
Abstract
The Betula genus contains pentacyclic triterpenoid betulin known for its environmental adaptation and medicinal properties. However, the mechanisms underlying betulin biosynthesis responding to climate change remain unclear. In this study, the role of epigenetic modification (DNA methylation) in betulin biosynthesis was examined and how climatic factors influence it. Whole-genome bisulfite sequencing was performed for greenhouse-grown Chinese white birch (Betula platyphylla Sukaczev) treated with DNA methylation inhibitor zebularine (ZEB) and a natural birch population in Northeast China. ZEB treatment significantly affected the CHH methylation level of transposable elements and betulin content in a hormesis dose-dependent manner. The methylation and expression of bHLH9, a key transcriptional factor controlling betulin biosynthesis, were also consistently affected by ZEB treatment as a hormetic dose–response. In the natural population, there was a positive correlation between promoter methylation of bHLH9 and summer precipitation, while winter temperature was negatively correlated. Thus climate-dependent methylation of bHLH9 regulates the expression of downstream genes involved in betulin biosynthesis. This study highlights the role of environmental signals to induce epigenetic changes that result in betulin production, possibly helping to develop resilient plants to combat ongoing climate change and enhance secondary metabolite production.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Climate change is a complex subject that involves changes in various climatic factors such as rises in atmospheric greenhouse gases, increases in the Earth's temperature, changes in precipitation patterns, and increases in the frequency of extreme weather events. High temperatures cause geographic range shifts of forest tree species, resulting in migration towards cooler northern or higher altitudes, which may not be suitable for their growth and reproduction (Aitken et al. 2008). Several examples of geographic range shifts of tree species have been documented, either in latitude (Lavergne et al. 2010) or in elevation (Lenoir et al. 2008). Severe drought episodes have also led to the local extinction of numerous tree species (Allen et al. 2010). These trends are predicted to become more pronounced in the future climate (Thuiller et al. 2005; Morin et al. 2008), which would most possibly change the composition of plant communities (Bertrand et al. 2011) and affect species adaptation to altered biotic and abiotic factors (Suttle et al. 2007).
Epigenetic modifications are independent of DNA sequence variation and can facilitate rapid species adaptation by giving rise to alternative epigenomes. Various epigenetic processes, including DNA methylation, histone modification and RNA-mediated processes, influence gene expression at the transcription level. Thus, epigenetic modifications increase genetic plasticity and provide an additional layer of gene regulation to a changing environment (Grativol et al. 2012; Schulz et al. 2014). In addition, epigenetics shape plant phenotypes that contribute to adaptation under natural selection (He et al. 2018).
DNA methylation is a conserved epigenetic mark, which involves the addition of a methyl group to the fifth position of a cytosine nucleotide catalysed by DNA methyltransferases (Cervera et al. 2002; Zhang et al. 2018). In plants, DNA methylation occurs in three different contexts, CG, CHG, and CHH sites where H = A, C, T “not G”, especially in repeat sequences and transposable elements (Schulz et al. 2014). De novo DNA methylation in different sequence contexts is executed by DNA methyltransferase DRM2 (Domains Rearranged Methyltransferase 2), which is guided to target loci by RNA-directed DNA methylation pathways (RdDM) that include two plant-specific RNA polymerases (Pol IV and Pol V) and small interfering RNAs (siRNAs) (He et al. 2011; Matzke and Mosher 2014). Distinct pathways regulate the maintenance of methylation patterns in different sequence contexts. METHYLTRANFERASE-1 (MET1) maintains the CG methylation and CHROMOMETHYLASE-3 (CMT3) maintains the CHG methylation. The primary H3K9 methyltransferase KRYPTONITE (KYP/SUVH4), SUVH5, and SUVH6 are SET domain proteins required for CMT3 activity (Ebbs and Bender 2006; Stroud et al. 2014; Ning et al. 2020). The changes in DNA methylation may affect gene expression which contributes to trait variation as epialleles and are stably inherited over generations (Heard and Martienssen 2014; Yong-Villalobos et al. 2015; He et al. 2018). Therefore, the role of epigenetic mechanisms in the microevolution of plants to natural environments is emerging, including in long-lived organisms such as forest trees (Bräutigam et al. 2013). The first step towards studying the ecological importance of epigenetic variation is to investigate DNA methylation patterns in natural populations (Bossdorf et al. 2008; Richards et al. 2010; Herrera et al. 2012; Hirsch et al. 2013; Busconi et al. 2015).
Environmental stresses such as drought, salinity, cold, pathogens, and heavy metals are known to alter DNA methylation patterns in plants (Wada et al. 2004; Verhoeven et al. 2010; Liu et al. 2018). In maize, cold stress caused genome-wide demethylation of stress-responsive genes (Steward et al. 2002). Likewise, drought resulted in the decrease of DNA methylation of stress-responsive genes in rice, most of which reverted to their original state at recovery (Wang et al. 2011). To combat environmental stress, plants have developed an extensive chemical arsenal comprising a diverse array of temporally and spatially specific secondary metabolites. Plant secondary metabolites (PSM) are generated to protect against biotic and abiotic stresses (Bennett and Wallsgrove 1994; Bray et al. 2000; Hirt and Shinozaki 2003). DNA methylation has an important role in the production of secondary metabolites; for example, the accumulation of paclitaxel (the anticancer drug taxol) in the cultured cells of a hybrid of English and Japanese yew (Taxus × media) is linked to DNA methylation modulation (Li et al. 2013).
White birch (Betula platyphylla suk) is a common species growing in temperate and boreal forests. The wood is of high quality and bark extracts protect the species from environmental stress, and are also valued as anticancer and cosmetic agents. Birch bark is also a reliable source of betulin, (a pentacyclic triterpenoid), known for its defence against ultraviolet radiation and for its medicinal properties, including anti-inflammatory, antiviral and anticonvulsant functions (Zhao et al. 2007). Betulin content varies significantly (10% to 40%) in the bark of birch species (B. platyphylla), and depends on species, age, and climate (Zhao et al. 2007; Holonec et al. 2012). For example, betulin content ranged from 11.3 to 20.2% in birch species of northeastern China (Zhao et al. 2007), and 14.5% to 16.5% from species in the Western Carpathian Mountains in Romania (Holonec et al. 2012). The transcription factor bHLH9 (basic helix-loop-helix9), together with gene encoding enzymes such as FPS (farnesyl pyrophosphate synthase), SS (squalene synthase), SE (squalene epoxidase), and LUS (lupeol synthase), play a crucial role in betulin biosynthesis (Yin et al. 2017; Alonso-Serra et al. 2019). Betulin content is also influenced by drought and temperature (Yin et al. 2015, 2017; Zhang et al. 2016; Guo et al. 2017). However, the relationship between climatic factors, gene regulatory components, and betulin content has not been studied in birch at a broad geographical scale.
In this study, seeds of Chinese white birch (B. platyphylla) were treated with different concentrations of zebularine (a DNA methylation inhibitor), a nucleoside analogue of cytidine that inhibits DNA methyltransferase by covalent modifications (Zhou et al. 2002). The hormesis effects of zebularine on the dynamics of DNA methylation, gene expression, and betulin accumulation of birch seedlings have been confirmed. In addition, the DNA methylation level of the bHLH9 promoter and the betulin contents of natural birch populations from different locations were significantly associated with climatic factors. Overall, experimental evidence is provided to support the hypothesis that climatic factors alter the DNA methylation status of the promoter region of the bHLH9 transcription factor, which in turn regulates the expression of betulin biosynthetic genes that affect natural betulin production.
Materials and methods
The leaves of naturally growing 20–30 year-old birch were collected at various locations (Table S1). The sample were denoted as H1 and H2, their geographic location was the Northeast Forestry University Harbin, Heilongjiang (126.62 °E, 45.72 °N). The second sample, European white birch (Betula pendula Roth., Finland), was denoted as J1, J2, and J3, and the collection location was the Jiangshanjiao region of Heilongjiang (128.97 °E, 43.85 °N). The third sample denoted as B1 of European white birch (Sweden) was collected at the Binxi Forest Farm Binxian region of Heilongjiang (127.19 °E, 45.65 °N).
Betula platyphylla seeds from 20 to 30 year-old trees were provided by the State Key Laboratory of Tree Genetics and Breeding (TGB) of the Northeast Forestry University in 2014. In the greenhouse, the seeds were planted in pots and germination frequency counted after 15 days. In 2016, the two-year-old seedlings were transplanted into cultivated land at the Northeast Forestry University’s Institute of Floral Bioengineering for two years (Fig. S1). Morphological data were collected, specifically, height, stem diameter, and number of branches. After data collection, leaves were frozen in liquid nitrogen and stored at – 80 ºC.
Zebularine (ZEB) treatments
Betula platyphylla seeds (approximately 21,000) were immersed for 16 h in a 50 mL Erlenmeyer flask containing distilled water and then gently shaken overnight at 22 °C. The seeds were sterilized in a solution of 75% alcohol, 2% sodium hypochlorite, and 0.01 M phosphate-buffered saline (PBS). After sterilization, the seeds were stored at 22 °C overnight, then soaked in 20 mL of different concentrations of zebularine buffers such as 0.0 μM (ZEB0), 50 μM (ZEB50), 100 μM (ZEB100), 200 μM (ZEB200), 300 μM (ZEB300), 400 μM (ZEB400), and shaken overnight. Following rinsing with distilled water 3–4 times, the seeds were mixed with sterilized medium of peat moss, garden soil, and river sand (V/V = 5:2:1).
DNA extraction and bisulfite libraries
Young leaves of 4-year-old Chinese white birch seedlings and the naturally growing birch samples were used to extract DNA using the SP Plant DNA Kit (OMEGA Bio-Tek, Norcross, GA, USA). The bisulfite kit ZYMO EZD DNA methylation-GoldTM (Sigma) was used on the extracted DNA after desalting. The DNA was recovered from a sizing gel to select for fragment size, and PCR amplification was performed to construct the MethylC-seq library, and finally, an Illumina Hiseq2000 sequencer was used for PE150 sequencing.
Data analysis of birch DNA methylation
The raw data from the Illumina Hiseq was analysed using SeqPrep (https://github.com/jstjohn/SeqPrep) and Sickle (https://github.com/najoshi/sickle) software, following standard rules to filter the raw data and to obtain “clean reads”, where the N base content was < 10%, the base quality at the 3’ end > 20, and the length of reads > 70. The clean reads were mapped to the European birch genome (GoGe comparative genomics platform, https://genomevolution.org/CoGe/GenomeInfo.pl?gid=35079) through Bitmap (https://github.com/genome-vendor/bsmap). The Fisher exact test and sliding window algorithm were used to analyse the genome-wide, differentially methylated regions (DMRs). The minimum coverage of mapping reads for each cytosine was 4, according to Zhang et al. (2016). The fold change of DMR methylation level must be greater than 2, and the regions with a distance between the intervals should be less than 200 bp. Reads were merged to obtain the final DMRs. The goatools (https://github.com/tanghaibao/GOatools) software was used to get gene ontology enrichment at 9 levels for genes with DMR in the promoters. The Bonferroni method calculated the false discovery rate (FDR) to correct the P-value for multiple tests and therefore, an FDR < 0.05 is considered statistically significant.
Determination of betulin content by UPLC
Ultra-high-performance liquid chromatography (UPLC) and a C18 column (1.7 μm, 2.1 mm × 50 mm, Waters) at a flow rate of 0.3 mL/min was used to measure betulin (Yin et al. 2012). A mobile phase composed of 0.05% phosphoric acid as solvent A and acetonitrile as solvent B was pumped into the column equilibrated with 30% solvent B, and then eluted by a 30% to 60% solvent B linear gradient for 30 min. A 90% solvent B linear gradient was run for 1 min, and then by a 90% solvent B gradient for isocratic development with a 90% to 30% solvent B linear gradient. The eluents were scanned at 203 nm.
RNA extraction and real-time quantitative PCR
After seed treatment with different concentrations of ZEB, the bark cambium of a 4-year-old Chinese white birch and a control seedling were collected and ground separately into a frozen powder using liquid nitrogen. Total RNA was extracted using the RNA extraction kit from Eastep® Super (Promega, Beijing). PCR primers were designed (Yin et al. 2015) for the specific betulin biosynthesis-related genes based on the European white birch genome using Geneious (http://www.geneious.com) (Table S2). After RNA extraction, cDNA was synthesised, and a reverse transcription reaction carried out. The total reaction mixture was 10 μL containing 1 μL Primer Script RT enzyme, 1 μL RT Primer Mix, 4 μL 5 × Primer Script buffer and 4 μL RNase-free ddH2O. The reaction conditions were 15 min at 37 °C, 5 s at 85 °C, and finally 10 min at 0 °C. The tenfold diluted DNA product was used for fluorescence quantification. A tubulin gene was used as a control, and PCR analysis was performed using Applied Biosystems ABI7500 real-time PCR and the SYBR Premix Ex Taq™II. Each PCR was performed in a 15 μL reaction containing 6.3 μL 10 × diluted cDNA template, 7.5 μL SYBR Green Supermix, 0.6 μL forward primer, and a 0.6 μL reverse primer. The transcript abundance of each gene was calculated using the 2-( − ΔΔCt) CT method (Skern et al. 2005). All qRT-PCR experiments were performed as three independent technical replicates.
McrBC-qPCR
DNA was extracted from the bark cambium of 4-year-old Chinese white birch plants treated with different ZEB concentrations and controls. McrBC digestion was carried out in 20 μL containing 2 μL 10 × NE buffer, 1 μL 2 mg/mL BSA, 2 μL 10 mM GTP, 1 μL McrBC enzyme, 10 ng DNA to make the final 20 μL using ddH2O. The reaction was incubated at 37 °C for 16 h. A sample (tenfold diluted) of the digested DNA was used as a template for real-time quantitative PCR using the same machines and procedure as described in the qRT-PCR section. The primer design for McrBC-qPCR followed the same procedures as described above (Table S3).
Statistical and meteorological data analysis
R language (https://www.r-project.org) was used for statistical analysis and chart drawing; the Wilcoxon-test was used to look for significant differences in white birch groups. The R package raster was used to extract the bio1–bio19 climatic data from WorldClim (http://www.worldclim.org) based on known latitude and longitude (Table S4). The R package Maptools was used to draw the maps of the field-grown birch trees located in northeastern China.
Results
Birch biomass growth and ZEB treatments
To analyse the effects of DNA methylation on birch growth, the cytosine methylation inhibitor ZEB was used at different concentrations to treat the B. platyphylla seeds before sowing (Fig. 1a). The germination rate varied with different concentrations of ZEB; germination of 12.7% occurred at the highest concentration (400 μM) compared to the controls at 37.7% (Fig. 1b). Approximately 65.8% reduction in germination occurred at 400 μM of ZEB, indicating the effects ZEB treatments (Table S5). To further investigate whether the ZEB- treated plants have morphological deformities, plant height, stem diameter, number of branches on the first side, and number on the second side was measured from one to 4-year-old seedlings (Figs. S1, S2). Compared to the controls, the different concentrations of ZEB- treated plants showed no significant differences in morphological traits (Fig. 1c–f). Therefore, the highest concentration of ZEB significantly inhibited seed germination but had no effect on birch plant biomass (Table S6).

Germination rate and morphological traits of birch treated with different concentrations of ZEB; a and b germination after treatment with 0, 50, 100, 200, 300, 400 µM concentrations of ZEB. Boxplot diagrams show morphological parameters, c height, d diameter, e first lateral branch, and f second lateral branch of 4-year-old birch treated with different concentrations of ZEB; Unmarked box = non-significant, *p < 0.05, **p < 0.01, ***p < 0.001 indicates levels of significance
Hormesis effect of ZEB treatment on CHH methylation of transposable elements underlying betulin biosynthesis
To assess the impact of different ZEB concentrations on birch genome methylation status, WGBS deeply sequenced one sample from each treatment. The genomic landscape for cytosine in each context of DNA methylation was plotted (Fig. 2a), and there was a clear correlation between methylated cytosine density and DNA methylation level (Fig. 2b). The four samples were used for bisulfite sequencing, such as ZEB0 (0 μM), ZEB50 (50 μM), ZEB200 (200 μM), and ZEB400 (400 μM) from 4-year-old birch seedlings to isolate DNA for the construction of bisulfite-treated DNA libraries. In total, 154,117,670– 181,942,768 raw reads were generated for each of the samples (Tables S7, S8). After eliminating adapter contaminates and low-quality reads, the number of reads aligned to the birch genome accounted for approximately 60% and was further reserved for analysis of cytosine methylation at single-base resolution with 30-fold genomic coverage (Table S9). The genome's high coverage also brings at least four reads close to 60% of the cytosine in the genome, thereby providing high-quality methylation levels and methylated sites (Fig. S3).

DNA methylation landscape and patterns in gene and TE in birch treated with different ZEB concentrations. a landscape of DNA methylation in the birch genome treated with different concentrations of ZEB. TEs and genes are indicated by orange and purple in the first two circles; methylation levels of the birch sample (ZEB0, ZEB50, ZEB200, ZEB400) are shown by mCG, mCHG, and mCHH circles. Maximum methylation levels are 0.96 (mCG), 0.80 (mCHG), and 0.16 (mCHH); b methylcytosine distribution of two DNA strands and the density of gene and TE throughout chromosome 1 in each context. Lines in each upper part show cytosine methylation levels in birch treated with different ZEB concentrations; black boxes indicate the centromere positions; c Each context of DNA methylation levels throughout the TE and Gene regions with a 1 kb flanking sequence. The lines in different colours represent birch samples with different concentrations of ZEB treatment
Over 99% CT conversion was observed for all samples by calculating the bisulfite mapping rate of unmethylated chloroplast and mitochondria genome, indicating a highly reliable methylation calling result (Table S9). Eventually, approximately 48%, 36%, and 16% of cytosine were methylated in CG, CHG, and CHH context (Fig. S4), (Table S10), respectively. This is quite different from the results from poplar (Liang et al. 2014), possibly due to differences in transposons contents. All DNA methylation was abundant in the heterochromatin region of the chromosome but relatively lower in the euchromatin region (Fig. 2a, Fig. S5), which mainly represses the jumping of the transposon to maintain the stability of the genome (Zhang et al. 2006). The distribution pattern of genomic features revealed that the repetitive sequence, such as TEs, was spread uniformly across the entire birch genome relative to Arabidopsis thaliana (Zeng et al. 2015). In Arabidopsis, the transposon ratio is less than 15% and is mainly enriched in the centromere's heterochromatic regions. In contrast, in the birch genome, the transposon content was very high, up to 50%, with a similar distribution of transposon as in the tomato genome (Salojärvi et al. 2017; Corem et al. 2018), suggesting that species with high transposon content are prone to higher epialleles.
To study how ZEB alters the degree of DNA methylation across chromosomes, the genomic features of all contexts of DNA methylation were examined, and it was found that TEs are the main target of methylation without context specificity. The CHH methylation level of birch treated with ZEB varied significantly compared to the controls, and there were no such observations in CG CHG (Fig. 2c). Maintenance of CG methylation throughout the genome is greater than in mCHG and mCHH (Zhang et al. 2006) and consistently affected the entire genome. Conversely, the depletion of CHG and CHH methylation was mostly affected in the chromosome pericentric regions. Patterns of DNA methylation throughout the gene regions were analysed, 1 kb upstream and downstream flanking sequence, and the rate of DNA methylation rapidly increased when departing from the transcription starting sites and termination sites in all the contexts. This CG methylation also showed a higher level in the gene body; in contrast, CHG and CHH methylation levels were fewer in the body region than in the flanking regions.
DNA methylation levels in the mCHH context of TEs showed a hormesis effect in the ZEB- treated plant, in which the CHH methylation level was decreased in ZEB50 treatment but increased in ZEB200 treatment and ZEB400 treatment compared to ZEB0 treatment(Fig. 2c). To confirm this, the methylation variation of the whole genome was further analysed in the genes and TEs through DMRs (differentially methylated regions). This is consistent with the trend of DNA methylation level on TE, i.e., low concentrations of ZEB (Z50) induced more hypo-DMRs (66.0%) compared with the controls (ZEB0), and high concentration of ZEB will produce more hyper-DMRs (66.3%), which is most evident in the CHH context. As described previously, ZEB treatments caused significant changes in the methylation levels of TEs. Therefore, the number of DMRs on the TEs is higher than on the genes (Fig. 3a, b, Figs. S6 and S7).

Counts of each context DMRs on genomic features and the GO enrichment of genes with CHH-DMRs; a genes b TEs c GO enrichment of genes located with hypo CHH-DMRs; d Betulin content at different concentrations of ZEB. Unmarked box = non-significant; * p < 0.05, ** p < 0.01, and ***p < 0.001 the significance level
Furthermore, the DMRs in the TEs showed no substantial variations in the number of DMRs in the context of mCG/mCHG, while the number of CHH-DMRs in the different ZEB treatments was considerably fivefold higher relative to mCG and mCHG (Fig. 3b), suggesting that CHH methylation, rather than CG and CHG methylation, is involved in biological functions of birch after ZEB treatments. To examine this, the mCHH-DMRs related genes, located within 2 kb in up and downstream flanking regions of the mCHH-DMRs, were enriched to gene ontology database to acquire the most important functions. Using the Fischer exact test (P-value < 0.05) to get the significance of the enrichment, it was found in the hypo-DMRs of CHH context. There was an obvious and clear biological role that influences birch triterpenoid biosynthesis (Fig. 3c, Table S11). Thus, the CHH methylation, with a hormesis pattern after ZEB treatments, may be involved in betulin biosynthesis.
Betulin content with hormesis caused by ZEB treatments
To verify the effects of ZEB on the content of betulin, ultra-high performance liquid chromatography measured betulin content in the 4-year-old bark of B. platyphylla. The low concentration of 50 μM ZEB significantly increased the amount of betulin in the bark relative to controls; for other concentrations of ZEB from 100 to 300 μM, the content of betulin was similar to controls. There was no substantial difference between treated plants and the controls (p > 0.05, t-test). The concentration of ZEB 400 μM- treated plants reduced betulin content significantly compared to controls (Fig. 3d). The low concentration of ZEB50 significantly enhanced the betulin content (+ 32%), while a higher concentration of ZEB400 substantially decreased the content of betulin ( − 31.6%). Thus, consistent with the hormesis pattern of CHH methylation regarding ZEB treatments, hormetic dose–response curves were also found for the betulin content under different ZEB treatments (Fig. S8), implying that DNA methylation regulates betulin biosynthesis as a hormesis pattern subjected to ZEB treatment.
Hormesis effects of ZEB treatment on DNA methylation and expression of bHLH9 transcription factor
McrBC-qPCR and qRT-PCR tests were carried to detect DNA methylation modification in the promoter regions of betulin biosynthetic transcription factor gene bHLH9, as well as the downstream enzymes, including FPS, SS, SE, LUS and their expression level among ZEB0, ZEB50, and ZEB400 samples. It was found that the expression patterns of all the genes were consistent. Moreover, their expression levels significantly increased with the ZEB50 treatment compared to controls, while the expression level of these genes significantly decreased at ZEB400- treated samples (Fig. 4a), demonstrating the same hormesis pattern with betulin contents, in which ZEB50 was the highest level and ZEB400 the lowest (Fig. 3d). While the DNA methylation was inconsistent, in which only DNA methylation of bHLH9 showed the reverse of its expression (Fig. 4b). In ZEB50 concentrations, DNA methylation was significantly lower in the promoter region of bHLH9 than in the control. In contrast, with ZEB400 treatment, DNA methylation was significantly higher compared to controls, which ultimately affects gene expression (Fig. 4b). As a result, DNA methylation confirmed a reverse hormesis pattern compared to expression level (Fig. 4a) and betulin contents (Fig. 3d). Therefore, the expression pattern and DNA methylation changes of bHLH9 are positively and negatively consistent with the pattern of betulin content under ZEB treatments, respectively; they all showed a trend of hormesis effects (Fig. 3d, Fig. 4a, b). Thus, variations in the expression of betulin biosynthetic genes with different ZEB concentrations, and hence betulin contents, are due to changes in the DNA methylation level at the promoter region of the bHLH9 transcription factor gene, but not to downstream enzymes.

Expression and methylation analysis of betulin biosynthesis genes; a qRT-PCR used to measure gene expression, and b McrBC-qPCR used to measure the methylation level of putative gene promoters’ regions, * p < 0.05, ** p < 0.01, and ***p < 0.001 indicate level of significance
DNA methylation of bHLH9 associated with climatic factors regarding betulin biosynthesis
The abundance of secondary metabolites such as betulin is often affected by climatic factors and geographic distribution, as reported by Yin et al. (2016), Guo et al. (2017), and Bont et al. (2020). To investigate DNA methylation involved in the regulation of betulin in natural field adaptation to climatic factors and geographic distribution, six samples were collected from naturally growing birch distributed in three locations, Harbin, Jiangshanjiao and Binxi (Fig. S9). The H1 and H2 samples from Harbin had the highest levels of betulin, with H1 somewhat lower than H2. Betulin levels of samples from Jiangshanjiao (J1,J2 and J3) and Binxi (B1) were similar and significantly lower than the Haribin samples. The DNA methylation level, located in a TIR (terminal inverted repeat) transposon of bHLH9 promoter regions of Harbin was the lowest, while Jiangshanjiao and Binxi samples showed similar DNA methylation levels in all three contexts of DNA methylation, CG, CHG and CHH (Fig. 5b). This implies that DNA methylation is involved in betulin biosynthesis in nature.

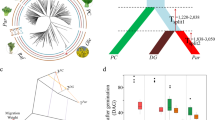
The correlation between bHLH9 promoter methylation and polymorphic climate factors. a The determination of betulin in natural population; x-axis is the natural birch samples (H, J, B), the y-axis the betulin content; N = 9, technique replicates; P-values and Rho indicate the Spearman correlation of betulin contents with CHH methylation level of bHLH9 promoter. b The IGB snapshot of CG/CHG/CHH methylation in the promoter region of the bHLH9 gene in the natural population; TIR, terminal inverted repeat. c Heatmap showing spearman significance and coefficient between the DNA methylation level in the promoter region of the betulin biosynthesis genes and climatic factors in the natural birch population. The x-axis represents the bio variables from the WorldClim v1.4 database. The left side of the y-axis represents the betulin contents (as shown in a) and genes in the betulin biosynthesis pathway, and the right side shows each context of DNA methylation. The box with brown (p-value < 0.01) and blue (p-value < 0.05) color represent the high significance of correlation in the left part of the heatmap. The box with green (Rho < 0) and the pink (Rho > 0) color in the right part represent the correlation coefficient, respectively. d Quantification of bHLH9 promoter methylation level and climatic data of the natural birch population
To evaluate the association of DNA methylation with betulin content in natural plants, Spearman correlation significance and coefficient of the methylation level of 2 kb regions upstream of transcription factor gene bHLH9 and the betulin contents were calculated (Fig. 5a, b) as in the previous analysis (He et al. 2018). As a result, the content of betulin is negatively associated with DNA methylation levels (Spearman Rho = − 0.94, P = 0.017, Fig. 5a), demonstrating that DNA methylation represses betulin biosynthesis in a natural population, consistent with the ZEB treatments (Fig. 3d, 4a, b). To exclude genetic variations involved in bHLH9 controlling betulin biosynthesis in nature, we searched the SNPs (single nucleotide polymorphisms) in the bHLH9 genes in the samples. The phylogenetic study indicated that J1, H1, and H2 were the same clade, and B1 and J2 were in the same clade (Fig. S10), demonstrating that there was no correlation of genetic variation with betulin contents across species in the corresponding bHLH9 genes. Thus, the epigenetic alterations are inextricably connected to betulin biosynthesis in nature.
Previous studies (Yin et al. 2016; Guo et al. 2017) confirm that betulin content is affected by climatic conditions such as temperature and precipitation, implying that DNA methylation is likely to be involved in the adaptation of birch through the accumulation of betulin, which acts as a natural secondary metabolite. To verify the relationship between climate factors, DNA methylation, and betulin biosynthesis, Spearman correlation analysis of climatic factors from WorldClim with betulin contents was carried out as well as DNA methylation levels of betulin biosynthesis genes as in our previous analysis (He et al. 2018). It was found that the DNA methylation level of the bHLH9 promoter is negatively correlated with winter temperatures, such as bio6 (minimum temperature of coldest month), bio9 (mean temperature of driest quarter), and bio11 (mean temperature of coldest quarter). When temperatures increase, the DNA methylation level of the bHLH9 promoter decreased, but the decrease in the level of DNA methylation is independent of sequence methylation contexts. Further, the DNA methylation level of the bHLH9 promoter has been found to have a strong positive correlation with summer precipitation such as bio12 (annual precipitation), bio16 (precipitation of wettest quarter), and bio18 (precipitation of warmest quarter). Conversely, betulin levels in natural populations are positively correlated with winter temperatures and negatively with summer precipitation (Fig. 5c, d; Table S12, S13). This is consistent with ZEB treatment (Guo et al. 2017). In contrast, the other genes involved in betulin biosynthetic pathways do not correspond with climates (Fig. 5c).
The siRNA (small interfering RNA) analysis of bHLH9 genes indicates the high density of the 24 nt siRNA in the TIR regions, consistent with the CHH methylation enrichments. But the DNA methylation level variations in the promoter region of bHLH9 were not correlated with 24 nt siNRA changes among different samples (Fig. S11). It suggests that the RdDM pathway may de novo establish the DNA methylation of the region, but the maintenance of the DNA methylation subject to mitosis or meiosis is somehow affected by the natural environment (Zhang et al. 2018; Johannes and Schmitz 2019). Therefore, environmental factors influence the betulin biosynthesis genes to regulate betulin content by affecting the level of methylation in the promoter regions of the bHLH9 transcription factor.
Discussion
It has been highly debated that DNA methylation, as well as other epigenetic modifications, may be involved in the adaptation of plant plasticity to environmental factors and/or climate changes, but solid evidence is lacking (Springer and Schmitz 2017; Johannes and Schmitz 2019). Previously, it was confirmed that the global DNA methylation of Arabidopsis accessions are negatively associated with the temperatures in Europe, in which the accessions with hypomethylome are enriched in regions with higher temperatures (Kawakatsu et al. 2016). A naturally occurring epiallele controlling leaf senescence negatively associated with temperatures in Arabidopsis was also uncovered (He et al. 2018). Therefore, leaf senescence is accelerated in higher temperatures mediated by hypo DNA methylation, which shortens the life cycle of Arabidopsis and gives rise to flowering and rapid seed production. Since it is safer and more efficient for Arabidopsis to avoid abiotic stresses by producing seeds, ‘death’ is the best way to survive for annual plants dealing with severe environments. Perennial plants, with a different adaptation strategy of annual plants, adapt to be resistant to seasonal alterations and climate changes. However, it has been poorly understood how epigenetic modifications are involved in the adaptation of trees to environmental factors. In this study, it was confirmed that betulin biosynthesis is controlled by DNA methylation of bHLH9 associated with temperature and precipitation.
To determine the causal effects of DNA methylation on betulin biosynthesis and expression of key genes, ZEB treatments, a common inhibitor of methylcytosine, were carried out on birch seed and the phenotypes and methylomes examined in 4-year-old seedlings. Unexpectedly, there were no significant differences between control and ZEB treated samples in terms of growth and biomass. However, the CHH methylome is affected by the ZEB as a hormesis pattern, in which ZEB50 decreases the whole genome methylation, but ZEB200 and ZEB400 increase the whole genome methylation. The hormesis pattern is consistent with chemical treatments of the study by Jones and Taylor (1980). In addition, the CHH-DMRs also support that compared to control; the hypo-DMR of ZEB50 is higher than the hyper-DMRs, whereas the patterns are reversed in ZEB200 and ZEB400. GO enrichment analysis demonstrated that the genes flanking CHH-DMRs are related to betulin biosynthesis. The betulin contents also consistently show a hormesis pattern under ZEB treatments. To verify the molecular mechanism, the focus was on the key genes of betulin biosynthesis, including bHLH9 transcriptional factors as well as the downstream enzymes (Yin et al. 2017; Alonso-Serra et al. 2019). It was found that the expression of bHLH9 genes, rather than the downstream genes, are negatively associated with DNA methylation of bHLH9 promoters.
Both the expression of and DNA methylation of bHLH9 exhibited hormesis patterns under the ZEB treatments (Fig. 4). Although CYP716A12 encodes an enzyme that directly converts lupeol into betulin in B. platyphylla (Fukushima et al. 2011), there is no clear evidence that bHLH9 influences or regulates CYP716A12 through environmental factors to regulate betulin content. It is suggested that the CYP716A12 gene expression will be consistent with other betulin biosynthetic genes. Therefore, DNA methylation may indirectly regulate betulin biosynthesis through bHLH9, an upstream transcription factor that directly regulates downstream genes involved in betulin biosynthesis. Thus, ZEB does not explicitly influence the methylation of betulin biosynthetic enzymes but acts indirectly by affecting methylation in the promoter regions of bHLH9 to regulate the expression of these genes. The specific effects of ZEB on DNA methylation of bHLH9 may be related to the TIR transposons of its promoter, which need to be further clarified. Betulin is a special metabolite produced from birch bark, and has a broad range of pharmacological and biological properties, preventing the birch from damage to UV light in nature. The natural birch material was mainly collected from northeast China which has a continental climate at high latitudes. The coldest and driest period is in the winter season; the warmest and wettest periods occur in the summer. The present study investigated Spearman correlations between the methylation levels of important gene promoter regions, which encode enzymes for betulin biosynthesis, and their relation with climate. There was a significant negative correlation with winter temperatures from WorldClim, for example, bio6 (minimum temperature of coldest month), bio9 (mean temperature of driest quarter), and bio11 (mean temperature of coldest quarter). When temperatures increased, the methylation level of bHLH9 decreased, but the decrease in the level of DNA methylation was independent of sequence contexts. In addition, the methylation level of bHLH9 has been found to have a strong positive correlation with summer precipitation such as bio12 (annual precipitation), bio16 (precipitation of wettest quarter), and bio18 (precipitation of warmest quarter), but no correlation with other climatic factors.
From the association analysis, it was inferred that the high temperatures from high-density sunlight in winter may induce low DNA methylation in the promoter region of the bHLH9 transcription factor and increase gene expression as well as betulin biosynthesis, promoting the capacity of birch resistance to the high density of UV light. Large amounts of rainfall, leading to low density sunlight in summer, may increase the DNA methylation of bHLH9 and decrease the expression of related genes and betulin content, adapting to the low density of UV light in sunlight (Fig. 5c, d). Previously, we reported that seasonal and environmental factors significantly influence betulin accumulation in the stem bark (Yin et al. 2015). The betulin content was at its peak in the warmest summer months from July to August in Harbin. Guo et al. (2017) also documented the site-based data on triterpenoid concentrations. Betulin levels increased with increased temperatures and declined with increased relative humidity. Nevertheless, more climatic factors, especially UV radiation and sunlight parameters, should be considered in future in terms of betulin biosynthesis associated with climate changes and environmental responses.
There is no clear association between betulin biosynthetic gene expression and environmental factors in the field samples. However, there is a relationship between the methylation of the bHLH9 promoter region and environmental factors. The evidence indicates a relationship between the epigenetic regulation of DNA methylation and climate. It has been previously reported (He et al. 2018) that epigenetic variation facilitates plant adaptation to changing environments. A DNA methylation variant region (NMR19-4) was identified whose methylation status was stably inherited and independent of underlying DNA sequence variations and may have a role in local climatic adaptation by plants (He et al. 2018). Furthermore, SNP analysis of selected birch species in different locations revealed no genetic differences. As a result, changes in bHLH9 gene expression are connected to epigenetic regulation (Fig. S11).
It was also confirmed that bHLH9 is an important upstream transcription factor in the regulation of betulin biosynthesis. The methylation in the promoter region of this transcription factor is inversely correlated with winter temperatures (Fig. 5c, d). CHROMOMETHYLASE 2 (CMT2) regulates temperature-dependent DNA methylation, whereas it is independent of the RNA-directed DNA methylation (RdDM) pathways involving 24 nt-siRNA. Previous studies have elaborated on the role of DNA methylation in response to temperature. Shen et al. (2014) showed that the absence of DNA methyltransferase in a cmt2 mutant increased heat-stress resistance, whereas nrpd2 mutants are hypersensitive to temperature. Such findings indicate a specific role for CMT2-dependent CHH methylation and the RdDM pathways in response to heat. Thus, the DNA methylation regulating the adaptation of forest species to climate changes needs to be further confirmed using more samples and sites.
Summarily, a model of betulin biosynthesis controlled by DNA methylation adapting to climate changes has been proposed (Fig. 6). In temperate and boreal forests with high sunlight in winter, high temperatures decrease DNA methylation levels and increase the expression of bHLH9 and the downstream enzymes, resulting in a high level of betulin biosynthesis. High levels of betulin promote birch resistance to high levels of sunlight in winter. In forests with high summer precipitation, high humidity accompanied by low levels of sunlight increase DNA methylation levels of bHLH9 and decrease the expression of related genes as well as low-level betulin contents, which is coincident with low density sunlight. However, whether DNA methylation of bHLH9 shows seasonal alterations, or is changed in winter, or is heritable, and how epimutations are associated with the environment and how environmental factors affect epigenetic modifications should be clarified. Future research should also determine the involvement of ecological interactions and natural selection in epigenetic adaptation to clarify the mechanism of epigenetics involved in adaptations in natural forests under climate changes.

Schematic model of DNA methylation involved in betulin biosynthesis regulating the bHLH9 gene expression; small blue balloons represent highly methylated cytosines; white ones represent low methylated cytosines; with increasing winter temperatures or decreasing summer precipitation, the methylation level of the promoter region of the bHLH9 gene will decrease, thereby promoting the expression of bHLH9, achieving the up-regulation of downstream betulin synthase and increasing betulin content
Conclusions
Our study revealed that temperature and precipitation influence DNA methylation, directly affecting the expression of the gene encoding the bHLH9 transcription factor and indirectly affecting betulin content in birch, providing additional evidence that DNA methylation in woody plants is a respond to climate change, new evidence for epigenetic ecological relevance in Betula. Winter temperature and summer precipitation increase betulin content by decreasing DNA methylation. High winter temperature or aridity in summer decrease DNA methylation in the promoter of the bHLH9 transcription factor gene and increase its expression, which acts as an upstream upregulation on the expression of the downstream enzymes of betulin biosynthesis, resulting in activation of betulin biosynthesis.
Further research is needed to address the following issues: (1) To what extent is the methylation-induced differential accumulation of betulin heritable? (2) What mechanism regulates the methylation of the promoter region of bHLH9 through temperature and precipitation? (3) To what extent is methylation of the bHLH9 promoter region genetically independent? and, (4) Which one is the driver of epigenetic adaptation, ecological interactions, or natural selection? More samples and climatic factors from wild birch populations from various geographical areas are required to characterize the population substructure and subsequently create an ecotype population library. To offer a firm foundation for forest ecological epigenetics in the future, more omics sequencing and mixed linear model analysis are needed to clarify the chemical adaptability of forest Betula to betulin.
References
Aitken SN, Yeaman S, Holliday JA, Wang TL, Curtis-McLane S (2008) Adaptation, migration or extirpation: climate change outcomes for tree populations. Evol Appl 1(1):95–111. https://doi.org/10.1111/j.1752-4571.2007.00013.x
Allen CD, Macalady AK, Chenchouni H, Bachelet D, McDowell N, Vennetier M, Kitzberger T, Rigling A, Breshears DD, Hogg EH(, Gonzalez P, Fensham R, Zhang Z, Castro J, Demidova N, Lim JH, Allard G, Running SW, Semerci A, Cobb N (2010) A global overview of drought and heat-induced tree mortality reveals emerging climate change risks for forests. Forest Ecol Manag 259(4):660–684. https://doi.org/10.1016/j.foreco.2009.09.001
Alonso-Serra J, Safronov O, Lim KJ, Fraser-Miller SJ, Blokhina OB, Campilho A, Chong SL, Fagerstedt K, Haavikko R, Helariutta Y, Immanen J, Kangasjärvi J, Kauppila TJ, Lehtonen M, Ragni L, Rajaraman S, Räsänen RM, Safdari P, Tenkanen M, Yli-Kauhaluoma JT, Teeri TH, Strachan CJ, Nieminen K, Salojärvi J (2019) Tissue-specific study across the stem reveals the chemistry and transcriptome dynamics of birch bark. New Phytol 222(4):1816–1831. https://doi.org/10.1111/nph.15725
Bennett RN, Wallsgrove RM (1994) Secondary metabolites in plant defence mechanisms. New Phytol 127(4):617–633. https://doi.org/10.1111/j.1469-8137.1994.tb02968.x
Bertrand R, Lenoir J, Piedallu C, Riofrío-Dillon G, de Ruffray P, Vidal C, Pierrat JC, Gégout JC (2011) Changes in plant community composition lag behind climate warming in lowland forests. Nature 479(7374):517–520. https://doi.org/10.1038/nature10548
Bont Z, Züst T, Arce CCM, Huber M, Erb M (2020) Heritable variation in root secondary metabolites is associated with recent climate. J Ecol 108(6):2611–2624. https://doi.org/10.1111/1365-2745.13441
Bossdorf O, Richards CL, Pigliucci M (2008) Epigenetics for ecologists. Ecol Lett 11(2):106–115. https://doi.org/10.1111/j.1461-0248.2007.01130.x
Bräutigam K, Vining KJ, Lafon-Placette C, Fossdal CG, Mirouze M, Marcos JG, Fluch S, Fraga MF, Guevara MÁ, Abarca D, Johnsen O, Maury S, Strauss SH, Campbell MM, Rohde A, Díaz-Sala C, Cervera MT (2013) Epigenetic regulation of adaptive responses of forest tree species to the environment. Ecol Evol 3(2):399–415. https://doi.org/10.1002/ece3.461
Bray EA, Bailey-Serres J, Weretilnyk E (2000) Responses to abiotic stresses. Biochemistry and Molecular Biology of Plants, American Society of Plant Physiologists, Rockville, 1158–1203
Busconi M, Colli L, Sánchez RA, Santaella M, De-Los-Mozos Pascual M, Santana O, Roldán M, Fernández JA (2015) AFLP and MS-AFLP analysis of the variation within saffron crocus (Crocus sativus L.) germplasm. PLoS ONE 10(4):e0123434. https://doi.org/10.1371/journal.pone.0123434
Cervera MT, Ruiz-García L, Martínez-Zapater J (2002) Analysis of DNA methylation in Arabidopsis thaliana based on methylation-sensitive AFLP markers. Mol Genet Genomics 268:543–552. https://doi.org/10.1007/s00438-002-0772-4
Corem S, Doron-Faigenboim A, Jouffroy O, Maumus F, Arazi T, Bouché N (2018) Redistribution of CHH methylation and small interfering RNAs across the genome of tomato ddm1 mutants. Plant Cell 30(7):1628–1644. https://doi.org/10.1105/tpc.18.00167
Ebbs ML, Bender J (2006) Locus-specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell 18(5):1166–1176. https://doi.org/10.1105/tpc.106.041400
Fukushima EO, Seki H, Ohyama K, Ono E, Umemoto N, Mizutani M, Saito K, Muranaka T (2011) CYP716A subfamily members are multifunctional oxidases in triterpenoid biosynthesis. Plant Cell Physiol 52(12):2050–2061. https://doi.org/10.1093/pcp/pcr146
Grativol C, Hemerly AS, Ferreira PC (2012) Genetic and epigenetic regulation of stress responses in natural plant populations. Biochem Biophys Acta 1819(2):176–185. https://doi.org/10.1016/j.bbagrm.2011.08.010
Guo SL, Zhang DH, Wei HY, Zhao YN, Cao YB, Yu T, Wang Y, Yan XF (2017) Climatic factors shape the spatial distribution of concentrations of triterpenoids in barks of white birch (Betula platyphylla Suk.) trees in northeast China. Forests 8(9):334. https://doi.org/10.3390/f8090334
He XJ, Chen TP, Zhu JK (2011) Regulation and function of DNA methylation in plants and animals. Cell Res 21(3):442–465. https://doi.org/10.1038/cr.2011.23
He L, Wu WW, Zinta G, Yang L, Wang D, Liu RY, Zhang HM, Zheng ZM, Huang H, Zhang QZ, Zhu JK (2018) A naturally occurring epiallele associates with leaf senescence and local climate adaptation in Arabidopsis accessions. Nat Commun 9(1):460. https://doi.org/10.1038/s41467-018-02839-3
Heard E, Martienssen RA (2014) Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157(1):95–109. https://doi.org/10.1016/j.cell.2014.02.045
Herrera CM, Pozo MI, Bazaga P (2012) Jack of all nectars, master of most: DNA methylation and the epigenetic basis of niche width in a flower-living yeast. Mol Ecol 21(11):2602–2616. https://doi.org/10.1111/j.1365-294X.2011.05402.x
Hirsch S, Baumberger R, Grossniklaus U (2013) Epigenetic variation, inheritance, and selection in plant populations. Cold Spring Harb Symp Quant Biol 77:97–104. https://doi.org/10.1101/sqb.2013.77.014605
Hirt H, Shinozaki K (2003) Plant Responses to Abiotic Stress. Springer Science & Business Media, Berlin
Holonec L, Ranga F, Crainic D, Truta A, Socaciu C (2012) Evaluation of betulin and betulinnic acid content in birch bark from different forestry areas of Western Carpathians. Notulae Botanicae Horti Agrobotanici Cluj-Napoca 40(2):99–105. https://doi.org/10.15835/NBHA4027967
Johannes F, Schmitz RJ (2019) Spontaneous epimutations in plants. New Phytol 221(3):1253–1259. https://doi.org/10.1111/nph.15434
Jones PA, Taylor SM (1980) Cellular differentiation, cytidine analogs and DNA methylation. Cell 20(1):85–93. https://doi.org/10.1016/0092-8674(80)90237-8
Kawakatsu T, Huang SC, Jupe F, Sasaki E, Schmitz RJ, Urich MA, Castanon R, Nery JR, Barragan C, He Y, Chen H, Dubin M, Lee CR, Wang C, Bemm F, Becker C, O’Neil R, O’Malley RC, Quarless DX, Schork NJ, Weigel D, Nordborg M, Ecker JR (2016) Epigenomic diversity in a global collection of Arabidopsis thaliana accessions. Cell 166(2):492–505. https://doi.org/10.1016/j.cell.2016.06.044
Lavergne S, Mouquet N, Thuiller W, Ronce O (2010) Biodiversity and climate change: integrating evolutionary and ecological responses of species and communities. Annu Rev Ecol Evol Syst 41:321–350. https://doi.org/10.1146/annurev-ecolsys-102209-144628
Lenoir J, Gégout JC, Marquet PA, de Ruffray P, Brisse H (2008) A significant upward shift in plant species optimum elevation during the 20th century. Science 320(5884):1768–1771. https://doi.org/10.1126/science.1156831
Li LQ, Li XL, Fu CH, Zhao CF, Yu LJ (2013) Sustainable use of Taxus media cell cultures through minimal growth conservation and manipulation of genome methylation. Process Biochem 48(3):525–531. https://doi.org/10.1016/j.procbio.2013.01.013
Liang D, Zhang ZJ, Wu HL, Huang CY, Shuai P, Ye CY, Tang S, Wang YJ, Yang L, Wang J, Yin WL, Xia XL (2014) Single-base-resolution methylomes of Populus trichocarpa reveal the association between DNA methylation and drought stress. BMC Genet 15(1):S9. https://doi.org/10.1186/1471-2156-15-S1-S9
Liu GF, Xia YD, Liu TK, Dai SJ, Hou XL (2018) The DNA methylome and association of differentially methylated regions with differential gene expression during heat stress in Brassica rapa. Int J Mol Sci 19(5):1414. https://doi.org/10.3390/ijms19051414
Matzke MA, Mosher RA (2014) RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat Rev Genet 15(6):394–408. https://doi.org/10.1038/nrg3683
Morin X, Viner D, Chuine I (2008) Tree species range shifts at a continental scale: new predictive insights from a process-based model. J Ecol 96(4):784–794. https://doi.org/10.1111/j.1365-2745.2008.01369.x
Ning YQ, Liu N, Lan KK, Su YN, Li L, Chen S, He XJ (2020) DREAM complex suppresses DNA methylation maintenance genes and precludes DNA hypermethylation. Nat Plants 6(8):942–956. https://doi.org/10.1038/s41477-020-0710-7
Richards CL, Bossdorf O, Verhoeven KJF (2010) Understanding natural epigenetic variation. New Phytol 187(3):562–564. https://doi.org/10.1111/j.1469-8137.2010.03369.x
Salojärvi J, Smolander OP, Nieminen K, Rajaraman S, Safronov O, Safdari P, Lamminmäki A, Immanen J, Lan T, Tanskanen J, Rastas P, Amiryousefi A, Jayaprakash B, Kammonen JI, Hagqvist R, Eswaran G, Ahonen VH, Serra JA, Asiegbu FO, de Dios B-L, Blande D, Blokhina O, Blomster T, Broholm S, Brosché M, Cui F, Dardick C, Ehonen SE, Elomaa P, Escamez S, Fagerstedt KV, Fujii H, Gauthier A, Gollan PJ, Halimaa P, Heino PI, Himanen K, Hollender C, Kangasjärvi S, Kauppinen L, Kelleher CT, Kontunen-Soppela S, Koskinen JP, Kovalchuk A, Kärenlampi SO, Kärkönen AK, Lim KJ, Leppälä J, Macpherson L, Mikola J, Mouhu K, Mähönen AP, Niinemets Ü, Oksanen E, Overmyer K, Palva ET, Pazouki L, Pennanen V, Puhakainen T, Poczai P, Possen BJHM, Punkkinen M, Rahikainen MM, Rousi M, Ruonala R, van der Schoot C, Shapiguzov A, Sierla M, Sipilä TP, Sutela S, Teeri TH, Tervahauta AI, Vaattovaara A, Vahala J, Vetchinnikova L, Welling A, Wrzaczek M, Xu E, Paulin LG, Schulman AH, Lascoux M, Albert VA, Auvinen P, Helariutta Y, Kangasjärvi J (2017) Genome sequencing and population genomic analyses provide insights into the adaptive landscape of silver birch. Nat Genet 49(6):904–912. https://doi.org/10.1038/ng.3862
Schulz B, Eckstein RL, Durka W (2014) Epigenetic variation reflects dynamic habitat conditions in a rare floodplain herb. Mol Ecol 23(14):3523–3537. https://doi.org/10.1111/mec.12835
Shen X, De Jonge J, Forsberg SK, Pettersson ME, Sheng ZY, Hennig L, Carlborg Ö (2014) Natural CMT2 variation is associated with genome-wide methylation changes and temperature seasonality. PLoS Genet 10(12):e1004842. https://doi.org/10.1371/journal.pgen.1004842
Skern R, Frost P, Nilsen F (2005) Relative transcript quantification by quantitative PCR: roughly right or precisely wrong? BMC Mol Biol 6:10. https://doi.org/10.1186/1471-2199-6-10
Springer NM, Schmitz RJ (2017) Exploiting induced and natural epigenetic variation for crop improvement. Nat Rev Genet 18(9):563–575. https://doi.org/10.1038/nrg.2017.45
Steward N, Ito M, Yamaguchi Y, Koizumi N, Sano H (2002) Periodic DNA methylation in maize nucleosomes and demethylation by environmental stress. J Biol Chem 277(40):37741–37746. https://doi.org/10.1074/jbc.M204050200
Stroud H, Do T, Du JM, Zhong XH, Feng SH, Johnson L, Patel DJ, Jacobsen SE (2014) Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat Struct Mol Biol 21(1):64–72. https://doi.org/10.1038/nsmb.2735
Suttle KB, Thomsen MA, Power ME (2007) Species interactions reverse grassland responses to changing climate. Science 315(5812):640–642. https://doi.org/10.1126/science.1136401
Thuiller W, Lavorel S, Araújo MB, Sykes MT, Prentice IC (2005) Climate change threats to plant diversity in Europe. Proc Natl Acad Sci U S A 102(23):8245–8250. https://doi.org/10.1073/pnas.0409902102
Verhoeven KJ, Jansen JJ, van Dijk PJ, Biere A (2010) Stress-induced DNA methylation changes and their heritability in asexual dandelions. New Phytol 185(4):1108–1118. https://doi.org/10.1111/j.1469-8137.2009.03121.x
Wada Y, Miyamoto K, Kusano T, Sano H (2004) Association between up-regulation of stress-responsive genes and hypomethylation of genomic DNA in tobacco plants. Mol Genet Genomics 271(6):658–666. https://doi.org/10.1007/s00438-004-1018-4
Wang WS, Pan YJ, Zhao XQ, Dwivedi D, Zhu LH, Ali J, Fu BY, Li ZK (2011) Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J Exp Bot 62(6):1951–1960. https://doi.org/10.1093/jxb/erq391
Yin J, Ren CL, Zhan YG, Li CX, Xiao JL, Qiu W, Li XY, Peng HM (2012) Distribution and expression characteristics of triterpenoids and OSC genes in white birch (Betula platyphylla suk.). Mol Biol Rep 39(3):2321–2328. https://doi.org/10.1007/s11033-011-0982-0
Yin J, Liang T, Wang SY, Zhang MY, Xiao JL, Zhan YG, Li CX (2015) Effect of drought and nitrogen on betulin and oleanolic acid accumulation and OSC gene expression in white birch saplings. Plant Mol Biol Rep 33:705–715. https://doi.org/10.1007/s11105-014-0778-1
Yin J, Li CX, Zhan YG, Sun HR, Gong Y, Zhang MY, Xiao JL (2016) The response of physiological characteristics, expression of OSC genes, and accumulation of triterpenoids in Betula platyphylla Suk to MeJA and SA treatment. Plant Mol Biol Rep 34:427–439. https://doi.org/10.1007/s11105-015-0931-5
Yin J, Li X, Zhan YG, Li Y, Qu ZY, Sun L, Wang SY, Yang J, Xiao JL (2017) Cloning and expression of BpMYC4 and BpbHLH9 genes and the role of BpbHLH9 in triterpenoid synthesis in birch. BMC Plant Biol 17(1):214. https://doi.org/10.1186/s12870-017-1150-z
Yong-Villalobos L, González-Morales SI, Wrobel K, Gutiérrez-Alanis D, Cervantes-Peréz SA, Hayano-Kanashiro C, Oropeza-Aburto A, Cruz-Ramírez A, Martínez O, Herrera-Estrella L (2015) Methylome analysis reveals an important role for epigenetic changes in the regulation of the Arabidopsis response to phosphate starvation. Proc Natl Acad Sci U S A 112(52):E7293-7302. https://doi.org/10.1073/pnas.1522301112
Zeng FS, Sun FK, Liang NS, Zhao XT, Luo W, Zhan YG (2015) Dynamic change of DNA methylation and cell redox state at different micropropagation phases in birch. Trees 29:917–930. https://doi.org/10.1007/s00468-015-1174-7
Zhang XY, Yazaki J, Sundaresan A, Cokus S, Chan SW, Chen HM, Henderson IR, Shinn P, Pellegrini M, Jacobsen SE, Ecker JR (2006) Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 126(6):1189–1201. https://doi.org/10.1016/j.cell.2006.08.003
Zhang MY, Wang SY, Yin J, Li CX, Zhan YG, Xiao JL, Liang T, Li X (2016) Molecular cloning and promoter analysis of squalene synthase and squalene epoxidase genes from Betula platyphylla. Protoplasma 253(5):1347–1363. https://doi.org/10.1007/s00709-015-0893-3
Zhang HM, Lang ZB, Zhu JK (2018) Dynamics and function of DNA methylation in plants. Nat Rev Mol Cell Biol 19(8):489–506. https://doi.org/10.1038/s41580-018-0016-z
Zhao GL, Yan WD, Cao D (2007) Simultaneous determination of betulin and betulinic acid in white birch bark using RP-HPLC. J Pharmaceut Biomed 43(3):959–962. https://doi.org/10.1016/j.jpba.2006.09.026
Zhou L, Cheng X, Connolly BA, Dickman MJ, Hurd PJ, Hornby DP (2002) Zebularine: a novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J Mol Biol 321(4):591–599. https://doi.org/10.1016/s0022-2836(02)00676-9
Acknowledgements
We cordially thank Professors Benli Pan and Xianchang Zhou at Heilongjiang Academy of Forestry Science in Harbin for the sampling of natural birch populations. We thank Professor Guifeng Liu at State Key Laboratory of Tree Genetics and Breeding of Northeast Forestry University, Harbin for providing the birch seeds for ZEB treatments. We appreciate the assistance of Qijiang Xu, Bing Ding, Chang Li, Tingting Zhou, Lei Chen, and Jianfei Yang at the College of Life Sciences of Northeast Forestry University at Harbin.
Author information
Authors and Affiliations
Contributions
JW and BC contributed equally to this work. QZ, YL, JW, BC designed the research, JW, YW, HZ, LW, YZ, LX, TJ, JY performed the field manipulations. BC, TZ analysed the data, QZ, SA, JW, LX, BC interpreted the results, SA, BC, QZ wrote the original manuscript with contribution HWS, GZ, RRS.
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Project funding: The work was supported by the National Non-profit Institute Research Grant of the Chinese Academy of Forestry (CAFYBB2019ZY003); the National Natural Science Foundation of China (31871220 and 31801444); the Innovation Project of State Key Laboratory of Tree Genetics and Breeding (Northeast Forestry University) (2013A06); the Fundamental Research Funds for the Central Universities (2572017DA06 and 2572020DP01); and, Heilongjiang Provincial Natural Science Foundation of China (LH2021C005).
The online version is available at http://www.springerlink.com.
Corresponding editor: Yu Lei.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Chen, B., Ali, S. et al. Epigenetic modification associated with climate regulates betulin biosynthesis in birch. J. For. Res. 34, 21–35 (2023). https://doi.org/10.1007/s11676-021-01424-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11676-021-01424-7