structure of SDL2 tends to be healthy or less diseased compared with other samples in severely diseased leaves. Bacterial network structures are weaker than fungal networks but they are also more resilient (Vries et al. 2018). Therefore, it was hypothesized that SDL2 is due to more severe leaf disease in its samples, resulting in a large increase in the relative abundance of phytopathogenic and saprophytic fungi. However, because the bacterial network was more resilient, the structure quickly recovered to converge to a healthy or less diseased state.
The majority of Paeonia lactiflora interleaf microorganisms came from soil and air, and endophytic microorganisms form a complex microbial network structure in the leaves and aid the host in various ways. Their importance cannot be overstated (Webster 1969; Brader et al. 2017). Because antioxidant enzyme activity and MDA content reflect the degree of plant stress and the plant's performance in response to plasma membrane peroxidation, combining leaf physiological index data with microbial diversity provides a more comprehensive picture of the host's ability to regulate and restore health.
In the correlation analysis (Fig. 5), Fusarium, Aspergillus, Mortierella, Aureobasidium, Penicillium, Neosetophoma, Botryotrichum, Sclerostagonospora, Zygosaccharomyce, Kazachstania, Ruminococcus_1 were significantly correlated with the powdery mildew pathogen, Erysiphe. Aureobasidium, Neosetophoma and Sclerostagonospora showed a significant positive correlation with Erysiphe and MDA, and have been regarded as biocontrol fungi or have biocontrol potential (Wachowska et al. 2016).
High-throughput sequencing led to the discovery of the fungus Ampelomyces quisqualis in the leaves of peonies, a powdery mildew parasitic fungus as a tool for controlling powdery mildew (Anna et al. 2020, 2021; Balendres et al. 2020; Carbó et al. 2021). Despite being present in this microbial diversity sequencing, the relative abundance of Ampelomyces quisqualis was low and was not in the top 30 (Table S1). By analyzing the relative abundance data of Ampelomyces quisqualis, this species increased steadily with the level of powdery mildew infection. Because both Ampelomyces quisqualis and Erysiphe are parasitic, there was a positive correlation between the relative abundance of Ampelomyces quisqualis and powdery mildew levels. Additionally, the strains discovered through correlation analysis showed a strong relationship with Erysiphe and leaf physiological markers, making them valuable for research on plant-beneficial bacteria and powdery mildew disease prevention.
Conclusion
Malondialdehyde levels rose as powdery mildew disease increased. Antioxidant enzyme activity rose and then drops. According to microbial diversity sequencing, the diversity of the fungal community decreased after the onset of the disease, while the bacterial community did not show any significant changes. As infection increased, the abundance of the bacterial community decreased. The relative abundance of more than half of the fungal species decreased after disease onset. Bacterial genera showed both enriched and reduced communities, with less pronounced changes in community structure following the spread of the disease. Different taxa critical to the organization of each microbiome were identified. According to correlation analysis, Erysiphe was significantly associated with 11 fungal genera compared to one bacterial genus. Among them, Aureobasidium, Neosetophoma and Sclerostagonospora showed significant positive correlations with Erysiphe and malondialdehyde. The study of the physiological indexes and leaf microbial community structure of peony leaves would contribute to a better understanding of the changes in the microbial community and leaf internal environment following disease initiation. It would serve as a guide in the ecological control and healthy growth regulation of peony powdery mildew.
References
Anna C, Rosario T, Josep U, Jordi B, Neus T (2020) Biocontrol potential of Ampelomyces quisqualis strain CPA-9 against powdery mildew: Conidia production in liquid medium and efficacy on zucchini leaves. Sci Hortic 267:109337. https://doi.org/10.1016/j.scienta.2020.109337
Anna C, Neus T, Josep U, Cristina S, Rosario T (2021) Formulated Ampelomyces quisqualis CPA-9 applied on zucchini leaves: influence of abiotic factors and powdery mildew mycoparasitization. Eur J Plant Pathol 161:37–48. https://doi.org/10.1007/S10658-021-02302-Y
Balendres M, Taguiam JD, Evallo E, Sison ML (2020) Premature defoliation in Zinnia sp. and mycoparasitism of Ampelomyces quisqualis against powdery mildew pathogen Golovinomyces cichoracearum from the Philippines. Indian Phytopathol 74:283–284
Brader G, Compant S, Vescio K, Mitter B, Trognitz F, Ma LJ, Sessitsch A (2017) Ecology and genomic insights into plant-pathogenic and plant-nonpathogenic endophytes. Annu Rev Phytopathol 55:61–83. https://doi.org/10.1146/annurev-phyto-080516-035641
Carbó A, Torres R, Usall J, Marin A, Contreras C, Chiralt A, Teixidó N (2021) Dehydration of Ampelomyces quisqualis CPA-9 conidia by adding biodegradable coatings: biocontrol activity against powdery mildew and physical characterization of the formulated product. Biol Control 161:104701
Chapelle E, Mendes R, Bakker PAHM, Raaijmakers JM (2016) Fungal invasion of the rhizosphere microbiome. ISME J 10:265–268. https://doi.org/10.1038/ismej.2015.82
Chen H, Boutros PC (2011) VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform 12:35. https://doi.org/10.1186/1471-2105-12-35
Claassen BJ, Wolfenbarger SN, Gent GD (2021) Fungicide Physical mode of action: impacts on suppression of hop powdery mildew. Plant Dis 106:1244–1252. https://doi.org/10.1094/PDIS-10-21-2131-RE
Durán P, Reinstädler A, Rajakrut AL, Hashimoto M, GarridoOter R, SchulzeLefert P, Panstruga R (2021) A fungal powdery mildew pathogen induces extensive local and marginal systemic changes in the Arabidopsis thaliana microbiota. Environ Microbiol 23:6292–6308. https://doi.org/10.1111/1462-2920.15768
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998. https://doi.org/10.1038/nmeth.2604
Han L, Zhou X, Zhao Y, Wu L, Ping X, He Y, Peng S, He X, Du Y (2020) First Report of Plectosphaerella Plurivora Causing Root Rot Disease in Panax Notoginseng in China. J Phytopathol 168:375–379. https://doi.org/10.1111/jph.12901
Helaoui S, Mkhinini M, Boughattas I, Alphonse V, Giusti-Miller S, Livet A, Banni M, Bousserrhine N (2020) Assessment of changes on rhizospheric soil microbial biomass, enzymes activities and bacterial functional diversity under nickel stress in presence of Alfafa plants. Soil Sediment Contam 29:1–21. https://doi.org/10.1080/15320383.2020.1771276
Kim BR, Shin J, Guevarra R, Lee JH, Kim DW, Seol KH, Lee JH, Kim HB, Isaacson R (2017) Deciphering diversity indices for a better understanding of microbial communities. World J Microbiol 27:2089–2093. https://doi.org/10.4014/jmb.1709.09027
Li X, Liu M, Liu Y, Zhao W, Li S, Liu W, Lin C, Miao W (2022) A putative effector of the rubber-tree powdery mildew fungus has elicitor activity that can trigger plant immunity. Planta 255:33. https://doi.org/10.1007/s00425-021-03818-7
Liu D, Perez-Moreno J, Zhang P, Wang R, Chater CC, Yu F (2021a) Distinct compartmentalization of microbial community and potential metabolic function in the fruiting body of Tricholoma matsutake. J Fungi 7:586. https://doi.org/10.3390/JOF7080586
Liu S, Wang J, Guo N, Sun H, Ma H, Zhang H, Shi J (2021b) Talaromyces funiculosus, a novel causal agent of maize ear rot and its sensitivity to fungicides. Talaromyces Funiculosus Plant Dis 105:3978–3984. https://doi.org/10.1094/PDIS-04-21-0686-RE
Manching HC, Balint-Kurti PJ, Stapleton AE (2014) Southern leaf blight disease severity is correlated with decreased maize leaf epiphytic bacterial species richness and the phyllosphere bacterial diversity decline is enhanced by nitrogen fertilization. Front Plant Sci 5:403. https://doi.org/10.3389/fpls.2014.00403
Mao XW, Wu ZW, Chen FR, Zhou MG, Hou YP (2022) FfCOX17 is Involved in Fumonisins Production, Growth, Asexual Reproduction, and Fungicide Sensitivity in Fusarium fujikuroi. Toxins 14:427. https://doi.org/10.3390/TOXINS14070427
Martínez-Diz MP, Díaz-Losada E, Andrés-Sodupe M, Bujanda R, Maldonado-González MM, Ojeda S, Yacoub A, Rey P, Gramaje D (2021) Field evaluation of biocontrol agents against black-foot and Petri diseases of grapevine. Pesticide Sci 77:697–708. https://doi.org/10.1002/ps.6064
Mit N, Cherednichenko O, Mussayeva A, Khamdiyeva O, Amirgalieva A, Begmanova M, Tolebaeva A, Koishekenova G, Zaypanova S, Pilyugina A, Amandykova M, Tlenshieva A, Nurzhanova A, Mamirova A, Bekmanov B, Djansugurova L (2021) Ecological risk assessment and long-term environmental pollution caused by obsolete undisposed organochlorine pesticides. J Environ Sci Health, Part B 56:11–13. https://doi.org/10.1080/03601234.2021.1913931
Mosher JJ, Bowman B, Bernberg EL, Shevchenko O, Kan J, Korlach J, Kaplan LA (2014) Improved performance of the PacBio SMRT technology for 16S rDNA sequencing. J Microbiol 104:59–60. https://doi.org/10.1016/j.mimet.2014.06.012
Mychele BS, Richard FD, Hung KD, Robert LN, Robert CK, Hannah CH, Marin TB, Ganpati J, Peng WC (2019) Fusarium wilt of cotton may commonly result from the interaction of Fusarium oxysporum f sp vasinfectum with Belonolaimus longicaudatus. J Nematol 51:1–10. https://doi.org/10.21307/jofnem-2019-015
Niu J, Chao J, Xiao Y, Chen W, Zhang C, Liu X, Rang Z, Yin H, Dai L (2017) Insight into the effects of different cropping systems on soil bacterial community and tobacco bacterial wilt rate. J Basic Microbiol 57:3–11. https://doi.org/10.1002/jobm.201600222
O’Dea C, Huerlimann R, Masters N, Kuballa A, Veal C, Fisher P, Stratton H, Katouli M (2021) Microbial diversity profiling of gut microbiota of Macropus giganteus using three hypervariable regions of the bacterial 16S Rrna. Microorganisms 9:1721. https://doi.org/10.3390/Microorganisms9081721
Opalski KS, Tresch S, Kogel K, Grossmann K, Köhle H, Hückelhoven R (2006) Metrafenone: studies on the mode of action of a novel cereal powdery mildew fungicide. Pest Manag Sci 62:393–401. https://doi.org/10.1002/ps.1176
Ren F, Dong W, Shi S, Dou G, Yan DH (2020) Chinese chestnut yellow crinkle disease influence microbiota composition of chestnut trees. Microb Pathog 152:104606. https://doi.org/10.1016/j.micpath.2020.104606
Ricotta C, Podani J (2017) On some properties of the Bray-Curtis dissimilarity and their ecological meaning. Ecol Complex 31:201–205. https://doi.org/10.1016/j.ecocom.2017.07.003
Saghaï A, Wittorf L, Philippot L, Hallin S (2022) Loss in soil microbial diversity constrains microbiome selection and alters the abundance of N-cycling guilds in barley rhizosphere. Appl Soil Ecol 169:104424
Sha SP, Jani K, Sharma A, Anupma A, Pradhan P, Shouche Y, Tamang JP (2017) Analysis of bacterial and fungal communities in Marcha and Thiat, traditionally prepared amylolytic starters of India. Sci Rep 7:10967. https://doi.org/10.1038/s41598-017-11609-y
Shi X, Zhao X, Ren J, Dong J, Zhang H, Dong Q, Jiang C, Zhong C, Zhou Y, Yu H (2021) Influence of Peanut, Sorghum, and soil salinity on microbial community composition in interspecific interaction zone. Front Microbiol 12:678250. https://doi.org/10.3389/FMICB.2021.678250
Steinrucken TV, Bissett A, Powell JR, Raghavendra AKH, Klinken RD (2016) Endophyte community composition is associated with dieback occurrence in an invasive tree. Plant Soil 405:311–323. https://doi.org/10.1007/s11104-015-2529-y
Takamatsu S, Limkaisang S, Kom-un S, Bolay A, To-anun C (2006) Identity of a powdery mildew fungus occurring on Paeonia and its relationship with Erysiphe hypophylla on oak. Mycoscience 47:367–373. https://doi.org/10.1007/S10267-006-0317-5
Tauqeer HM, Rizwan SAM, Ali Q, Saeed R, Iftikhar U, Ahmad R, Farid M, Abbasi GH (2016) Phytoremediation of heavy metals by Alternanthera bettzickiana: growth and physiological response. Ecotoxicol 126:138–146. https://doi.org/10.1016/j.ecoenv.2015.12.031
Vielba-Fernández A, Polonio Á, Ruiz-Jiménez L, Vicente A, Pérez-García A, Fernández-Ortuño D (2020) Fungicide resistance in powdery mildew fungi. Microorganisms 8:1431. https://doi.org/10.3390/microorganisms8091431
Vries FT, Griffiths RI, Bailey M, Craig H, Girlanda M, Gweon HS, Hallin S, Kaisermann A, Keith AM, Kretzschmar M, Lemanceau P, Lumini E, Mason KE, Oliver A, Ostle N, Prosser JI, Thion C, Thomson B, Bardgett BD (2018) Soil bacterial networks are less stable under drought than fungal networks. Nat Commun 9:1–12. https://doi.org/10.1038/s41467-018-05516-7
Wachowska U, Głowacka K, Mikołajczyk W, Kucharska K (2016) Biofilm of Aureobasidium pullulans var. pullulans on winter wheat kernels and its effect on other microorganisms. Microbiology 85:523–530. https://doi.org/10.1134/S0026261716050192
Wang Y, Sheng H, He Y, Wu J, Jiang Y, Tam NF, Zhou H (2012) Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Appl Environ Microb 78:8264–8271. https://doi.org/10.1128/AEM.01821-12
Wang C, Liu D, Bai E (2018a) Decreasing soil microbial diversity is associated with decreasing microbial biomass under nitrogen addition. Soil Boil Biochem 120:126–133. https://doi.org/10.1016/j.soilbio.2018.02.003
Wang L, Ma R, Yin Y, Zhen J (2018b) Antioxidant response of Arabidopsis thaliana seedlings to oxidative stress induced by carbon ion beams irradiation. J Environ Radioact 195:1–8. https://doi.org/10.1016/j.jenvrad.2018.09.018
Wang QS, Yan K, Li KD, Gao LN, Wang X, Liu H, Zhang Z, Li K, Cui YL (2021a) Targeting hippocampal phospholipid and tryptophan metabolism for antidepressant-like effects of albiflorin. Phytomedicine 92:153735
Wang Z, Ji X, Wang S, Wu Q, Xu Y (2021b) Sugar profile regulates the microbial metabolic diversity in Chinese Baijiu fermentation. Int J Food Microbiol 359:109426
Webster JM (1969) The host-parasite relationships of plant-parasitic nematodes. Adv Parasitol 7:1–40. https://doi.org/10.1016/S0065-308X(08)60433-9
White JR, Nagarajan N, Pop M (2009) Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput 5:e1000352. https://doi.org/10.1371/journal.pcbi.1000352
Wu C, Wang F, Ge A, Zhang H, Chen G, Deng Y, Yang J, Chen J, Ge T (2021) Enrichment of microbial taxa after the onset of wheat yellow mosaic disease. Agr Ecosyst Environ 322:107651. https://doi.org/10.1016/J.AGEE.2021.107651
Xi H, Zhang X, Qu Z, Yang D, Alariqi M, Yang Z, Nie X, Zhu L (2021) Effects of cotton–maize rotation on soil microbiome structure. Mol Plant Pathol 22:673–682. https://doi.org/10.1111/MPP.13053
Xie D, Cai X, Yang C, Xie L, Qin G, Zhang M, Huang Y, Gong G, Chang X, Chen H (2021) Studies on the control effect of Bacillus subtilis on wheat powdery mildew. Pest Manag Sci 77:4375–4382. https://doi.org/10.1002/PS.6471
Xu X, Wu B, Zhao W, Lao F, Chen F, Liao X, Wu J (2021) Shifts in autochthonous microbial diversity and volatile metabolites during the fermentation of chili pepper (Capsicum frutescens L.). Food Chem 335:127512. https://doi.org/10.1016/j.foodchem.2020.127512
Yang J, Wei S, Su D, Zhang Z, Chen S, Luo Z, Shen X, Lai Y, Jamil A, Tong J, Cui X (2020) Comparison of the rhizosphere soil microbial community structure and diversity between powdery mildew-infected and noninfected strawberry plants in a greenhouse by high-throughput sequencing technology. Curr Microbiol 77:1724–1736. https://doi.org/10.1007/s00284-020-01948-x
Zakir H, Michelle H, Yantai G, Luke DB (2021) Root rot alters the root-associated microbiome of field pea in commercial crop production systems. Plant Soil Environ 460:593–607. https://doi.org/10.1007/S11104-020-04779-8
Zeng P, Guo Z, Xiao X, Cao X, Peng C (2018) Response to cadmium and phytostabilization potential of Platycladus orientalis in contaminated soil. Int J Phytoremediat 20:1337–1345. https://doi.org/10.1080/15226514.2018.1501338
Zhang JW, Li LX, Wu WZ, Pan TZ, Yang Z, Yang ZS (2018) Anti-tumor effects of paeoniflorin on epithelial-to-mesenchymal transition in human colorectal cancer cells. Med Sci Monit 24:6405–6413
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Project funding: This research was supported by grants from “Cataloguing, flora study and database establishment of mini-type fungi in Northeast Asia” from the Northeast Asia Biodiversity Research Center
The online version is available at https://link.springer.com/.
Corresponding editor: Yu Lei.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, X., Wang, Z., Liu, R. et al. Effect of powdery mildew on interleaf microbial communities and leaf antioxidant enzyme systems. J. For. Res. 34, 1535–1547 (2023). https://doi.org/10.1007/s11676-023-01597-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11676-023-01597-3