
Abstract
Populus euphratica Oliv., the Euphrates poplar, is the tallest tree species in the arid desert areas of Northwest China. Investigation of its drought-resistance genes is valuable to increase understanding of drought resistance mechanisms. RNA-seq of leaves and roots under drought simulation by 25% polyethylene glycol-6000 (PEG 6000) were performed at 0, 4, and 12 h. Leaves and roots responded differently to drought via differentially upregulated and downregulated genes; more genes were downregulated than upregulated in both leaves and roots. Additionally, these differentially expressed genes were enriched in different GO terms and KEGG pathways. For example, GO term ‘response to organic substance’ and KEGG pathway ‘nitrogen metabolism’ were enriched for drought-stressed leaves, while GO term ‘cell wall organization or biogenesis’ and KEGG pathway ‘zeatin biosynthesis’ were enriched for drought-stressed roots. The enrichment of the GO term ‘enzyme linked receptor protein signalling pathway’ in both leaf and root drought responses suggests that these tissues may also have similar mechanisms. However, roots under drought stress for four hs responded by activating programed cell death. The KEGG pathway ‘plant hormone signal transduction’ was detected for 4- and 12-h drought-stressed leaves and 12-h drought-stressed roots, suggesting that plant hormone signal transduction plays an important role in both roots and leaves. GO enrichment of upregulated and downregulated genes for leaves and roots reflect differentially regulatory mechanisms of response to drought stress via different biological processes such as the regulation of photosynthesis and auxin signalling pathway in leaves, and the regulation of defence response and water homeostasis in roots. Fifteen candidate genes, including transcription factors, protein kinase, transporter, late embryogenesis abundant protein and mannitol dehydrogenase, were further selected to determine their response to drought using qRT-PCR. The results show that the expression patterns of 13 of the 15 genes correspond to the RNA-seq results. This study provides new insights into the drought response mechanism of P. euphratica and suggests new candidate gene resources.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Drought, salinity, and cold are abiotic stresses that significantly influence plant growth and productivity (Mahajan and Tuteja 2005), and plants have developed strategies to cope with these adverse environmental conditions (Chinnusamy et al. 2004). Forests are important terrestrial ecosystems and dominate much of the landscape. However, most forest species cannot tolerate saline soils or extreme drought conditions. Poplus euphratica, a species of Populus, is native to desert regions ranging from western China to North Africa that are arid and saline (Ma et al. 2013). P. euphratica is considered to be an ideal model species for studying resistance to abiotic stress (Li et al. 2011; Iqbal et al. 2017). Therefore, research into the resistance mechanisms to abiotic stress by P. euphratica will help to improve the resistance of this species in arid and semi-arid regions. During long-term drought stress of P. euphratica, altered transcript abundances of the transcription factor families, AP2/EREPB, bZIP, NAC, NF-Y, WRKY, MYB and Homeobox, and the small HSP, HSP70 and HSP90 heat shock protein families, indicated that these families are involved in the response to drought stress (Yan et al. 2012). Additionally, at the biological process level, genes involved in the inhibition of stomatal closure, the ascorbate–glutathione pathway and the ubiquitin–proteasome system may modulate the drought stress response of P. euphratica (Tang et al. 2013). However, previous studies have focused on long-term drought stress in P. euphratica. Short-term responses to biotic or abiotic stresses at the transcriptional level are vital for growth and development, and plants adopt various strategies to cope with these stresses in order to survive. For example, short-term osmotic stress in watermelons prompted a series of biological processes such as osmotic adjustment, signal transduction, and cell cycle and ribosome-related processes; the watermelons adapted to osmotic stress via repressing root growth (Yang et al. 2016). Additionally, exposure to short-term drought stress can induce a memory response in rice, which aids the plants in adapting to recurrent stress environments (Li et al. 2019). The coping mechanisms of P. euphratica seedling roots and leaves in response to short-term drought stress have not been reported.
In this study, RNA-seq was used to investigate the transcriptome profiles of roots and leaves of hydroponically grown P. euphratica at the start (0 h, the control), and at 4 and 12 h of drought stress created by adding 25% PEG 6000. This study presents new insights into the molecular mechanisms underlying short-term water deficit tolerance by P. euphratica.
Materials and methods
Plant material and osmotic stress treatment
Seeds of P. euphratica were planted in pots with nutrient soil, mulched and placed in a 30 °C chamber for germination and culturing. After two months of cultivation, 5–7 seedlings per pot were transplanted into nutrient soil and grown for a further two months under a 16 h light/8 h dark photoperiod. The seedlings were then washed and transferred to distilled water to recover until new seedling roots emerged. ROS-scavenging enzymes such as malondialdehyde (MDA), peroxidase (POD), catalase (CAT), and chlorophyll contents play important roles in drought tolerance responses (Wang 2014). In addition, the inhibition of photosynthesis and energy dissipation are common features under drought stress in many plant species, and which reflect photosystem II (PSII) thermostability and electron transport changes (Zivcak et al. 2014). Hence, changes in chlorophyll contents indicate that drought reshaped the structure of chloroplasts and influenced photosynthesis. In this study, six concentrations of PEG 6000 were used (weight/volume, 5%, 10%, 15%, 20%, 25%, and 30%) under five time periods (0 h, the controls, and at 4, 8, 12, and 24 h) to evaluate biochemical responses (MDA, POD, CAT and chlorophyll contents) to drought stress. Enzyme activities of MDA, POD and CAT, as well as chlorophyll contents, increased gradually with increasing concentrations and treatment times. PEG 6000 at a low concentration such as 5% is insufficient to induce a response to drought stress. Using 25% PEG 6000, POD activity, leaf chlorophyll contents and root CAT activity began to decrease for 24 h. When using 30% PEG 6000 for 4 h, the POD, SOD, CAT activities of roots and leaves and the chlorophyll content of leaves decreased. At the concentrations of 5–25%, CAT activity increased with the increase of stress time from 4 to 24 h, which eliminated the hydrogen peroxide and protected the cells from damage. At the concentration of 30%, CAT activity increased first and then decreased with the increase of stress time, which indicated that PEG stress with high concentration caused damage that CAT could not maintain high activity level. Therefore, 30% PEG 6000 caused irreversible damage to P. euphratica seedlings for 12 h and 24 h, and we concluded that 25% PEG 6000 was the critical concentration for triggering drought resistance in P. euphratica seedlings. For five time periods under 25% PEG 6000, treatments under 4 and 12 h were selected for representation of response to drought stress (Fig. S1). Leaves and roots were collected and frozen immediately in liquid nitrogen for further drought-response analysis after treatment with 25% PEG 6000 solution at the beginning and at 4 and 12 h.
RNA-seq and quality control of sequence data
Total RNA from six samples of leaves and roots at three time periods were extracted and digested with DNase I to remove DNA, and mRNAs were then purified and enriched with magnetic beads of oligo (dT). Each constructed library of enriched mRNAs was evaluated for fragment size and concentration using an Agilent 2100 Bioanalyzer and sequenced using ion proton sequencing at the Beijing Genomics Institute (BGI, Shenzhen, China).
The original image data, i.e., raw data, generated by the sequencer were converted into sequence data by base calling. Prior to data analysis, the raw data were pre-processed as follows: (1) reads were trimmed by removing adaptors; (2) average quality of reads was calculated with a sliding window of 15 bp, and reads with an average quality value ˃ 9 were retained; and, (3) additionally processed reads whose lengths were ˂ 30 were removed. The retained reads were separately aligned to the reference genome of P. trichocarpa (Tuskan et al. 2006) (allowing two base mismatches) using the short-read alignment software TMAP (3.4.1).
Identification of differentially expressed genes
Gene expression was calculated using the RPKM (reads per kilobase per million mapped reads) method. Based on the Poisson distribution model test, DEGSeq (Wang et al. 2010) was used to identify differentially expressed genes. These are defined as genes having a fold change ≥ 2 and a statistical FDR ≤ 0.001, and were retained for subsequent analysis. The Gene Ontology (GO) database was used to perform significant enrichment analysis of functional annotations by mapping the differentially expressed genes to each term of the database to calculate the number of associated genes. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed using the KEGG database, and the differentially expressed genes were mapped to the database to determine the number of genes and the degree of enrichment in each pathway.
Expression pattern of drought-related genes quantified by qRT-PCR
Combining these results with reports in the literature (Ke et al. 2009; Kushwaha et al. 2009; Marshall et al. 2012; De Abreu-Neto et al. 2013; Wu et al. 2015, 2017), fifteen candidate genes were selected for further analysis (Table 1). The specific primers of drought-related genes used for qRT-PCR were designed using Primer Premier 5.0. The qRT-PCR system consisted of 5 μL of SYBR® Premix Ex Taq II (2 ×), 0.4 μL of the upstream and downstream primers (10 μM), 1 μL of cDNA, and RNase-free ddH2O. The reaction procedure was: 95 °C for 30 s, 40 cycles of 95 °C for 5 s, 55 °C for 30 s, and 72 °C for 30 s, followed by a dissolution curve analysis: 95 °C for 0 s (20 °C/s), 60 °C for 15 s (20 °C/s), 95 °C for 0 s (0.1 °C/s). The data were analysed by the 2−ΔΔCT method and the specificity of the amplified products was based on the dissolution curve.
Results
Differentially expressed genes involved in drought stress
After quality control, a total of 156,663,152 clean reads were produced from six samples (Pe_L0, Pe_L4, Pe_L12, Pe_R0, Pe_R4, and Pe_R12). After alignment to the gene sets of the reference genome (PRJNA10772) with a maximum of two mismatches, only uniquely matched reads with greater than 70% mapping rates (71.5–80.6%) were used for the subsequent analysis of differentially expressed genes (Table S1). To ensure adequate reads for differential gene analysis, a saturation analysis of sequencing depth was evaluated with RNA-SeQC (De Luca et al. 2012). When the quantity of clean reads is less than 9 Mb, the number of detected genes is proportional to the number of sequenced reads; when it approaches 27 Mb, the number of detected genes tends to be saturated. The amounts of sequencing data for six samples, Pe_L0, Pe_L4, Pe_L12, Pe_R0, Pe_R4, and Pe_R12, were 26 Mb, 24 Mb, 25 Mb, 27 Mb, 26 Mb, and 27 Mb, respectively. These amounts were close to saturation, indicating that the sequencing depths of all samples could be expected to cover nearly all expressed genes (Fig. S2). To ensure uniform coverage of each gene from the 5’ end to the 3′ end, the randomness of mRNA disruption was assessed by read distribution on the reference genes. Since the lengths of the reference genes differed, the positions of reads on the reference genes were normalized to their relative positions, and the number of reads aligned to each different gene position was then counted. If the randomness is good, the reads should be evenly distributed on all parts of each gene without bias. The reference genes were normalized to the relative positions of a total of 100 windows and divided into five equal parts. Each window corresponded to the number of reads in the alignment. The reads of each sample were basically randomly distributed on each gene, indicating that the six constructed libraries were homogeneous (Fig. S3). Gene sequencing coverage refers to the percentage of each gene covered by reads, namely, the ratio of base numbers from uniquely mapped reads in certain genes to the total base numbers in all genes. The gene coverage was divided into ten levels: approximately 60% of each gene had a gene coverage of more than 60%, and the gene coverage was 90–100% of the number of reads. The absolute advantage reached 21–42% of the total reads (Fig. S4). This indicates that the proportion of high-coverage genes was high, and the sequencing results met the experimental needs.
Each group had many upregulated and downregulated genes. For leaves at different times, the number of downregulated genes was much greater than that of upregulated genes. Compared with the leaves of the controls (0 h) and at 12 h, there were 4260 upregulated genes and 8938 downregulated genes (Pe_L0-vs-Pe_L12). The comparison of leaves at nearby time periods (Pe_L0-vs-Pe_L4 and Pe_L4-vs-Pe_L12) showed that the leaves had a similar number of downregulated genes, but there were more upregulated genes (3047) under Pe_L0-vs-Pe_L4 than under Pe_L4-vs-Pe_L12 (2660), suggesting a stronger response to short-term drought stress. For roots, the total number of differentially expressed genes was lower than that of leaves. In contrast to the leaves, there were more upregulated than downregulated genes when comparing roots at 12 h of drought stress to those at 4 h (Pe_R4-vs-Pe_R12). Additionally, the comparison of roots at nearby time periods (Pe_R0-vs-Pe_R4 and Pe_R4-vs-Pe_R12) showed that roots had a stronger upregulated response at 12 h than at other times, with more upregulated genes (3141, Pe_R4-vs-Pe_R12). During the early response at 4 h (Pe_R0-vs-Pe_R4), there were fewer upregulated genes than downregulated genes in roots (Fig. 1). The results suggest that roots and leaves produced differential responses to short-term drought. Moreover, differential analysis of genes at different time periods within the same tissues show that the differentially expressed genes in the leaves underwent greater alterations in expression than those in the roots, indicating that the leaves responded more strongly to drought.

Statistics of differentially expressed genes in leaves and roots after 4-h and 12-h drought stress treatments; abbreviations are: Pe_L0 the 0-h control of leaf, Pe_L4 the 4-h treated leaf; Pe_L0-VS-Pe_L4 of the x-axis indicates the differential genes with Pe_L0 as denominator and Pe_L4 as numerator. DEGs of the y-axis is the abbreviation of differential expressed genes
GO classification and differential enrichment of leaf and root genes in response to drought
GO is an international standardization system for the classification of gene functions that provides a dynamically updated vocabulary to fully describe the properties of genes and gene products in organisms (Ashburner et al. 2000). GO has three categories describing the molecular function of the gene, the cellular component, and the biological process involved. According to the annotation information of the Non-Redundant Protein Sequence Database (NR database), GO annotations of all differentially expressed genes were obtained using Blast2GO software (Conesa and Gotz 2008). GO functional classification was then performed by WEGO software (Ye et al. 2006). In the three ontologies of cellular components, molecular function and biological processes, GO annotations of leaf and root samples showed a similar response to drought stress. In the cellular components, the genes were mainly distributed in cells, cell parts and organelles; in the molecular function, the gene functions mainly included ‘transport activity’, ‘catalytic activity’ and ‘molecular binding’; and in the biological process, the gene functions were mainly ‘metabolic process’ and ‘cell process’, followed by ‘response to stimulus’ (Fig. 2). These results indicate that the roots and leaves of P. euphratica resist drought via metabolic processes mediated by the binding and catalysis of multiple enzymes. At the same time, the increase in transporter genes may help the cells to change the osmotic pressure through the transport of the substance, thereby improving the ability to respond to drought.

Gene Ontology functional annotations of differentially expressed genes following 4-h and 12-h drought stress in leaves (a) and roots (b) by WEGO 2.0 analysis; each Gene Ontology annotation contains three levels, cellular component, molecular function and biological process
The GO annotation and enrichment analysis of differentially expressed genes generally reflected the drought response of both leaves and roots. There were identically enriched GO terms for both 4-h and 12-h treated leaves. For example, two of these shared GO terms were ‘enzyme-linked receptor protein signalling pathway’ and ‘cell surface receptor signalling pathway’, which suggests that leaves respond to drought via sensing signals from cell surface receptors. However, compared with the response of leaves to 4-h drought, differentially expressed genes in leaves under 12-h treatment showed many more enriched GO terms. This indicates that other biological processes, such as ‘response to hormone stimulus’ and ‘cellular nitrogen compound catabolic process’, are involved in the response to persistent drought (Fig. 3a, b). For roots under drought stress at different times, the GO terms enriched in the response to 12-h drought differed from those enriched in response to 4-h drought. After four hs, the GO terms contained only five enriched biological processes, three of which were ‘death’, ‘cell death’ and ‘programed cell death’. This suggests that, unlike leaves, roots respond to drought by activating programed cell death under short-term (4-h) drought. After 12 h of drought, roots take advantage of multiple cell wall-related biological processes to respond to drought, such as ‘cell wall organization or biogenesis’ and ‘cell wall macromolecule metabolic processes’. Similar to leaves under 12 h of drought, roots were also enriched in the ‘cell surface receptor signalling pathway’ and ‘enzyme-linked receptor protein signalling pathway’ (Fig. 3c, d). Together, these results reflect the commonalities and differences of roots and leaves in response to drought at different time periods. Despite the different responses of leaves and roots to 4-h drought, the enzyme-linked receptor protein signalling pathway may play an essential role in the response of P. euphratica to drought under 12 h, regardless of the tissue.

Enriched Gene Ontology terms of differentially expressed genes in the biological process ontology in 4-h treated leaves (a), 12-h treated leaves (b), 4-h treated roots (c) and 12-h treated roots (d); x-axis is the number of genes annotated with each Gene Ontology term, the top right bar represents the enriched − log10 P value
Differential KEGG enrichment of leaf and root genes in response to drought
As noted above, P. euphratica responded to drought via a large number of genes involved in metabolic processes. KEGG is the main public database for identifying metabolic pathways. Those with a Q-value (the corrected P value after multiple hypothesis tests) ≤ 0.05 are defined as significantly enriched in differentially expressed genes. The results of KEGG enrichment analysis are represented graphically; the RichFactor refers to the ratio of the number of genes in the pathway entry of the differentially expressed gene to the total number of genes in the pathway entry of all annotated genes. The larger the RichFactor, the greater the degree of enrichment. For leaves, the uniquely enriched KEGG pathways under 4-h drought were ‘flavonoid biosynthesis’, ‘ABC transporters’, ‘stilbenoid, diarylheptanoid and gingerol biosynthesis’, ‘limonene and pinene degradation’ and ‘starch and sucrose metabolism’, while the uniquely enriched KEGG pathways under 12-h drought were ‘nitrogen metabolism’, ‘porphyrin and chlorophyll metabolism’ and ‘amino sugar and nucleotide sugar metabolism’. Common KEGG pathways were ‘plant hormone signal transduction’, ‘photosynthesis’, ‘photosynthesis-antenna proteins’, ‘plant-pathogen interaction’, and ‘circadian rhythm-plant’ (Fig. 4a, b). For roots, ‘RNA polymerase’, ‘plant-pathogen interaction’, ‘pyrimidine metabolism’ and ‘purine metabolism’ were unique KEGG pathways under 4-h drought; of the top 20 enriched KEGG pathways in roots under 12-h drought, most were metabolism-related. Only one KEGG pathway, ‘flavonoid biosynthesis’, was common in roots under both 4-h and 12-h drought periods (Fig. 4c, d). The results suggest that leaves and roots adopt different pathways to protect against drought over different time periods, and roots under 12-h drought showed enrichment in most pathways, yielding the strongest response among the tissues and time periods.

Enriched Kyoto Encyclopedia of Genes and Genomes terms for differentially expressed genes in 4-h treated leaves (a), 12-h treated leaves (b), 4-h treated roots (c) and 12-h treated roots (d); x-axis is the RichFactor of each Kyoto Encyclopedia of Genes and Genomes term, the top right bar represents the enriched -log10 P value, and the bottom right dot the gene number
Expression patterns of differentially expressed genes in leaves and roots
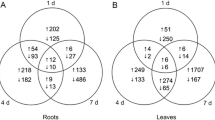
To explore the differential and concordant responses to drought stress, four Venn diagrams were drawn for upregulated and downregulated genes in both leaves and roots. In addition to being specifically and separately regulated in leaves and roots, there were 146 simultaneously upregulated genes and 680 simultaneously downregulated genes compared to the control for leaves. For roots, there were eight simultaneously upregulated and 34 consecutively downregulated genes in adjacent comparisons (Fig. 5). The number of simultaneously downregulated genes was obviously much higher than that of upregulated genes in any comparison, suggesting that P. euphratica may slow the development of roots and leaves via inhibiting gene transcription, thereby reducing energy consumption. The expression patterns of these co-regulated genes showed significant spatiotemporal specificity (Fig. 6).

Venn diagram of differentially expressed genes in leaves and roots after 4-h and 12-h drought treatments; Venn diagram of upregulated genes in the 0-h control (a), Venn diagram of downregulated genes in the 0-h control (b), Venn diagram of consecutively up-regulated genes between adjacent times (c), Venn diagram of consecutively down-regulated genes between adjacent times (d). The integral number and percentage at the corresponding bottom in each represent the gene number and proportion of part genes to total genes, respectively

Expression pattern of co-regulated genes in leaves and roots after 4-h and 12-h drought treatments showed by heatmap; 146 simultaneously upregulated genes under the 0-h control between leaves and roots (a), 680 simultaneously downregulated genes under the 0-h control between leaves and roots (b), eight consecutively upregulated genes between adjacent times from leaves and roots (c), 34 consecutively downregulated genes between adjacent times from leaves and roots (d)
To confirm the candidate genes involved in the drought response, 146 simultaneously upregulated and 680 simultaneously downregulated genes were annotated with BlastP against the NR database (Altschul et al. 1997). There were 42 genes that may be related to drought resistance, and six with large changes in expression (RPKM) but with annotation functions that may be unrelated to drought resistance. Among the fifteen candidate genes for qRT-PCR analysis, 13 showed consistent expression patterns as those from RNA-seq (Table 1, Fig. 7, Fig. S5), suggesting the reliability of the identification of the differentially expressed genes and implicating these gene in drought response. These genes include transcription factors (Pe MYB, Pe ZnF1, Pe ZnF2, Pe ZnF3, Pe WRKY1, Pe WRKY2), receptor kinase (Pe LRR1), transporter (Pe AQU), late embryogenesis-rich proteins (Pe LEA) and mannitol dehydrogenase (Pe MTD), and provide a good foundation for further exploration of the molecular mechanisms of drought resistance genes.

qRT-PCR confirmation of 13 candidate drought resistance genes with consistent expression patterns with RNA-seq; top right DGE and qRT-PCR represent results from RNA-seq and qRT-PCR confirmation, respectively
Discussion
To survive, drought stress induces a series of responses in leaves and roots at the morphological, physiological and biochemical levels (Wang et al. 2016). Eventually, these responses are reflected at the genome-wide transcriptional and post-transcriptional levels. Regardless of the length of time, the response of leaves to drought was more dramatic than that of roots, as indicated by the number of differentially expressed genes. When comparisons of the treated groups with the control group were performed, except for Pe_R4 vs. Pe_R12, the number of downregulated genes was greater than that of upregulated ones in roots and leaves, indicating that the inhibition of gene expression is more critical than gene activation under drought stress (Fig. 1). This may be the result of the inhibition of energy dissipation, a common feature under drought stress. This inhibition in leaves, as reflected by the number of downregulated genes, is more serious than in roots at different time periods, and also indicates the inhibition of photosynthesis under drought stress (Wang 2014).
Differential GO and KEGG terms reflect the biological processes and pathways involved in drought stress. In the short-term response of roots, GO enrichment mainly involved processes related to death. This is consistent with a report by Duan et al. (2010) for other plants under drought, for example, water stress- induced programed cell death in the root tips of Arabidopsis. In addition, cell wall-related GO terms were also detected in the drought response of roots. The influence of drought stress on cell wall formation has also been reported in black spruce (Picea mariana (Mill.) BSP) saplings (Balducci et al. 2015). Receptor-like kinases (RLKs) can contribute to improving plant performance under drought stress (Marshall et al. 2012). In this study, the common GO terms ‘cell surface receptor signalling pathway’ and ‘enzyme-linked receptor protein signalling pathway’ were enriched in both leaves and roots. Therefore, we speculate that RLK pathways play an important role in the responses of P. euphratica to drought.
The expression of flavonoid biosynthesis genes has been reported in leaves and roots in response to drought (Yuan et al. 2012; Ma et al. 2014). In this study, the ‘flavonoid biosynthesis’ pathway was enriched in leaves under four hs of drought and in roots under four and 12 h. The signal transduction pathway is the bridge mediating the sensing mechanism and the genetic response. Plants cope with environmental stress via activating signal transduction cascades that coordinate the physiological and biochemical responses necessary for adaptation (Huang et al. 2012). The pathway ‘plant hormone signal transduction’ pathway was enriched in leaves subjected to 4-h and 12-h drought, and in roots subjected to 12-h drought but to 4-h drought. This suggests that plant hormone signal transduction may not be activated when roots are exposed to drought stress for a short time. Such opposite metabolic responses of leaves and roots to drought have been reported in other plants (Gargallo-Garriga et al. 2014).
Upregulated and downregulated genes indicated the activation and inactivation of specific biological processes under stress. To further explore the possible differential response of leaves and roots to drought stress, GO and KEGG analysis was carried out for downregulated and upregulated genes for both roots and leaves, respectively (Table S6). We found ‘transcription, DNA-templated (GO:0006351)’ was only significantly enriched for upregulated genes for leaves under 4-h treatment. For downregulated genes under 4-h treatment, seven GO terms were enriched at the biological process level: ‘photosynthesis, light harvesting in photosystem I (GO:0009768)’, ‘photosynthesis (GO:0015979)’, ‘response to light stimulus (GO:0009416)’, ‘secondary metabolite biosynthetic process (GO:0044550)’, ‘protein-chromophore linkage (GO:0018298)’, ‘flavonoid biosynthetic process (GO:0009813)’ and ‘flavonoid glucuronidation (GO:0052696)’. Seven KEGG pathways similar to GO terms were enriched: ‘pop00195: photosynthesis’, ‘pop00196:photosynthesis-antenna proteins’, ‘pop01100:metabolic pathways’, ‘pop00710:carbon fixation in photosynthetic organisms’, ‘pop01200:carbon metabolism’, ‘pop01110:biosynthesis of secondary metabolites’ and ‘pop00941:flavonoid biosynthesis’. These results indicate that more photosynthesis-related genes were inhibited in their response to leaf drought stress. When compared genes under 12-h stress to those under four hs, seven GO terms at the biological process level and two KEGG pathways were enriched for upregulated genes, while ten GO terms at the biological process level and two KEGG pathways were enriched for downregulated genes. Among the enriched GO terms for downregulated genes, the ‘auxin-activated signalling pathway (GO:0009734)’ and more photosynthesis-related GO terms (GO:0009768, GO:0009416 and GO:0015979) were enriched. This suggests that, besides inhibition of photosynthesis, the auxin signalling pathway, (as a positive pathway in the developmental process), was inhibited to help P. euphratica to adapt to drought stress.
For root response to drought, there were less enriched GO terms and KEGG pathways than for leaves. Enriched GO term of ‘defense response (GO:0006952)’ in downregulated genes in roots with four h drought stress was also enriched in upregulated genes in 12-h stressed roots showed differential regulation of the same biological processes for response to drought at different times. In addition, for downregulated genes in 12-h treated roots compared to 4-h treated roots, the three most enriched GO terms, ‘xylan biosynthetic process (GO:0045492)’, ‘ion transmembrane transport (GO:0034220)’ and ‘cellular water homeostasis (GO:0009992)’, suggests that the suppression of these biological processes may help roots adapt to drought. These results indicate the differential regulation of leaves and roots to respond to drought.
Conclusion
A transcriptome analysis of leaves and roots under 0-h (control, no stress), and 4-h and 12-h drought stress treatment simulated by 25% PEG 6000 identified differentially expressed genes at different periods of time. Seedlings responded to drought via inhibition rather than by activation of gene expression, possibly resulting in the inhibition of energy dissipation, a common feature by many plants under drought stress. Differentially enriched GO and KEGG terms were found for the responses of leaves and roots to short-term (4-h treatment) and relatively long-term (12-h treatment). This indicates that leaves and roots over different time periods respond differently to drought through different biological processes and pathways. Enrichment analysis of GO and KEGG for upregulated and downregulated genes for leaves and roots reflect the differentially regulatory mechanisms of response to drought, such as the regulation of photosynthesis and auxin signalling pathway in leaves, and the regulation of defense response and water homeostasis in roots. Confirmation of 13 of 15 candidate drought-related genes by qRT-PCR further validated our RNA-seq results. This study provides new insights into the drought response of P. euphratica seedlings and provides candidate gene resources for engineered improvement of P. euphratica.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res 25:3389–3402
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. Gene Ontol Consort Nat Genet 25:25–29
Balducci L, Deslauriers A, Giovannelli A, Beaulieu M, Delzon S, Rossi S, Rathgeber CB (2015) How do drought and warming influence survival and wood traits of Picea mariana saplings? J Exp Bot 66:377–389
Chinnusamy V, Schumaker K, Zhu JK (2004) Molecular genetic perspectives on cross-talk and specificity in abiotic stress signalling in plants. J Exp Bot 55:225–236
Conesa A, Gotz S (2008) Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 2008:619832
De Abreu-Neto JB, Turchetto-Zolet AC, De Oliveira LF, Zanettini MH, Margis-Pinheiro M (2013) Heavy metal-associated isoprenylated plant protein (HIPP): characterization of a family of proteins exclusive to plants. FEBS J 280:1604–1616
De Luca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, Reich M, Winckler W, Getz G (2012) RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 28:1530–1532
Duan YF, Zhang WS, Li B, Wang YN, Li KX, Sodmergen Han CY, Zhang YZ, Li X (2010) An endoplasmic reticulum response pathway mediates programmed cell death of root tip induced by water stress in Arabidopsis. New Phytol 186:681–695
Gargallo-Garriga A, Sardans J, Perez-Trujillo M, Rivas-Ubach A, Oravec M, Vecerova K, Urban O, Jentsch A, Kreyling J, Beierkuhnlein C, Parella T, Penuelas J (2014) Opposite metabolic responses of shoots and roots to drought. Sci Rep 4:6829
Huang GT, Ma SL, Bai LP, Zhang L, Ma H, Jia P, Liu J, Zhong M, Guo ZF (2012) Signal transduction during cold, salt, and drought stresses in plants. Mol Biol Rep 39:969–987
Iqbal A, Wang TX, Wu GD, Tang WS, Zhu C, Wang DP, Li Y, Wang HF (2017) Physiological and transcriptome analysis of heteromorphic leaves and hydrophilic roots in response to soil drying in desert Populus euphratica. Sci Rep 7:12188
Ke YQ, Han GQ, He HQ, Li JX (2009) Differential regulation of proteins and phosphoproteins in rice under drought stress. Biochem Biophys Res Commun 379:133–138
Kushwaha HR, Singh AK, Sopory SK, Singla-Pareek SL, Pareek A (2009) Genome wide expression analysis of CBS domain containing proteins in Arabidopsis thaliana (L.) Heynh and Oryza sativa L. reveals their developmental and stress regulation. BMC Genom 10:200
Li BS, Qin YR, Duan H, Yin WL, Xia XL (2011) Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J Exp Bot 62:3765–3779
Li P, Yang H, Wang L, Liu HJ, Huo HQ, Zhang CJ, Liu AZ, Zhu AD, Hu JY, Lin YJ, Liu L (2019) Physiological and transcriptome analyses reveal short-term responses and formation of memory under drought stress in rice. Front Genet 10:55
Ma T, Wang JY, Zhou GK, Yue Z, Hu QJ, Chen Y, Liu BB, Qiu Q, Wang Z, Zhang J, Wang K, Jiang DC, Gou CY, Yu LL, Zhan DL, Zhou R, Luo WC, Ma H, Yang YZ, Pan SK, Fang DM, Luo YD, Wang X, Wang GN, Wang J, Wang Q, Lu X, Chen Z, Liu JC, Wan DS, Li J, Yin TM, Lascoux M, DiFazio SP, Tuskan GA, Wang J, Liu JQ (2013) Genomic insights into salt adaptation in a desert poplar. Nat Commun 4:2797
Ma DY, Sun DX, Wang CY, Li YG, Guo TC (2014) Expression of flavonoid biosynthesis genes and accumulation of flavonoid in wheat leaves in response to drought stress. Plant Physiol Biochem 80:60–66
Mahajan S, Tuteja N (2005) Cold, salinity and drought stresses: an overview. Arch Biochem Biophys 444:139–158
Marshall A, Aalen RB, Audenaert D, Beeckman T, Broadley MR, Butenko MA, Cano-Delgado AI, de Vries S, Dresselhaus T, Felix G, Graham NS, Foulkes J, Granier C, Greb T, Grossniklaus U, Hammond JP, Heidstra R, Hodgman C, Hothorn M, Inze D, Ostergaard L, Russinova E, Simon R, Skirycz A, Stahl Y, Zipfel C, De Smete I (2012) Tackling drought stress: receptor-like kinases present new approaches. Plant Cell 24:2262–2278
Tang S, Liang HY, Yan DH, Zhao Y, Han X, Carlson JE, Xia XL, Yin WL (2013) Populus euphratica: the transcriptomic response to drought stress. Plant Mol Biol 83:539–557
Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, Schein J, Sterck L, Aerts A, Bhalerao RR, Bhalerao RP, Blaudez D, Boerjan W, Brun A, Brunner A, Busov V, Campbell M, Carlson J, Chalot M, Chapman J, Chen GL, Cooper D, Coutinho PM, Couturier J, Covert S, Cronk Q, Cunningham R, Davis J, Degroeve S, Dejardin A, Depamphilis C, Detter J, Dirks B, Dubchak I, Duplessis S, Ehlting J, Ellis B, Gendler K, Goodstein D, Gribskov M, Grimwood J, Groover A, Gunter L, Hamberger B, Heinze B, Helariutta Y, Henrissat B, Holligan D, Holt R, Huang W, Islam-Faridi N, Jones S, Jones-Rhoades M, Jorgensen R, Joshi C, Kangasjarvi J, Karlsson J, Kelleher C, Kirkpatrick R, Kirst M, Kohler A, Kalluri U, Larimer F, Leebens-Mack J, Leple JC, Locascio P, Lou Y, Lucas S, Martin F, Montanini B, Napoli C, Nelson DR, Nelson C, Nieminen K, Nilsson O, Pereda V, Peter G, Philippe R, Pilate G, Poliakov A, Razumovskaya J, Richardson P, Rinaldi C, Ritland K, Rouze P, Ryaboy D, Schmutz J, Schrader J, Segerman B, Shin H, Siddiqui A, Sterky F, Terry A, Tsai CJ, Uberbacher E, Unneberg P, Vahala J, Wall K, Wessler S, Yang G, Yin T, Douglas C, Marra M, Sandberg G, Van de Peer Y, Rokhsar D (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604
Wang LF (2014) Physiological and molecular responses to drought stress in rubber tree (Hevea brasiliensis Muell. Arg.). Plant Physiol Biochem 83:243–249
Wang LK, Feng ZX, Wang X, Wang XW, Zhang XG (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26:136–138
Wang XL, Cai XF, Xu CX, Wang QH, Dai SJ (2016) Drought-responsive mechanisms in plant leaves revealed by proteomics. Int J Mol Sci 17:1706
Wu HL, Lv H, Li L, Liu J, Mu SH, Li XP, Gao J (2015) Genome-wide analysis of the AP2/ERF transcription factors family and the expression patterns of DREB genes in moso bamboo (Phyllostachys edulis). PLoS ONE 10:e0126657
Wu J, Chen JB, Wang LF, Wang SM (2017) Genome-wide investigation of WRKY transcription factors involved in terminal drought stress response in common bean. Front Plant Sci 8:380
Yan DH, Fenning T, Tang S, Xia XL, Yin WL (2012) Genome-wide transcriptional response of Populus euphratica to long-term drought stress. Plant Sci 195:24–35
Yang YC, Mo YL, Yang XZ, Zhang HF, Wang YQ, Li H, Wei CH, Zhang X (2016) Transcriptome profiling of watermelon root in response to short-term osmotic stress. PLoS ONE 11:e0166314
Ye J, Fang L, Zheng HK, Zhang Y, Chen J, Zhang ZJ, Wang J, Li ST, Li RQ, Bolund L, Wang J (2006) WEGO: a web tool for plotting GO annotations. Nucl Acids Res 34:W293–297
Yuan Y, Liu YJ, Wu C, Chen SQ, Wang ZY, Yang ZC, Qin SS, Huang LQ (2012) Water deficit affected flavonoid accumulation by regulating hormone metabolism in Scutellaria baicalensis Georgi roots. PLoS ONE 7:e42946
Zivcak M, Kalaji HM, Shao HB, Olsovska K, Brestic M (2014) Photosynthetic proton and electron transport in wheat leaves under prolonged moderate drought stress. J Photochem Photobiol B Biol 137:107–115
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Project funding: The work was supported by the National Natural Sciences Foundation of China (31860198), the Innovation Team Construction Plan Project of Xinjiang Production and Construction Group (2018CB003), and the Scientific and Technological Planning Project of Xinjiang Production and Construction Group (2012BB045).
The online version is available at http://www.springerlink.com
Corresponding editor: Tao Xu.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S2
List of differentially expressed genes for each group with FDR ≤ 0.001 and |log2 (fold change)|≥ 1 (XLSX 7254 kb)
Table S4
List of Kyoto Encyclopedia of Genes and Genomes enrichment terms for each group with Q-values ≤ 0.05 (XLSX 37 kb)
Table S6
Gene Ontology and Kyoto Encyclopedia of Genes and Genomes analysis for upregulated and downregulated genes in pair-wise comparasion for leaves and roots with Q-values ≤ 0.05, respectively (XLSX 49 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiao, P., Wu, Z., Wang, X. et al. Short-term transcriptomic responses of Populus euphratica roots and leaves to drought stress. J. For. Res. 32, 841–853 (2021). https://doi.org/10.1007/s11676-020-01123-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11676-020-01123-9