
Abstract
Thermodynamic assessment of the Pt-Pb binary system has been performed by combining first-principle calculations with the CALPHAD method. The formation enthalpies of the Pt3Pb and PtPb4 were calculated by using the projector-augmented-wave (PAW) method within the generalized gradient approximation (GGA). The CALPHAD assessment of the Pt-Pb system was then performed. The solution phases (liquid and fcc) were treated as substitutional solutions, the excess Gibbs energies of which were modeled using the Redlich-Kister polynomial. The three intermetallic compounds, Pt3Pb, PtPb, and PtPb4, were described as stoichiometric phases. Thermodynamic parameters of all the phases were optimized, which reproduces the most experimental data.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Platinum was shown to be the only active and stable pure metal catalyst for dissociative chemisorption of ethanol in acidic media.[1] However, Pt is prone to be poisoned by the intermediates such as carbon monoxide and thus becoming inactive for ethanol oxidation in the potential region of fuel cells of interest. Fortunately, many studies have shown that addition of other metals such as Sn,[2,3] Mo,[4] and Pb[5] into Pt can mitigate poisoning and increase the catalytic activity of Pt. In order to better understand the role of Pb in Pt-Pb catalysts, study of the Pt-Pb phase diagram is necessary.
The phase diagram of the Pt-Pb binary system has been established by Hansen and Anderko[6] based on the data reported by Doerinckel.[7] The thermodynamic properties of this system have been investigated.[8,9] Thermodynamic assessment of the Pt-Pb system has not been reported. This study calculates the formation enthalpies of the intermetallic compounds using the first principles based on the density functional theory (DFT) and then uses the data to evaluate the Pt-Pb binary phase diagram by means of the CALPHAD technique[10] so as to obtain a consistent and reliable description of the phase relations.
Evaluation of Experimental Data
Phase Diagram Data
The phase diagram of the Pt-Pb system reported by Hansen and Anderko[6] exhibits three intermediate compounds, Pt3Pb, PtPb, and PtPb4. These compounds decompose peritectially at 1188, 1068, and 633 K, respectively. Later, Amzil and Castanet[8] reported some liquidus data based on calorimetry. Subsequently, Lbibb et al.[9] calculated most of the liquidus values, and the results were in good agreement with the experimental phase diagram reported by Hansen and Anderko.[6] All the phase relations mentioned above are adopted in the present assessment.
Thermodynamic Data
Enthalpies of mixing of the liquid Pt-Pb alloys at 923, 1023, 1123, 1239, and 1316 K were measured using direct reaction calorimetry by Amzil and Castanet.[8] In light of these data, it was known that enthalpy of mixing of the liquid alloys is temperature dependent. This indicates the obvious tendency of formation of associates in the Pt-Pb liquid. So, the temperature dependence of mixing enthalpy was taken into account for the liquid.
Formation enthalpy of the PtPb compound was determined with reaction calorimetry at room temperature by Lbibb et al.[9] No experimental thermodynamic data were reported for Pt3Pb and PtPb4. First-principle calculations were utilized to determine the formation enthalpies of the compounds Pt3Pb and PtPb4.
First-Principle Calculations
The first-principle calculations were carried using the scalar relativistic all-electron Blöchl’s projector-augmented-wave (PAW)[11,12] method within the generalized gradient approximation (GGA), as implemented in the highly efficient Vienna ab initio simulation package (VASP).[13,14] The Perdew-Wang parameterization (PW91)[15,16] was employed in the GGA exchange-correlation function. For the two intermetallic compounds, i.e., Pt3Pb and PtPb4, the plane-wave energy cutoff was 400 eV. Brillouin zone integrations were performed using Monkhorst-Pack K-point meshes and reciprocal space (k-point) meshes are increased to achieve convergence to a precision of 0.3 kJ/mol of atom. The k-point meshes for Pt3Pb and PtPb4 were 15 × 15 × 15 and 11 × 11 × 11, respectively. The total energy was converged numerically to less than 1 × 10−6 eV/unit cell with respect to electronic, ionic, and unit cell degrees of freedom. The latter two relaxed using Hellmann-Feyman forces with a preconditioned conjugated gradient algorithm. After structural optimization, the total forces on each ion were less than 0.01 eV/Å. In order to avoid wrap-around errors, all the calculations are performed using the “high” setting within VASP.
The formation enthalpies for the intermetallic compounds were calculated by the following equation
where \( E_{\text{total}} ({\text{Pt}}{}_{x}{\text{Pb}}_{y} ), \) \( E_{\text{total}} ({\text{Pt}}), \) and E total (Pb) are the calculated total energies (per atom at T = 0 K) of the intermetallic compound, pure Pt, and pure Pb, respectively.
Thermodynamic Modeling
Solution Phases
A substitutional solution model based on random mixing of the constituent atoms is employed to describe liquid, fcc (Pt), and fcc (Pb). The molar Gibbs energy of a solution phase Φ (Φ = liquid, fcc (Pt), fcc (Pb)) can be represented as a sum of the weighted Gibbs energy for the pure components, the ideal entropy term describing a random mixing of the components, and the excess Gibbs energy describing the degree of deviation from ideal mixing, i.e.,
where \( G_{\text{m}}^{\Upphi } \) is the molar Gibbs energy of a solution phase Φ, \( {}^{0}G_{i}^{\Upphi } \) the Gibbs energy of pure element i (i = Pt, Pb) in the structural state of Φ, R the gas constant, and T the temperature. And the excess Gibbs energy, \( {}^{\text{E}}G_{\text{m}}^{\Upphi } , \) can be expressed by the Redlich-Kister polynomial functions as follows:
where x Pt and x Pb are the mole fractions of component Pb and Pt, respectively. \( {}^{(j)}L^{\Upphi } \) is the interaction parameter between Pt and Pb, which is formulated as
where A j , B j , and C j are model parameters to be optimized, and when j = 0, \( {}^{(j)}L^{\Upphi } \) means the nearest-neighbor interaction between atomic Pt and Pb. In order to model the temperature dependence of the mixing enthalpy of the liquid in this system, at least C 0 value should be of non-zero.
Intermetallic Compounds
The intermetallic compounds, Pt3Pb, PtPb, and PtPb4 are all treated as stoichiometric compounds since there are no experimental data to indicate any appreciable homogeneity range. Because of lack of heat capacity data of these compounds, the Neumann-Kopp rule is adopted to cope with the formation enthalpy of these compounds. Thus, the Gibbs energy per mole of molecules \( {\text{Pt}}_{m} {\text{Pb}}_{n} \) is calculated as
where A and B are the parameters to be optimized.
Results and Discussion
Crystal structures, calculated lattice constants, and formation enthalpies of the intermetallic compounds are listed in Table 1. The lattice constants of pure elements were previously calculated with first principles proposed by Wang et al.[17] It is clear that the results obtained in the present calculation are in good agreement with Wang et al.[17] The lattice constants of pure elements obtained from experiments[18,19] are also listed in Table 1. The calculated values agree reasonably well with the experimental data. In addition, the calculated lattice parameters of the two intermetallic compounds are in good agreement with the experimental data from literature.[20,21] In view of these, it is reasonable to accept the formation enthalpies of these compounds obtained by the first-principle calculations.
With the lattice stabilities for elements Pt and Pb adopted from Dinsdale,[22] thermodynamic parameters for all the phases occurring in the Pt-Pb binary system have been optimized using the PARROT module of the Thermo-Calc program developed by Sundman et al.,[23] and listed in Table 2.
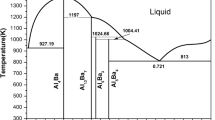
The phase diagram calculated in this study is shown in Fig. 1 and its comparison with experimental data is illustrated in Fig. 2, while Table 3 lists all the invariant reactions in Pt-Pb system. An agreement within 6 K between the calculated and the experimentally determined temperatures for these invariant reactions has been obtained.

The assessment phase diagram of the Pt-Pb system in this study

Comparison of the calculated phase boundaries with reported experimental data
Enthalpies of mixing of liquid Pt-Pb alloys at different temperatures are calculated in comparison with reported experimental data as shown in Fig. 3. We can see that not only most of the measured data but also the temperature dependence of mixing enthalpy of liquid alloys were reasonably reproduced.

Enthalpies of mixing of liquid Pt-Pb alloys: comparison between the assessed and experimental data. Reference states: liquid Pt and Pb
A further check on the reliability of the thermodynamic modeling is provided by Fig. 4, where the calculated enthalpies of formation of intermetallic phases at 298 K are compared with the experimental data and values by the first principles. It is demonstrated that the experimental and calculated data are well reproduced by the parameters obtained in this study.

Calculated enthalpies of formation of the compounds compared with experimental data in Ref 9 and values by the first principles. Reference states: fcc_A1 Pt and Pb
Conclusions
The phase diagram of the Pt-Pb binary system has been thermodynamically optimized based on experimental data and first-principle calculations of the phase equilibrium and thermodynamic properties. A set of self-consistent thermodynamic parameters have been obtained, which can reasonably reproduce the experimental and thermodynamic data.
References
C. Lamy, E.M. Belgsir, J.M. Leger, Electrocatalytic Oxidation of Aliphatic Alcohols: Application to the Direct Alcohol Fuel Cell (DAFC), J. Appl. Electrochem., 2001, 31, p 799–809.
A.O. Neto, E.G. Franco, E. Arico, M. Linardi, E.R. Gonzalez, Electro-oxidation of Methanol and Ethanol on Pt-Ru/C and Pt-Ru-Mo/C Electrocatalysts Prepared by Bönnemann’s Method, J. Eur. Ceram. Soc., 2003, 23, p 2987–2992.
W.J. Zhou, Z.H. Zhou, S.Q. Song, W.Z. Li, G.Q. Sun, Q.P. Tsiakaras, S. Xin, Pt Based Anode Catalysts for Direct Ethanol Fuel Cells, Appl. Catal. B, 2003, 46, p 273–285.
N. Fujiwara, K.A. Friedrich, U. Stimming, Ethanol Oxidation on PtRu Electrodes Studied by Differential Electrochemical Mass Spectrometry, J. Electroanal. Chem., 1999, 472, p 120–125.
G. Li, P.G. Pickup, The Promoting Effect of Pb on Carbon Supported Pt and Pt/Ru Catalysts for Electro-oxidation of Ethanol, Electrochim. Acta., 2006, 52, p 1033–1037.
[6] M. Hansen, K. Anderko, Constitution of Binary Alloys, 2nd Edition, McGraw-Hill, New York, 1958.
F. Doerinckel, Metallographische Mitteilungen aus dem Institut für anorganische Chemie der Universität Göttingen über einige Platinlegierungen, Z. Anorg. Chem., 1907, 54, p 358–365, in German.
A. Amzil, R. Castanet, Thermodynamic Investigation of the Pt-Pb Binary Alloys, Ber. Bunsengesel. Phys. Chem., 1992, 96, p 1872–1876.
R. Lbibb, R. Castanet, A. Rais, Thermodynamic Investigation of Pt-Pb Binary Alloys, J. Alloys Compd., 2000, 302, p 155–158.
[10] L. Kaufman, H. Bernstein, Computer Calculation of Phase Diagrams, Academic Press, New York, 1970.
P.E. Blöchl, Projector Augmented-wave Method, Phys. Rev. B, 1994, 50, p 17953–17979.
G. Kresse, J. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-wave Method, Phys. Rev. B, 1999, 59, p 1758–1775.
G. Kresse, J. Furthmuller, Efficient Iterative Schemes for Ab Initio Total-energy Calculations Using a Plane-wave Basis Set, Phys. Rev. B, 1996, 54, p 11169–11186.
G. Kresse, J. Furthmuller, Efficiency of Ab initio Total Energy Calculations for Metals and Semiconductors Using a Plane-wave Basis Set, Comput. Mater. Sci., 1996, 6, p 15–50.
J.P. Perdew, Y. Wang, Accurate and Simple Analytic Representation of the Electron-gas Correlation Energy, Phys. Rev. B, 1992, 45, p 13244–13249.
J.P. Perdew, J.A. Chevary, S.H. Vosko, K.A. Jackson, M.R. Pederson, D.J. Singh, et al., Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation, Phys. Rev. B, 1992, 46, p 6671–6687.
Y. Wang, S. Curtarolo, C. Jiang, R. Arroyave, T. Wang, G. Ceder, L.Q. Chen, Z.K. Liu, Ab Initio Lattice Stability in Comparison with CALPHAD Lattice Stability, CALPHAD, 2004, 28, p 79–90.
J. Häglund, A.F. Guillermet, G. Grimvall, M. Körling, Theory of Bonding in Transition-metal Carbides and Nitrides, Phys. Rev. B, 1993, 48, p 11685–11691.
H.K. Mao, Y.Wu, J.F. Shu, J.Z. Hu, R.J. Hemley, D.E. Cox, High-pressure Phase Transition and Equation of State of Lead to 238 GPa, Solid State Commun., 1990, 74, p 1027–1029.
M. Ellner, Relationship between Structural and Thermodynamic Properties in Phase of the Copper Family in T**1**0-B**4 Systems, J. Less-Common Metals, 1981, 78, p 21–32, in German.
U. Rösler, K. Schubert, Kristallstruktur von PtPb4, Naturwissenschaften., 1951, 38, p 331–331, in German.
A.T. Dinsdale, SGTE Data for Pure Elements, CALPHAD, 1991, 15, p 317–425.
B. Sundmann, B. Jansson, J.O. Anderson, The Thermo-Calc Databank System, CALPHAD, 1985, 9, p 153–190.
Acknowledgments
This study was financially supported by the National Science Foundation of China (No. 50671122) and Hunan Provincial Innovation Foundation for Postraduate (No. 1343-74236000007).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Long, Z.H., Tao, X.M., Liu, H.S. et al. First-Principle Calculation Assisted Thermodynamic Assessment of the Pt-Pb System. J. Phase Equilib. Diffus. 30, 318–322 (2009). https://doi.org/10.1007/s11669-009-9530-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11669-009-9530-1