
Abstract
Phosphate-based glasses (PBGs) are promising materials for biomedical applications due to their biocompatible and fully resorbable characteristics in aqueous environments. These glasses can be coated onto metal substrate via the technique of suspension high-velocity oxy-fuel (SHVOF) thermal spraying to produce nanostructured coatings with improved physical and mechanical properties. PBGs coatings were produced using SHVOF thermal spray process at 50 and 75 kW flame power. The 75 kW coating was rougher (Ra = 3.6 ± 0.1 µm) than the 50 kW coating (Ra = 2.7 ± 0.1 µm), whereas the 50 kW coating was much thicker (24.6 ± 2.3 µm) than the 75 kW coating (16.0 ± 3.4 µm). Due to the rougher surface, the 75 kW coating showed high degradation and ion release rates. Moreover, structural changes were observed by Raman analysis, and the initial glass formulation contained Q1 (phosphate tetrahedra with one-bridging oxygen) and Q2 (phosphate tetrahedra with two-bridging oxygen) species. However, the coatings showed a reduction of Q2 species and higher concentrations of Q1 and Q0 (phosphate tetrahedra with no-bridging oxygen) species, which led to lower degradation rates and reduced ion release profiles in the glass coating compared to the initial glass.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Phosphate-based glasses (PBG) are considered as a third-generation biomaterials, because these are fully resorbable in aqueous environments and these release ions (inorganic phosphate, Ca2+, Mg2+ and Na+) that are easily assimilated by the human body, which makes these glasses highly suitable for biomedical applications, such as degradable implant materials (Ref 1). In PBGs, P2O5 is the network former, and the orthophosphate (PO43−) tetrahedron represents the basic unit. These tetrahedrons bond through the bridging oxygen atoms, creating the P-O-P network that forms the glass structure. The number of bridging oxygen atoms linked to the adjacent tetrahedra is represented by Qn (where ‘n’ is the number of bridging oxygen per tetrahedra), and this parameter is used to describe the structure of PBGs (Ref 2). Na2O and CaO are usually present in PBGs as network modifiers to control their dissolution in aqueous media (Ref 3). Ions such as Ca (Ref 4), F (Ref 5), and Sr (Ref 6) were added to increase bone regeneration, and other ions such as Cu (Ref 7) and Ag (Ref 8) were used to improve the antibacterial properties. In addition, Ti (Ref 9) and Fe (Ref 10) were incorporated into PBGs to tailor their durability and control their degradation for use in specific end applications (Ref 11, 12).
The dissolution mechanism of PBGs in aqueous environments involves hydration, followed by hydrolysis of the PBGs chains (Ref 13). In the first step, hydration, the glass exchanges Na+ ions with H+ ions, resulting in a hydrated layer at the interface between the glass surface and media. The second stage involves the hydrolytic attack of the phosphate chains that are released into the solution (Ref 13). Bunker et al. proposed that the hydration is the dominating step in the glass dissolution process and involves two kinetic stages according to the dissolved profiles (q) in time (t) (Ref 14). In the first stage, the dissolution occurs linearly (q \(\propto\) t) until the saturation of the network, after which hydration of the glass occurs by diffusion following an exponential (q ∝ t1/2) degradation profile (Ref 14, 15). In addition, it was also reported that the degradation rate of PBGs is highly influenced by the pH of the media, since higher degradation would occur at lower pH (Ref 13,14,15,16).
The degradation of PBGs can easily be tailored and controlled by changing the glass compositions to balance the tissue growth; therefore, they have been proposed for different biomedical applications, using varying geometries such as glass fibers (Ref 16) or porous microspheres (Ref 17, 18). In addition, they could potentially be exploited as coatings on metallic implants to enhance bone tissue growth (Ref 2). Physical vapor deposition has been previously explored to produce thin-film PBGs coatings via radio-frequency magnetron sputtering (Ref 15). In the process of sputtering, the solid target is bombarded with ions or neutral particles, resulting in the removal of material from the target via atomization and its deposition onto a substrate to form a thin film. Stuart et al. have studied PBG coatings obtained on Ti6Al4V through magnetron sputtering, obtaining coatings with similar composition to the initial glass; however, the structure of these coatings suffered significant changes establishing that the glass structure of the coatings was highly depolymerized, and the coatings contained a higher percentage of Q2 (having two bridging oxygen) species (up to 45.2%) than the starting melt-quenched glass (which had Q2 species up to 31.0%) (Ref 15, 19). These structural differences between magnetron-sputtered PBG coatings and the initial melt-quenched glass represent a noticeable limitation of this technique.
The coatings had reduced phosphate content than the melt-quenched glass due to the lower sputtering rates of phosphate ions; at the same time, the network-modifying ions such as Ca, Na, and Mg deposited at higher rates. It was shown that the order of sputtering was P < Fe < Ca < Mg < Na due to the decreasing dissociation energy (Ref 20). Also, higher sputtering powers (>100 W) could cause cracking of the target (Ref 2). Moreover, the thickness of the PBGs coatings produced through radio-frequency magnetron sputtering is only few microns (2.5, 2.7 µm) (Ref 14, 18) and the process takes much longer to produce sufficient thickness coatings. For example, some metals were deposited at 1 µm/h or even slower (50 nm/h) if RF sputtering was used (Ref 21).
The emerging technique of suspension high-velocity oxy-fuel (SHVOF) thermal spray could represent an alternative to produce PBGs coatings. It has been used to deposit coatings of a wide variety of materials such as ceramics, bioceramics, bioactive glasses, metals and cermets (Ref 22). This technology allows for rapid deposition (usually only a few minutes) of micro- and nanometric particles in suspension form. A modified gas-fuelled high-velocity oxy-fuel (HVOF) torch is employed in SHVOF thermal spraying to process the liquid suspension. As the suspension is axially injected inside of the combustion chamber of the torch, complete mixing of the suspension droplets with the gas takes place. Depositing particles are heated by the gas in the combustion chamber and in the expansion nozzle. Upon expansion to ambient pressure, the gas attains supersonic velocity and projects the solid particles at high velocities toward the target. Extensive flattening of the particles occurs on the substrate resulting in dense microstructure with excellent cohesive and adhesive strength (Ref 23). With SHVOF, thin and thick coatings can be deposited, which can preserve the properties (chemical composition and phase) of the feedstock. The coatings deposited by SHVOF usually exhibit fine microstructure having excellent mechanical properties (Ref 22,23,24,25).
This work proposes the production of PBG coatings using SHVOF thermal spray to assess the potential development of resorbable coatings. The aim was to study the effect of the processing parameters (flame power) on the microstructure and chemical composition of the coatings. Moreover, the degradation and the ion release profiles of these coatings in phosphate buffer saline solution (PBS) and Milli-Q water were also investigated to establish how the degradation of the coatings was affected by the surface topography and the glass structure obtained with the different parameters.
Materials and Methods
Feedstock Material
A phosphate-based glass with nominal composition of 40 mol.% P2O5, 16 mol.% CaO, 24 mol.% MgO and 20 mol.% Na2O, later referred to as P-40 (Ref 11), was prepared using the following precursors: sodium dihydrogen phosphate (NaH2PO4), calcium hydrogen phosphate (CaHPO4), magnesium hydrogen phosphate trihydrate (MgHPO4 3H2O), and phosphorous pentoxide (P2O5) (Sigma-Aldrich, UK). Amounts of each precursor were weighed and mixed manually. After mixing these were transferred to a platinum-rhodium crucible (Birmingham Metal Company, Birmingham, UK) and the crucible was then placed in a furnace at 350 °C for 30 min to remove moisture. Then, the mixture was melted at the temperature of 1150 °C for 1.5 h. The molten glass was then quenched at room temperature using a steel slab. The size of the P-40 glass was reduced using a PM-100 ball mill (Retsch1-5, Germany) in a zirconia jar with 5 mm zirconia balls at 450 rpm for 20 min. The resultant powder had D10 = 1.5 μm, D50 = 12 μm and D90 = 48 μm and was again ball-milled with 2 mm zirconia beads at 350 rpm for 20 min to reduce the size of the particles further, to allow the preparation of the suspension that will be used in the spray process.
The powder suspension was prepared by adding 10 wt.% ball-milled P-40 glass powder in 90 wt.% water. The ball-milled powder was observed by scanning electron microscopy (SEM) (JEOL 6490, Tokyo, Japan) and energy-dispersive x-ray spectroscopy (EDX) (INCA 350, Oxford Instruments, Abingdon, UK). The size distribution of the P-40 glass particles in suspension was measured by laser diffraction method (Beckman Coulter, Inc., California, USA) using a 750 nm laser.
Suspension Thermal Spray Processing
304 stainless steel substrates were grit-blasted using F100 brown alumina with a size range of 0.125-0.149 mm. An ultrasonic bath was used to clean the substrates in industrial methylated spirit (IMS) for the removal of any embedded alumina particles. After cleaning, the substrates were mounted onto a carousel rotating at 73 rpm with a vertical axis of rotation. A modified Top Gun HVOF thermal spray torch (GTV gmbH, Germany) with axial injection was used to spray the suspension. The diameter of the suspension injector was 0.3 mm, while the combustion chamber was 22 mm long with 135 mm long barrel nozzle. The suspension was fed from a pressurized vessel at a flow rate of 50 ml/min. The nozzle was cleaned after each spray run with deionized water. The torch was mounted onto a traverse unit moving at a speed of 5 mm/s at a stand-off distance of 85 mm to the substrates on the carousel. Twenty traverse gun passes were used to build up the coating. Compressed air jets were used to provide cooling to substrates during and after spray.
P-40 suspension was sprayed by following the parameters given in Table 1, which were selected in order to obtain coatings with an appropriate thickness, porosity and adhesion to the substrate based on the deposition of a typical bioactive glass (45S5) coatings by SHVOF thermal spray investigated in a previous research (Ref 22). Theoretical flame energy (in kW) was calculated, and the coatings were labeled according to their theoretical flame power.
Coatings Characterization
An SEM (Jeol 6049) was used at 20 kV accelerating voltage to examine the surface morphology and cross section of the coatings. For cross section examination, the coatings were cut transversally with a SiC cutting disk using a precision cutting machine (Brilliant 220, ATM GmbH, Mammelzen, Germany). After cutting, the samples were mounted in Bakelite resin and then ground and polished to 1 µm surface finish. Before SEM examination, all samples were carbon-coated as these were non-conductive. EDX surface and line scan across the cross section were done using INCA software.
The surface roughness of the coatings was analyzed with a Talysurf Profilometer (Taylor Hobson Ltd., UK) with stylus radius of 2 µm. The sampling distance was kept 0.5 µm in the scan direction, while the stylus was moving with a speed of 0.25 mm/s. Porosity and thickness of the coatings were determined by ImageJ software (NIH, USA) with thresholding technique to highlight features of interest with five SEM secondary electron (SE) images from different regions of the cross section.
An x-ray diffractometer (D500, Siemens) with a Cu K-α radiation source in Bragg–Brentano θ-2θ configuration was used to determine any phase change in P-40 from powder to coatings. The scan was performed over a 2θ range of 20°-70° with step of 0.05° and 7 s dwell time. Raman analysis was conducted to investigate the structural connectivity of P-40 powder and coatings. A LabRAM HR spectrometer (Horiba–Jobin–Yvon) was used for Raman analysis, with a 532 nm laser with a 100X objective, 300 µm pinhole and diffraction grating of 600 lines/mm. All spectra were acquired for 20 accumulations of 30 seconds each.
Ion Release Profiles
The ion release profiles of the coatings were determined by immersing 9 mm disks coatings samples in 15 ml of Milli-Q water at 37 °C (Ref 11, 15). The samples were kept in the medium for 1, 3, 7 days, and the medium was refreshed at each time point. After each time point, the medium was analyzed for Ca, Mg, Na, P ions concentration using an Inductively Coupled Plasma Mass Spectrometry (ICP-MS, Thermo-Fisher Scientific iCAP-Q) coupled with a 2 × 250 mm Ion- Pac1 CS12A separator column.
Degradation Study
P-40 coated discs (diameter around 9 mm) were placed in 15 ml phosphate buffer saline solution (PBS) solution (pH ≈ 7.40) and Milli-Q water and incubated at 37 °C. The samples were left in the solution for 1, 3, 7 and 14 days. After each time point, the samples were removed from the suspension, dried in the oven for one hour at 50 °C to remove surface moistures and the mass loss of the samples was calculated using Equation 1.
where Mo is the mass of the starting sample and Mt is mass at a particular time ‘t’ during the study.
Then, the samples were immersed in PBS solution and Milli-Q water until the next time point. A microprocessor pH meter (Mettler Toledo, Schwerzenbach, Switzerland) was used for pH measurement of PBS, and pH meter was calibrated using standard pH buffer solutions of pH 4.0, pH 7.0, and pH 10.0 (Fisher Scientific, UK).
Results
Powder Characterization
The size distribution of the glass particles used for suspension preparation and measured by laser diffraction particle size analyzer is given in Fig. 1(a). The powder had D10 = 1.1 µm, D50 = 3.6 µm, D90 = 14 µm, with a wide distribution range from 2 µm to 27 µm. SEM micrograph in Fig. 1(b) also showed the angular morphologies of the particles and confirmed a mixture of fine and coarser particles.

Particle size distribution (a) and SEM (SE) image (b) of ball-milled P-40 glass
Morphology and Microstructure of the Coatings
The surface morphology of the P-40 coatings deposited at 50 kW and 75 kW is shown in Fig. 2. Both coatings surfaces were covered with large globules that were evenly distributed throughout the surface of the coatings (see Fig. 2a and d). On both coatings, the globules were the same diameter measuring approximately 20 µm. However, the morphology of the globules observed revealed significant differences depending on the flame power used for the coating deposition. The globules present on the 50 kW coating (Fig. 2b) revealed the presence of fine particles that provided a rough exterior to the globules. In contrast, the globules present on the 75 kW coating (Fig. 2e) presented an even skin. In the hump-free areas, the molten splats could clearly be observed in both cases, as can be seen in Fig. 2(c) and (f). The material had clearly melted in both cases so that the size of individual splats could not be estimated as the ends of each splat were joined to other splats. However, the finer partially molten particle can also be seen; these unmolten particles seem more abundant in the 50 kW than in the coating sprayed at 75 kW.

SEM (SE) images of the surface morphology of P-40 coatings (a), (b) and (c) at 50 kW, and (d), (e) and (f) at 75 kW, at increasing magnification
The cross-sectional images of the P-40 coatings are shown in Fig. 3. As can be seen, both coatings were well adhered, and no delamination was observed at the interface between coating and substrate. However, the thickness of these coatings was not uniform along the cross section. From Fig. 3(a) and (c), it can be seen that the coating deposited at 50 kW was 1.5 times thicker than the coating deposited 75 kW. However, the 75 kW coating was less porous (1.5 ± 0.4 %) in comparison to the 50 kW coating, which had a porosity of 2.9 ± 0.2 % (see Table 2). Both coatings were rough (Table 2), with the 75 kW coating being comparatively rougher (3.6 ± 0.1 µm) than the 50 kW coating (at 2.7 ± 0.1 µm).

SEM (SE) images of the cross sections of (a) and (b) for coating deposited at 50 kW, and (c) and (d) for coating deposited at 75 kW at different globules and globules-free regions of the coatings
To explore any compositional changes in P-40 glass after thermal spray, EDX scans were carried out along the cross section of the coatings and can be seen in Fig. 4, where elemental wt.% is presented as function of distance from the interface between coating and substrate to the top of the coatings (along with their corresponding BSE images).

BSE images for the cross section of P-40 phosphate glass coating deposited (a) at 50 kW and (c) at 75 kW, (b) and (d) the corresponding EDX line scan along the cross section of the coatings starting at the interface between coating and substrate to the top of the coating
In the P-40 coating deposited at 50 kW, the composition was approximately constant along the cross section, while going from the coating–substrate interface to the top of coating. However, slight variation was observed in comparison to the starting powder composition (Table 3). The P content was 26.4 wt.% (Table 3) in the starting powder, which had slightly reduced to 24.5 wt.% at the coating–substrate interface. However, the same starting value at 26.4 wt.% was observed at the top of the coating. Moreover, 12.5 wt.% Na was present in the powder before spraying, which had been reduced to approximately 5.5 wt.% at the coating–substrate interface and remained at this value through all the thickness of the coating. Ca content present in the powder was 6.2 wt.% and was around 9.2 wt.% at the interface and did not vary until the top of the coating. Mg wt.% was increased to 10 wt.% at the interface between coating and substrate from the 8.2 wt.% present in the starting powder and remained at this value to the top of the coating. Some Fe and Cr were observed near the coating–substrate interface, which originated from the metal substrate used.
For the 75 kW P-40 glass coating, the same trend as for the 50 kW coating was observed. The composition was nearly the same along the cross section of the coating. However, some compositional variations relative to the starting material were observed. For example, 22.6 wt.% of P was observed at the coating–substrate interface, which increased to approximately 25 wt.% at the top of the coating. Reduction in Na content was slightly more, which had reduced to 3.53 wt.% at the interface from the starting 12.5 wt.% (in the powder) and remained at this value till the top of the coating. 12 wt.% Ca was observed along the cross section of the coating. However, Ca content was less in the starting powder (6.2 wt.%) before spray. Similarly, Mg wt.% was observed to be increased to 11.74 wt.% along the coating cross section till the top of the coating from 8.2 wt.% which was present in the powder.
Moreover, it can be seen (Table 3) that post-thermal spraying the P2O5 content of the starting glass powder decreased significantly from 36 mol.% to 29 mol.% (for the 50 kW coating) and 27 mol.% (for the 75 kW coating) as analyzed via EDX at the top surfaces of the coatings.
The amorphous nature of the P-40 glass did not seem to have changed and remained amorphous after thermal spraying at both flame powers, as shown by the XRD analysis in Fig. 5. The broad diffraction halos centered at 2θ ≈ 30° were identified in the starting powder and in both coatings produced. The only crystalline peaks visible were from the substrate confirmed to be austenite (PDF card no. 00-023-0298) and ferrite (PDF card no. 00-006-0696).

XRD diffractograms of P-40 glass powders and coatings deposited at 50 and 75 kW flame power
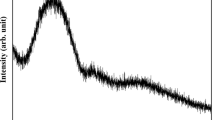
Raman spectroscopy analysis of the starting P-40 powder and the produced coatings are given in Fig. 6. For the P-40 powder, the peak at 351 cm−1 was related to bending vibrations of the O-P-O bonds associated with Q2 tetrahedral units (Ref 26). The low intensity peaks in the range of 470-620 cm−1 were suggested to be due to in-chain O-P-O stretching vibrations (Ref 26,27,28). The Raman peaks at 670-790 cm−1 were associated with the P-O-P symmetric stretching of the bridging oxygen in Q2 and Q1 units, respectively (Ref 29). In the previous study, it has been shown that P-40 formulation contained 50% Q2 and Q1 units (Ref 30). The peak at 1040 cm−1 was related to symmetric stretching of PO2 non-bridging oxygen in Q1 unit (Ref 30), whereas the peak at 1160 cm−1 was associated to the symmetric stretching modes O-P-O non-bridging oxygen in Q2 phosphate units (Ref 29).

Raman spectra of P-40 phosphate glass powder and coatings deposited at 50 and 75 kW flame power
Post-thermal spraying, analysis of the P-40 glass coating at flame power 50 and 75 kW revealed that the peak at 670 cm−1 (which had been assigned to P-O-P symmetric stretching of the bridging oxygen in Q2 units) had disappeared, while the peak at 790 cm−1 had appeared for both coatings. A new peak was also observed at 960 cm−1 for both the 50 and 75 kW coatings which was associated with the symmetric stretching of orthophosphate group (PO4−3) in Q0 unit (Ref 2, 28). The intensity of the band at 1046 cm−1 (associated with Q1 unit) also increased for both coatings, while the peak intensity at 1160 cm−1 (associated with Q2 unit) decreased for the 50 kW coating and appeared as a shoulder in the Raman spectrum of the 75 kW coating (see Fig. 6).
Ion Release Profiles
Ion release profiles for the coatings were investigated using ICP-MS for the measurement of Na+, Mg2+, Ca2+, and total P ions in Milli-Q water. The cumulative ion release profiles of both coatings are presented in Fig. 7. The ion release rates were calculated as the slope from the linear interpolation of the values and are given in Table 4.

Na+, Mg2+, P5+, and Ca2+ ion release profile for P-40 coatings (a) deposited at 50 kW and (b) 75 kW
For the 50 kW coating, the highest ion release was that of total P ions with the release rate of 1.4 ± 0.1 ppm/day, while the other ions such as Na+, Ca2+ and Mg2+ were released in similar amounts with the release rate of 0.38 ± 0.04 ppm/day for Na+ and a rate of 0.49 ± 0.06 ppm/day for Ca2+ and Mg2+ (while R2 values for ions of P, Ca, Na and Mg were 0.95, 0.98, 0.99 and 0.97). Similar to the 50 kW, the 75 kW coating also revealed total P ion released at the highest rate (1.7 ± 0.1 ppm/day) in comparison to other ions analyzed (Na+, Ca2+ and Mg2+) which were released at 0.57 ± 0.02 ppm/day for Mg2+ and the same rate of 0.59 ± 0.08 ppm/day and 0.59 ± 0.02 ppm/day for Na+ and Ca2+, respectively (while R2 values for ions of P, Ca, Na and Mg were 0.982, 0.996, 0.950 and 0.995). It can also be seen that the overall ion release rates were higher for 75 kW coating than for the 50 kW coating.
Degradation Studies
Degradation profiles of the P-40 coatings deposited at 50 and 75 kW are shown in Fig. 8. The degradation rates revealed linear mass loss profiles for both coatings in Milli-Q water and PBS.

(a) Degradation of P-40 coating deposited at 50 kW and (b) 75 kW in PBS and MQ water
Degradation rate for the coatings was higher in MQ water compared to those in the PBS (see Fig. 8 and Table 5). The highest degradation rate of 1 × 10−4 mg/mm2·day was observed for the 75 kW coating in MQ water. The lowest degradation rate (1×10−5 mg/mm2·day) was observed for 50 kW in PBS, while this coating was degraded 5 times higher in Milli-Q water at the rate of 5×10−5 mg/mm2·day.
Discussion
Microstructure Formation in P-40 Coatings
SHVOF thermal spraying at 50 kW and at 75 kW flame power resulted in globules on the surface of the coatings (see Fig. 2). In our previous work, it was observed that spraying of the silicate based bioactive glass (45S5) at 75 kW also resulted in globules on the surface of the coating (Ref 22), which were attributed to the formation of large agglomerates inside the combustion chamber during spray and subsequent deposition onto the substrate. A flame power of 75 kW had sufficient energy to melt these large agglomerates, resulting in homogeneous globules with a smooth surface (Ref 31). However, P-40 glass coating at 50 kW had the same globules on the surface of the coating, which means that the 50 kW flame power had enough heat energy for this glass to generate a similar surface topography to the 45S5 coating deposited at 75 kW coating. This could be due to the low glass transition (448 ± 1 °C) and melting (764 ± 1 °C) temperature of the P-40 glass (Ref 11) in comparison to 45S5 bioactive glass for which glass transition temperature is 520 °C (Ref 32) and melting temperature is 1217 °C (Ref 33). In globule-free areas, individual splats were not seen in both coatings, as a result of better melting of the material at both flame powers (50 and 75 kW) due to the lower melting temperature of PBG.
Both P-40 glass coatings were dense and presented low porosity (Fig. 2), and this was attributed to the higher heat transfer at both flame powers, causing better melting of the P-40 particles and leading to denser coatings in comparison to the Bioglass® coatings studied in our previous work (Ref 22, 25). However, the 75 kW coating was two thirds the thickness of the 50 kW coating which may have been due to the fact that the high flame power of 75 kW caused the material to evaporate (due to lower melting temperature of the PBGs (Ref 25), resulting in a thinner coating (Ref 22, 34).
EDX analysis along the cross section of the coatings (Fig. 4) showed that the P-40 composition with respect to the initial bulk formulation had been slightly altered due to the thermal spraying process, thereby imparting a new thermal prehistory on the samples produced. These variations in composition were observed from EDX area scan at the top surface of the coatings. As in Table 3, it can be seen that thermal spray caused reductions in contents of P and Na while spraying at 50 and 75 kW. This might be due to the direct volatilization of P2O5 at high temperature (Ref 34). Reduction in the wt.% of Na2O is attributed to its volatile nature (Ref 35). Loss of Na2O from the glass melt is due to the heterogeneous chemical reaction between Na2O and water vapors present in the combustion chamber resulting in NaOH (Ref 36). A decrease in the content of Na2O was followed by an increase in CaO content (Ref 36).
The structure of the initial P-40 glass was altered due to the thermal spray process as shown by the Raman analysis (Fig. 6), which showed that the Raman spectra of the coatings were different from the Raman spectrum of the P-40 powder. A new peak was found at 960 cm−1 in the Raman spectra of coatings which was not present in the starting powder spectrum. In the literature, this band was associated with the symmetric stretching of orthophosphate groups (PO4−3) (Ref 28, 29, 37), which suggested that the P-40 glass had depolymerized forming Qo units (Ref 26). Higher intensity of the peak at 1046 cm−1 in coatings spectra compared to P40 powder indicated a higher concentration of Q1 units (Ref 26). Moreover, the intensity of the peak at 1160 cm−1 decreased in the spectra of the coatings, and the decreased intensity of this peak in the coating spectra could be due to the reduction in the concentration of Q2 units (Ref 26, 29). Finally, the evolution of the peak at 670-790 cm−1 as at 750 cm−1 indicated a reduction in the chain length. It can be concluded from the Raman spectra that the P-40 glass structure changed as a reduction in the concentration of Q2 units, increased in Q1 units and the potential formation of Qo units. In Raman analysis, the observation of orthophosphate (PO4−3) peak/ Q0 species in both coatings could be attributed to the reduction of P2O5 content (from 36 mol% for glass powder to 27-29 mol.% for coatings) post-thermal spray which is confirmed via EDX. In literature, it has been reported that the Q0 species was observed for the phosphate glasses, which contained P2O5 below 35 mol.% (Ref 38).
This structural analysis warrants further investigation, via NMR characterization. However, the challenges are ensuring that sufficient powder samples can be obtained from the coatings deposited for accurate analysis.
Ion Release Profiles and Degradation Studies
From the ion release studies conducted (Table 4), it can be seen that, for both coatings, ion release rates ranked P > Na > Mg > Ca. P-40 coatings suffered a structural change during spraying and present a higher ratio of Q1 and Q0 species than the initial glass, as observed from Raman analysis, and these changes in the glass structure had an effect on the glass solubility. In PBGs, Q2 species are more soluble than species with a lower degree of polymerization (i.e., Q1 and Q0), so the P-40 coatings with a higher ratio of depolymerized species (Q1 and Q0 species) will present reduced ion release profiles than the initial glass. Ion release study of P-40 bulk glass has been conducted by Islam et al. (Ref 11), who reported ion release rates of 0.8 ppm/day for Mg, 0.9 ppm/day for Ca and 1.55 ppm/day for Na ions, and these values were significantly higher than the ones obtained for the thermal sprayed P-40 coatings of this study, indicating that the produced coatings were more resistant to hydrolytic attack, due to the structural changes induced in the glass during spraying, as proved by Raman analysis (Fig. 6).
The ion release rates were also influenced by the spray conditions, and the intensity of the bands associated with Q1 and Q0 species was very similar for both spray conditions. However, a higher ratio of depolymerized species was present in the coatings deposited at 75 kW, whereas a band associated with Q2 units (1160 cm−1) was absent in the 75 kW coating while was present in the 50 kW coating (see Fig. 6). Therefore, according to the glass chemistry, higher dissolution rates should be expected on the 50 kW coating. However, the coating deposited at 75 kW presented higher release rates than the coating deposited at 50 kW. This could be due to the rougher surface of the 75 kW coating, which involves a larger surface area in contact with the liquid, contributing to an increased ion leaching in comparison to the 50 kW coating (Ref 25). This could indicate that small changes in the glass structure of the coating had a limited effect on the ion release rates.
The degradation rates of the coatings followed the same behavior as the ion release profiles. In Milli-Q water and PBS, both coatings were more stable and presented lower degradation rates than the PBGs thin films (produced via magnetron sputtering) reported in the literature (Ref 39), probably due to the presence of a higher rate of depolymerized species. Also, the enrichment of MgO in the coating composition from the initial glass, which is due to the volatilization of labile elements during spraying, has an important role because PBGs with higher MgO content tend to present lower degradation rates (Ref 11). The P-40 coating deposited at 75 kW showed higher degradation than the coating deposited at 50 kW (Fig. 8, Table 5), and this could be attributed to the higher surface roughness of the 75 kW coating.
Overall, the PBG thermal sprayed coatings showed lower degradation and ion release rates in comparison to the starting glass powder and to those reported in the literature for bulk PBG and PBG thin films. This behavior of the PBG thermal sprayed coatings could be attributed to the change in the structure of the glass as revealed via Raman analysis (although further analysis is still required).
Conclusion
The SHVOF thermal spray technique was used to deposit P-40 PBG on to stainless steel substrate at two flame powers of 50 and 75 kW. Surface topography of both coatings was very similar, showing globules on the top surface of the coatings. However, ion release profiles and degradation rates of both coatings were different, which might be due to the difference surface roughness of the coatings. Both coatings were amorphous as shown by XRD analysis; however, the structure of the coatings was different from that of the starting powder as revealed by Raman analysis with the reduction in the concentration of Q2 specie, increased of Q1 specie and the formation of Q0 specie in coatings.
75 kW coating showed more mass loss and ion leaching in comparison to 50 kW coating, which could be due to the rougher surface of the 75 kW coating. However, comparing these results with those reported in the literature for thin films of P-40 glass and bulk P-40 glass, both coatings showed lower degradation and ions release rates.
References
R.A. Velapoldi, L. Boehm and M.I. Shalom, Phosphate Glasses, 1971, 76, p 1–4.
I. Ahmed, H. Ren and J. Booth, Developing Unique Geometries of Phosphate-Based Glasses and their Prospective Biomedical Applications, Johnson Matthey Technol. Rev., 2018, 63(1), p 34–42. https://doi.org/10.1595/205651319x15426460058863
I. Ahmed, M. Lewis, I. Olsen and J.C. Knowles, Phosphate Glasses for Tissue Engineering: Part 1. Processing and Characterisation of a Ternary-Based P2O5-CaO-Na 2O Glass System, Biomaterials, 2004, 25(3), p 491–499. https://doi.org/10.1016/S0142-9612(03)00546-5
E.A. Abou Neel et al., Doping of a High Calcium Oxide Metaphosphate Glass with Titanium Dioxide, J. Non. Cryst. Solids, 2009, 355(16–17), p 991–1000. https://doi.org/10.1016/j.jnoncrysol.2009.04.016
D.S. Brauer, M.N. Anjum, M. Mneimne, R.M. Wilson, H. Doweidar and R.G. Hill, Fluoride-Containing Bioactive Glass-Ceramics, J. Non. Cryst. Solids, 2012, 358(12–13), p 1438–1442. https://doi.org/10.1016/j.jnoncrysol.2012.03.014
E.A. Abou Neel et al., Structure and Properties of Strontium-Doped Phosphate-Based Glasses, J. R. Soc. Interface, 2009, 6(34), p 435–446. https://doi.org/10.1098/rsif.2008.0348
J.C.K. Ensanya Ali Abou Neel and W. Chrzanowski, Effect of Increasing Titanium Dioxide Content on Bulk and Surface Properties of Phosphate-Based Glasses, Acta Biomater., 2008, 4(3), p 523–534. https://doi.org/10.1016/j.biomaterials.2004.07.024
I. Ahmed et al., The Structure and Properties of Silver-Doped Phosphate-Based Glasses, J. Mater. Sci., 2007, 42(23), p 9827–9835. https://doi.org/10.1007/s10853-007-2008-9
E.A. Abou Neel, W. Chrzanowski and J.C. Knowles, Effect of Increasing Titanium Dioxide Content on Bulk and Surface Properties of Phosphate-Based Glasses, Acta Biomater., 2008, 4(3), p 523–534. https://doi.org/10.1016/j.actbio.2007.11.007
I. Ahmed, C.A. Collins, M.P. Lewis, I. Olsen and J.C. Knowles, Processing, Characterisation and Biocompatibility of Iron-Phosphate Glass Fibres for Tissue Engineering, Biomaterials, 2004, 25(16), p 3223–3232. https://doi.org/10.1016/j.biomaterials.2003.10.013
M.T. Islam, K.M.Z. Hossain, N. Sharmin, A.J. Parsons and I. Ahmed, Effect of Magnesium Content on Bioactivity of Near Invert Phosphate-Based Glasses, Int. J. Appl. Glas. Sci., 2017, 8(4), p 391–402. https://doi.org/10.1111/ijag.12320
M.T. Islam, R.M. Felfel, E.A. Abou Neel, D.M. Grant, I. Ahmed and K.M.Z. Hossain, Bioactive Calcium Phosphate–Based Glasses and Ceramics and Their Biomedical Applications: A Review, J. Tissue Eng., 2017 https://doi.org/10.1177/2041731417719170
H. Gao, T. Tan and D. Wang, Dissolution Mechanism and Release Kinetics of Phosphate Controlled Release Glasses in Aqueous Medium, J. Control. Release, 2004, 96(1), p 29–36. https://doi.org/10.1016/j.jconrel.2003.12.031
B.C. Bunker, G.W. Arnold and J.A. Wilder, Phosphate Glass Dissolution in Aqueous Solutionsitle, J. Non. Cryst. Solids, 2021, 64(3), p 291–316.
B.W. Stuart, M. Gimeno-Fabra, J. Segal, I. Ahmed and D.M. Grant, Degradation and Characterization of Resorbable Phosphate-Based Glass Thin-Film Coatings Applied by Radio-Frequency Magnetron Sputtering, ACS Appl. Mater. Interfaces, 2015, 7(49), p 27362–27372. https://doi.org/10.1021/acsami.5b08957
E.A. Abou Neel et al., Effect of Iron on the Surface, Degradation and Ion Release Properties of Phosphate-Based Glass Fibres, Acta Biomater., 2005, 1(5), p 553–563. https://doi.org/10.1016/j.actbio.2005.05.001
M.T. Islam, L. Macri-Pellizzeri, K.M.Z. Hossain, V. Sottile and I. Ahmed, “Effect of Varying the Mg with Ca Content in Highly Porous Phosphate-Based Glass Microspheres, Mater. Sci. Eng. C, 2021, 120, p 111668. https://doi.org/10.1016/j.msec.2020.111668
K.M.Z. Hossain et al., Porous Calcium Phosphate Glass Microspheres for Orthobiologic Applications, Acta Biomater., 2018, 72, p 396–406. https://doi.org/10.1016/j.actbio.2018.03.040
B.W. Stuart, J.J. Titman, M. Gimeno-Fabra, I. Ahmed and D.M. Grant, Insights into Structural Characterisation and Thermal Properties of Compositionally Equivalent Vapour-Condensed and Melt-Quenched Glasses, Mater. Des., 2016, 111, p 174–184. https://doi.org/10.1016/j.matdes.2016.08.063
B. Stuart, M. Gimeno-Fabra, J. Segal, I. Ahmed and D.M. Grant, Preferential Sputtering in Phosphate Glass Systems for the Processing of Bioactive Coatings, Thin Solid Films, 2015, 589, p 534–542. https://doi.org/10.1016/j.tsf.2015.05.072
P.J. Kelly and R.D. Arnell, Magnetron Sputtering: A Review of Recent Developments and Applications, Vacuum, 2000, 56(3), p 159–172. https://doi.org/10.1016/S0042-207X(99)00189-X
S. Bano, I. Ahmed, D.M. Grant, A. Nommeots-Nomm and T. Hussain, Effect of processing on microstructure, mechanical properties and dissolution behaviour in SBF of Bioglass (45S5) coatings deposited by Suspension High Velocity Oxy Fuel (SHVOF) thermal spray, Surf. Coat. Technol., 2019, 372(May), p 229–238. https://doi.org/10.1016/j.surfcoat.2019.05.038
G. Bolelli et al., Comparison between Suspension Plasma Sprayed and High Velocity Suspension Flame Sprayed bioactive coatings, Surf. Coat. Technol., 2015, 280, p 232–249. https://doi.org/10.1016/j.surfcoat.2015.08.039
J.W. Murray, A.S.M. Ang, Z. Pala, E.C. Shaw and T. Hussain, Suspension High Velocity Oxy-Fuel (SHVOF)-Sprayed Alumina Coatings: Microstructure, Nanoindentation and Wear, J. Therm. Spray Technol., 2016, 25(8), p 1700–1710. https://doi.org/10.1007/s11666-016-0462-0
M. Bai, R. Khammas, L. Guan, J.W. Murray and T. Hussain, Suspension High Velocity Oxy-Fuel Spraying of a Rutile TiO2 Feedstock: Microstructure, Phase Evolution and Photocatalytic Behaviour, Ceram. Int., 2017, 43(17), p 15288–15295. https://doi.org/10.1016/j.ceramint.2017.08.068
B.W. Stuart, C.A. Grant, G.E. Stan, A.C. Popa, J.J. Titman and D.M. Grant, Gallium Incorporation into Phosphate Based Glasses: Bulk and Thin Film Properties, J. Mech. Behav. Biomed. Mater., 2018, 82(March), p 371–382. https://doi.org/10.1016/j.jmbbm.2018.03.041
J.E. Pemberton, L. Latifzadeh, J.P. Fletcher and S.H. Risbud, Raman Spectroscopy of Calcium Phosphate Glasses with Varying CaO Modifier Concentrations, Chem. Mater., 1991, 3(1), p 195–200. https://doi.org/10.1021/cm00013a039
A.K. Yadav and P. Singh, A Review of the Structures of Oxide Glasses by Raman Spectroscopy, RSC Adv., 2015, 5(83), p 67583–67609. https://doi.org/10.1039/c5ra13043c
S. Kaoua, S. Krimi, A. El Jazouli, E.K. Hlil and D. de Waal, Preparation and Characterization of Phosphate Glasses Containing Titanium and Vanadium, J. Alloys Compd., 2007, 429(1–2), p 276–279. https://doi.org/10.1016/j.jallcom.2006.04.004
M.T. Islam et al., The Effect of MgO/TiO2 on Structural and Crystallization Behavior of Near Invert Phosphate-Based Glasses. J, Biomed. Mater. Res. Part B Appl. Biomater., 2019, 3, p 1–13. https://doi.org/10.1002/jbm.b.34421
G. Bolelli, V. Cannillo, R. Gadow, A. Killinger, L. Lusvarghi and J. Rauch, Investigation of High-Velocity Suspension Flame Sprayed (HVSFS) , Ceram. Silikaty, 2010, 54(1), p 1–7. https://doi.org/10.1016/j.jeurceramsoc.2009.01.032
D. Groh, F. Döhler and D.S. Brauer, Bioactive Glasses with Improved Processing. Part 1. Thermal Properties, Ion Release and Apatite Formation, Acta Biomater., 2014, 10(10), p 4465–4473. https://doi.org/10.1016/j.actbio.2014.05.019
B. Pföss, M. Höner, M. Wirth, A. Bührig-Polaczek, H. Fischer and R. Conradt, Structuring of Bioactive Glass Surfaces at the Micrometer Scale by Direct Casting Intended to Influence Cell Response, Biomed. Glas., 2016, 2(1), p 63–71. https://doi.org/10.1515/bglass-2016-0008
R.G.C. Beerkens, Modeling the Kinetics of Volatilization from Glass Melts, J. Am. Ceram. Soc., 2001, 84(9), p 1952–1960. https://doi.org/10.1111/j.1151-2916.2001.tb00942.x
D. M. Sanders, W. K. Haller, Effect of Water Vapor on Sodium Vaporization from Two Silica-Based Glasses, Am. Soc. Ceram., 1977, 60(3-4), p 138–141. https://doi.org/10.1111/j.1151-2916.1977.tb15489.x
F. Pigeonneau, H. Kočárková and F. Rouyer, Stability of Vertical Films of Molten Glass Due to Evaporation, Colloids Surf. A Physicochem. Eng. Asp., 2012, 408, p 8–16. https://doi.org/10.1016/j.colsurfa.2012.04.014
A. Kiani et al., Structural Characterization and Physical Properties of P2O5-CaO-Na2O-TiO2 Glasses by Fourier Transform Infrared, Raman and Solid-State Magic Angle Spinning Nuclear Magnetic Resonance Spectroscopies, Acta Biomater., 2012, 8(1), p 333–340. https://doi.org/10.1016/j.actbio.2011.08.025
T. Moeller, Phosphorus and its Compounds. Volume I: Chemistry. J. Am. Chem. Soc., 1959, 81(11), p 2916. https://doi.org/10.1021/ja01520a086
B.W. Stuart, M. Gimeno-Fabra, J. Segal, I. Ahmed and D.M. Grant, Mechanical, Structural and Dissolution Properties of Heat Treated Thin-Film Phosphate Based Glasses, Appl. Surf. Sci., 2017, 416, p 605–617. https://doi.org/10.1016/j.apsusc.2017.04.110
Acknowledgments
This work was supported by the Engineering and Physical Sciences Research Council [EP/N50970X/1). Saira Bano acknowledges a fully funded Faculty of Engineering Research Excellence PhD scholarship for international students by University of Nottingham, UK (Sponsor License Number RFDX42DB2, Scholarship Ref Number: 16453). Saira Bano is a lecturer at the University of Engineering and Technology Peshawar, Pakistan. We acknowledge support from Mr. John Kirk for thermal spray experiments. We gratefully acknowledge technical support from Scott Young in ion leaching tests, as well as the Nanoscale and Microscale Research Centre (NMRC) at University of Nottingham for access to the SEM and Raman spectroscopy facilities.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bano, S., Rincon Romero, A., Islam, M.T. et al. Development and Characterization of Phosphate-Based Glass Coatings via Suspension High-Velocity Oxy-Fuel (SHVOF) Thermal Spray Process. J Therm Spray Tech 30, 1862–1874 (2021). https://doi.org/10.1007/s11666-021-01233-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11666-021-01233-8