
Abstract
Gallium antimonide (GaSb) is a III-V semiconductor of technological interest for low-power, high-mobility field-effect transistors, as well as for mid-wave infrared detectors. In such devices, GaSb interfaces with other III–V semiconductors with different lattice constants that can induce strain in the GaSb layers. Two dominant limiting factors in hot carrier relaxation are the intra-valley and the inter-valley electron–phonon (e-ph) scattering. In GaSb, these are sensitive to the \(\Gamma \)–L energy ordering, which depend intimately on the strain. Here, we report ab initio calculations of electronic structure, phonon dispersion, e-ph scattering and relaxation times for GaSb as a function of strain. As observed previously for other group IV and III-V semiconductors, our results show strong anisotropy, a strong contribution from LO phonons, and the need to go beyond the deformation potential scattering. For GaSb, the main finding is that a compressive strain between 0.4% and 0.6% converts GaSb from a direct-bandgap semiconductor to an indirect-bandgap semiconductor, with dramatic changes in the competing scattering rates and carrier relaxation times.
Similar content being viewed by others

Introduction
Gallium antimonide (GaSb) is a material with a wide range of applications including infrared \(1.2-3.0\) \(\mu \)m lasers,1 infrared detectors,2,3 photodiodes,4 and field-effect transistors with reduced size and power.5,6,7 It is also used as a substrate material with a lattice parameter that matches various ternary and quaternary compounds with a wide spectral range of bandgap.8 The unstrained GaSb has a small direct bandgap of 0.81 eV, accompanied by a secondary minima only 80 meV higher in energy. This allows GaSb to be converted into an indirect-bandgap semiconductor by applying lattice strain. Strain is ubiquitous in GaSb-containing heterostructures inside electronic and electro-optic devices, where materials typically have significant lattice mismatches at interfaces.
To understand hot carrier dynamics in this material, a detailed picture of the prominent scattering rates is necessary. The main contributions are electron–phonon (e-ph) scattering and dislocation scattering. To date, very little is known quantitatively about the e-ph scattering in GaSb, tracing back to early photoluminescence measurements9 which showed competition between \(\Gamma \)–\(\Gamma \) intra-valley scattering and L–\(\Gamma \) inter-valley scattering. Raman scattering has been used to determine strain in superlattices such as GaSb/AlSb, and it was found that the GaSb longitudinal optical (LO) frequency was lower by 2 cm\(^{-1}\) than bulk GaSb.10
In the case of highly lattice-mismatched III-V interfaces, the main carrier scattering mechanism has been associated with dislocations. For example, the effect of strain on carrier lifetime was measured in InAs/GaSb heterostructures with lattice mismatch of the order of 0.6%.11 This is qualitatively consistent with measurements in other strained heterostructures, such as GaAs\(_{1-x}\)Sb\(_{x}\) lattice matched to InP in high-speed electron transport in heterostructure based bipolar transistor or photodiodes.12 Other GaSb heterostructures, such as GaSb/GaAs\(_{1-x}\)Sb\(_{x}\), were grown and electrically characterized for optoelectronic applications by allowing relaxation of strain.13 Ab initio calculations have used molecular dynamics to study reconstruction of the surface when InSb is grown on GaSb,14 which have shown that by creating a single monolayer of InSb on GaSb, the large buckling between Ga and Sb is reduced and their atomic positions become similar to the second layer in the clean GaSb surface, whereas adding a second monolayer of InSb results in a disordered surface. A study of molecular beam epitaxy (MBE)-grown GaSb on GaAs revealed that a \(8\%\) lattice mismatch between the two materials resulted in a relaxed GaSb layer attributed to the existence of dislocations.15
Since strain influences mainly the e-ph scattering rates, here we focus on a comprehensive description of e-ph scattering in terms of all phonon modes as a function of strain. We follow the methodology used for AlAs, GaAs, diamond, Si and Ge.16,17,18,19 We apply the methodology16,17 to GaSb that includes the effects of strain on electronic structure, phonon dispersions and e-ph scattering rates which determines the carrier relaxation times. Recent work on first-principles prediction of mobility in many elemental and III-V semiconductors presented the scope of such calculations and a systematic comparison between theory and experimental results.20 This work demonstrated the capabilities and the effect of different approximations on the accuracy of mobility calculations wherein the e-ph scattering rates are also evaluated using the same framework that is used in our work. Although this work is a compendium of many III-V semiconductors, they have not discussed GaSb or the effects of strain. Hydrostatic strain ranging below \(1\%\) is artificially created by compressing or expanding the lattice constant of GaSb. In GaSb, the conduction band minimum (CBM) lies at \(\Gamma \) and the next minimum is at the L-point, which is at energy of 0.08 eV. On applying hydrostatic pressure, the relative positions of the minimum can change, and GaSb is found to convert to an indirect-bandgap semiconductor. This will impact the e-ph coupling as well as the resulting scattering rates. We discuss the significant features and observations in the following sections.
Methodology
The work reported here was performed using Quantum ESPRESSO (QE)21 with norm-conserving pseudopotentials within the local density approximation (LDA)22 for the exchange–correlation potentials defined by Hartwigsen–Goedekar–Hutter.23 Lattice dynamics were computed using density functional perturbation theory.24 The optimized lattice constant was found to be 5.98 Å, which is \(2\%\) lower than the experimental lattice constant. The energy cutoff for the plane-wave basis was optimized and chosen as 60 Ryd after verifying convergence. A uniform k-mesh of \(16\times 16\times 16\) was used to sample the reciprocal space for self-consistent calculation, and a q-grid of \(8\times 8\times 8\) was used for the calculations of phonons. Wannierization was performed starting with Ga-\(sp^3\) and Sb-p orbitals using the Wannier90 package.25 Convergence of the Wannierization process was constrained by choosing a cutoff for the spread at \(10^{-12}\) Å\(^2\). This ensures that the Hamiltonian, electron velocity and the e-ph matrix elements decay rapidly in the Wannier representation. The Heyd–Scuseria–Ernzerhof (HSE) hybrid functional is used to improve the bandgap with a Fock parameter of 0.05. Details of the resulting correction are discussed in the next sections, and the input files that can demonstrate the steps to evaluate the electronic structure for the hybrid functional using Wannier functions are given in the supplementary material. Electron–phonon matrix elements \(g_{mn}^{\nu }\), where m, n are electron band indices and \(\nu \) is the phonon eigenmode, are initially evaluated on a coarse k-grid of \(16\times 16\times 16\) and coarse q-grid of \(8\times 8\times 8\) points using the EPW (electron–phonon Wannier) package as implemented in QE.26,27 Wannier interpolation is used to evaluate the e-ph matrix elements on a fine q-grid of \(150\times 150\times 150\) and k-mesh discretized over the conduction band along the \(L \rightarrow \Gamma \rightarrow X\) path in 5000 points. The imaginary part of self-energy (\({{{\mathcal {I}}}}m(\Sigma ^{e-ph}_{n\textbf{k}})\)) is evaluated as a function of the phonon mode (\(\nu \)) to better understand the e-ph scattering mechanisms and to identify the dominant phonon modes. Overall scattering rates are given by \((\Gamma ^{e-ph}_{n\textbf{k}})=2[{{{\mathcal {I}}}}m(\Sigma ^{e-ph}_{n\textbf{k}})]/\hbar \), and the corresponding relaxation times (\(\tau _{n,\textbf{k}}\)) are also evaluated by taking the inverse of the scattering rates \(\tau _{n,\textbf{k}}=(\Gamma ^{e-ph}_{n\textbf{k}})^{-1}\). Hydrostatic strain is simulated by appropriately changing the lattice constant of GaSb such that the strain is within \(\pm 0.8\)% of the equilibrium lattice constant.
Results

The band structure of GaSb using LDA (black/continuous line) and HSE (red/broken line) hybrid functionals. HSE predicts the correct energy difference of 80 meV between the \(\Gamma \) and L points in the conduction band. The HSE bandgap is 0.69 eV and the LDA bandgap is 0.55 eV. \(X_m\) marks the conduction band minimum along [100] (Color figure online).
GaSb is a direct-bandgap semiconductor with an experimental bandgap of 0.81 eV which lies in the infrared range.8 The local density approximation (LDA) misses two important features of the GaSb electronic structure. First, it underestimates the bandgap as 0.56 eV, which is \(\sim 30\)% lower than the experimental value.28 Secondly, an important feature of the conduction band is that the energy difference between the \(\Gamma \) and L points is experimentally found to be 80 meV, whereas the same energy difference is calculated as 20 meV using LDA. In order to improve the bandgap and relative energies of the CBM, we use the HSE hybrid functional and tune the Fock mixing parameter such that the bandgap and the relative energies of secondary minima correspond to the experimental values. By using the hybrid functional, the bandgap is corrected to 0.69 eV, and more importantly, the energy difference between the \(\Gamma \) and L points is corrected to 80 meV, as shown in Fig. 1. The reader should note that the electronic structure shown for HSE hybrid functional is evaluated by first performing a self-consistent calculation using the hybrid functional, followed by using the Wannierization process that involves representation of the converged eigenfunctions using the maximally localized Wannier functions.25 In addition, the \(\Gamma \) and X-valley energy difference also improves from 28 meV determined using only LDA to 41 meV using the HSE functional, which compares well with the experimental value of 43 meV. The actual minimum is along [100] and not exactly at X as shown in Fig. 1 by label \(X_m\). Hence, the use of the hybrid functional improves the band structure considerably.

The effect of compressive strain on the conduction band of GaSb. Compressive strain over 0.4% converts GaSb into an indirect-bandgap semiconductor.
For the accuracy of the work proposed here, it is important that the energy difference between the various conduction band minima is as accurate as possible for GaSb under strain. To ensure that the band structure is consistent with the GaSb at equilibrium, the same Fock mixing parameter for the HSE hybrid functional is used for all the strain calculations performed here. Table I shows the bandgaps, relative energies of the different minima, and whether the bandgap is direct (D) or indirect (ID) for the different strains in GaSb. The corresponding experimental values for GaSb at equilibrium are shown for reference.

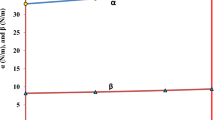
The effect of strain on zone center LO- (empty triangles) and TO-phonons (filled triangles) in GaSb with linear fitting shown for reference.

The contribution to the imaginary part of e-ph self-energy as a function of electron energy for GaSb at different strains from acoustic phonon modes. The strains shown are: tensile 0.6% (a); equilibrium (b) and (c) compressive 0.6%. Energy corresponding to LO and secondary minimum in the conduction band is shown for reference. The scattering is very similar as long as GaSb remains a direct-bandgap semiconductor where as for compressive strain of 0.6% all the phonon modes contribute to scattering. TA-1 and TA-2 correspond to transverse acoustic modes, LA: longitudinal acoustic. \(X_m\) marks the minimum along the X-symmetry direction in the Brillouin zone.

The contribution to the imaginary part of e-ph self-energy as a function of electron energy for GaSb at different strains from optical phonon (bottom row) modes. The strains shown are: tensile 0.6% (a); equilibrium (b) and (c) compressive 0.6%. Energy corresponding to LO and secondary minimum in the conduction band is shown for reference. The scattering is very similar as long as GaSb remains a direct-bandgap semiconductor where as for compressive strain of 0.6% all the phonon modes contribute to scattering. TO-1 and TO-2: transverse optical and LO: longitudinal optical phonon mode. \(X_m\) marks the minimum along the X-symmetry direction in the Brillouin zone.
Under a compressive strain of 0.2 and 0.4% GaSb remains a direct-bandgap semiconductor as indicated by the zero under the \(\Gamma \) column in Table I. The relative energy difference that is evaluated and tabulated for L minimum reduces for higher strains. The X-minimum also moves to a lower relative energy. At 0.6% and higher strain, the conduction band minimum is at L and the \(\Gamma \) valley moves to higher relative energy as shown in Fig. 2. The X-minimum continues to move to a relative lower energy with compressive strain. In comparison, for GaSb under tensile strain the CBM remains at \(\Gamma \) and the relative energies indicate that L- and X-minimum move higher in energy.
The effect of strain on zone center longitudinal optical (LO) and transverse optical (TO) phonons is shown in Fig. 3 along with the least square fit line as reference. The phonon dispersion for GaSb under equilibrium is shown in Fig. S1 in the supplementary material. Under compressive strain the LO and TO frequency shifts to a lower value and under tensile strain they move to higher value. Overall the change in frequency under strain can be considered within the linear regime.
Scattering Rates
Electron–phonon scattering by all phonon modes is not equal, as seen in other III-V semiconductors such as GaAs and AlAs.16,17,30 To understand which phonon modes contribute more to scattering at different energies, we evaluate and present the scattering rates for each phonon mode at different strains. GaSb under three strains is discussed in detail here: tensile strain of 0.6%, equilibrium (0% strain) and compressive 0.6%. Figure 4 shows the acoustic and Fig. 5 the optical phonon contributions to the total scattering rates at these strains. The zero of the energy axis corresponds to the CBM which is at \(\Gamma \) for GaSb under tensile strain and at equilibrium and at L for compressive strains of 0.6% or higher. LO-phonon energy and the energy corresponding to the secondary minimum in the conduction band is also shown.
First consider e-ph scattering rates at equilibrium which are shown in Figs. 4b and 5b where L-minima is at (\(E_L=80\) meV). Acoustic phonon scattering is negligible for electron energy below \(E_L\) in the conduction band. In this energy range LO-phonon scattering is the dominant mechanism. Below 29 meV, which corresponds to the LO-phonon energy (\(E_\textrm{LO}\)), only phonon absorption is allowed as there are no empty states available for phonon emission to be possible. For electrons with energy between \(E_\textrm{LO}\) and \(E_L\), intra-valley scattering within \(\Gamma \)-valley is the only permitted mechanism. When electron energy is more than \(E_L\), all phonon modes contribute to the scattering almost equally up to electron energy corresponding to \(X_m\). At these energies intra-valley scattering in \(\Gamma \) and L valleys, inter-valley scattering between \(L-\Gamma \) or \(\Gamma -L\) and scattering between equivalent L-valleys is possible. At room temperature, as we have shown here, the contribution to the scattering rates is dominated by acoustic phonons for electrons with energies above 0.36 eV which corresponds to the \(X_m\).
Scattering rates for GaSb under tensile strain of 0.6% are shown in Figs. 4a and 5a, where the CBM lies at \(\Gamma \). This allows only scattering within the \(\Gamma \)-valley up to electron energy corresponding to \(E_L\). Due to the applied strain, \(E_L\) is at 0.18 eV relative to the CBM. At low energy, LO-phonon scattering is the dominant mechanism. Inter-valley scattering can only occur above 0.18 eV. Acoustic and LO phonons dominate scattering above \(E_L\). This implies that the carrier relaxation time up to electron energy equal to E\(_L\) is longer than at higher energy where inter-valley scattering involving long-wavelength phonons is allowed. This case can be considered representative of the effect of tensile strain in GaSb, because under such strains it remains a direct-bandgap semiconductor and moves L- and X-minima to higher energy, as seen in Table I up to the strains we have shown here. So the LO-phonon scattering will be dominant as long as the electron energy lies below E\(_L\), and the other phonon modes will be involved in scattering for electrons at higher energy.
Finally, considering the case of compressive strain on GaSb, at compressive strain of 0.6% in GaSb, the line widths (scattering rates) are as shown in Figs. 4c and 5c, where the origin of the energy axis corresponds to the L-minimum, indicating the indirect-bandgap nature of GaSb at this strain. If the electron energy is less than \(E_{\Gamma }\), only LA- and LO-phonon scattering is observed, which includes intra-valley scattering within the L-valley and inter-valley scattering between equivalent L-valleys. For electrons with energy greater than \(E_{\Gamma }\), all except one TA and one TO mode contribute to scattering, and the magnitude is higher than the other two cases. At this strain, LO-energy and E\(_{\Gamma }\) are almost identical, which would suggest that both phonon emission and absorption would be possible. A comparison of acoustic scattering rates in GaSb shown in Fig. 4 at 0.1 eV clearly shows that the scattering is high at 0.6% compressive strain. It has been observed that the scattering rate is proportional to the density of states (DOS) available at any energy.17 At strains where the conduction band minimum is at \(\Gamma \), the DOS is smaller and the effective mass is small. When the CBM is at L, the DOS increases because of the presence of equivalent L-minima, and the effective mass is comparatively larger. This is attributed to the larger phase-space available for e-ph scattering. Near the conduction band edge, the largest contribution comes from LO-phonons. This conforms to the selection rules derived to predict the inter-valley scattering, which state that in III-V semiconductors, when the group V element is heavier than the group III element, LO scattering is allowed.31
Relaxation Times
Experimental methods such as time-resolved Raman spectroscopy or other methods to study ultrafast carrier dynamics are used to measure carrier lifetimes or relaxation times.32,33,34 Here, the e-ph relaxation times (RT) in GaSb can be evaluated as the inverse of the total scattering rates. The RT results for a few strains and temperatures are shown in Fig. 6: (a) GaSb under equilibrium conditions at 10 K, (b) GaSb under equilibrium conditions at 300 K, (c) GaSb under compressive strain of 0.6% at 300 K and (d) GaSb under tensile strain of 0.6% at 300 K for carriers in the conduction band up to an energy of 0.5 eV. The colors indicate the conduction band valley from which the points originate, where green represents the L-valley, red represents the \(\Gamma \)-valley and blue represents the X-valley, as shown in the inset of Fig. 6a–d, which is the lowest conduction band level. The energy corresponding to the LO-phonon (\(E_\textrm{LO}=\sim 28\) meV) is shown as reference, and the origin of the energy axis corresponds to the conduction band minimum at that strain. The next conduction band minimum is shown for the reference as well. The X-minimum does not always lie within \(0-0.5\) eV of the CBM and hence does not appear in all the figures.
The RT shows a large variation with strain. For carrier energy below \(E_\textrm{LO}\), the RT increases rapidly because they are close to the band edge. The relaxation times within the energy range \(E_\textrm{LO}\) to \(E_L\) correspond to intra-valley scattering in the \(\Gamma \)-valley. For carriers with energy above \(E_L\), relaxation by scattering between equivalent L-valleys and L-\(\Gamma \) would be the dominant mechanisms.
The effect of temperature on the RT is seen in Fig. 6a and b. The RT at 10 K is \(\sim 2.6\mu \)s, as compared to \(\sim 1.0\) ps at 300 K at the bottom of the conduction band. At the bottom of the L- and X-valley, the RT is \(\sim 16\) ps and \(\sim 230\) fs at 10 K and 500 fs and 50 fs at 300 K, respectively. At energy equivalent to \(E_\textrm{LO}\) in the conduction band, the RTs for 10 K and 300 K are 3 ps and 720 fs, respectively. This difference in RT with temperature is attributed to the population (or the lack) of acoustic phonons corresponding to that temperature. It was observed that the scattering rates by optical phonons are nearly constant with temperature in silicon, whereas scattering rates by acoustic phonons at low temperature are very low but at higher temperature they become comparable to scattering by optical phonons.35 Thus we conclude that acoustic phonon scattering is dominant at high temperatures, consequently lowering the RTs. The relaxation time for compressive strain of 0.6% is evaluated as 600 fs at the bottom of both the L- and \(\Gamma \)-valleys and 54 fs at the bottom of the X-valley. In comparison, for tensile strain of 0.6%, the RT is 2 ps at the bottom of the \(\Gamma \) -valley and 0.6 ps at the bottom of the L-valley.
Electrons in the \(\Gamma \)-valley near the band edge under equilibrium conditions or under tensile strain of 0.6% have large relaxation times as a result of reduced DOS near the bottom of the conduction band. At 0 strain, the phase space near the \(\Gamma \)-valley is very small, and only intra-valley scattering is possible; hence, the relaxation time is high. It can thus be understood that RT for GaSb under tensile strain will always be high near the conduction band edge because of the reduced DOS near the band edge. In the case of 0.6% compressive strain or higher, where the CBM is at L, the phase space is higher because of the presence of equivalent L-minima available for scattering, resulting in shorter RT.
Comparisons with experimentally measured relaxation times for GaSb at equilibrium are quite good in terms of temperature, where Maly et al.9 observed that on cooling GaSb, the relaxation times are much higher than at room temperature.

Electron–phonon relaxation times for carriers in the conduction band in GaSb at (a) equilibrium at 10 K, (b) equilibrium at 300 K, (c) 0.6% compressive strain and (d) 0.6% tensile strain. The data point colors show electrons originating from different valleys in the lowest conduction band level such that \(\Gamma \) (red), L (green) and X (blue) (Color figure online).
Concluding Remarks
In conclusion, this work presents a detailed investigation of the dependence on scattering direction, energy and strain for e-ph processes in GaSb. The crucial prerequisites for this accurate description for GaSb are a realistic first-principles band structure that quantitatively accounts for the relative positions of the closely spaced satellite valleys and a full Brillouin zone interpolation of the e-ph coupling matrix elements decomposed over all phonon modes. This analysis was enabled by the EPW interpolation method applied previously to group IV and III-V semiconductors. For low strain values, the electron lifetimes reflect the dependence in AlAs and diamond, whereas for large values of strain they reflect the energy and direction dependence characteristic of silicon and GaAs, which are indirect-bandgap semiconductors. The relative displacement between the L- and X-valleys distinguishes this material from the other semiconductors mentioned above. The detailed dependence presented here is relevant for hot electron dynamics in optoelectronic devices, where strain is always present in various amounts.
References
D.H. Chow, R.H. Miles, T.C. Hasenberg, A.R. Kost, Y. Zhang, H.L. Dunlap and L. West. 1995. Mid-wave infrared diode lasers based on GaInSb/InAs and InAs/AlSb superlattices. Appl. Phys. Lett. 67: 3700. https://doi.org/10.1063/1.115354.
H. Mohseni, E. Michel, J. Sandoen, M. Razeghi, W. Mitchel and G. Brown. 1997. Growth and characterization of InAs/GaSb photoconductors for long wavelength infrared range. Appl. Phys. Lett. 71: 1403. https://doi.org/10.1063/1.119906.
R.A. Carrasco, J. George, D. Maestas, Z.M. Alsaad, D. Garnham, C.P. Morath, J.M. Duran, G. Ariyawansa and P.T. Webster. 2021. Proton irradiation effects on InGaAs/InAsSb mid-wave barrier infrared detectors. J. Appl. Phys. 130: 114501. https://doi.org/10.1063/5.0064043.
A.H. Jones, S.D. March, S.R. Bank and J.C. Campbell. 2020. Low-noise high-temperature AlInAsSb/GaSb avalanche photodiodes for 2-\(\mu \)m applications. Nat. Photon. 14: 559. https://doi.org/10.1038/s41566-020-0637-6.
B.R. Bennett, M.G. Ancona, J.B. Boos, C.B. Canedy and S.A. Khan. 2008. Strained GaSb/AlAsSb quantum wells for p-channel field-effect transistors. J. Cryst. Growth 311: 47. https://doi.org/10.1016/j.jcrysgro.2008.10.025.
Y. Shao, and J.A. del Alamo. 2022. Sub-10-nm diameter vertical nanowire p-type GaSb/InAsSb tunnel FETs. IEEE Electron Device Lett. 43: 846. https://doi.org/10.1109/LED.2022.3166846.
P. Wang, S. Han and R. Quhe. 2021. Quantum transport simulation of the two-dimensional GaSb transistors. J. Semicond. 42: 122001. https://doi.org/10.1088/1674-4926/42/12/122001.
P.S. Dutta, H.L. Bhat and V. Kumar. 1997. The physics and technology of gallium antimonide: an emerging optoelectronic material. J. Appl. Phys. 81: 5821. https://doi.org/10.1063/1.365356.
P. Maly, A.C. Maciel, J.F. Ryan, N.J. Mason and P.J. Walker. 1994. Time-resolved Raman measurement of intervalley scattering in GaSb. Semicond. Sci. Technol. 9: 719. https://doi.org/10.1088/0268-1242/9/5s/086.
B. Jusserand, P. Voisin, M. Voos, L.L. Chang, E.E. Mendez and L. Esaki. 1985. Raman scattering in GaSb-AlSb strained layer superlattices. Appl. Phys. Lett. 46: 678. https://doi.org/10.1063/1.95527.
M.K. Hudait, S.W. Johnston, M. Meeker and G.A. Khodaparast. 2022. Carrier recombination dynamics and temperature dependent optical properties of InAs-GaSb heterostructures. J. Mater. Chem. C 10: 17994. https://doi.org/10.1039/D2TC03443C.
C.R. Bolognesiand and O.J.S. Ostinelli. 2021. \(\gamma \)-l intervalley separation and electron mobility in GaAsSb grown on InP: transport comparison with the GaInAs and GaInAsSb alloys. Appl. Phys. Lett. 119: 242103. https://doi.org/10.1063/5.0060423.
E. Chidley, S. Haywood, R. Mallard, N. Mason, R. Nicholas, P. Walker and R. Warburton. 1988. GaSb heterostructures grown by MOVPE. J. Cryst. Growth 93: 70. https://doi.org/10.1016/0022-0248(88)90508-8.
G.H. CocoletziandN, 2000. Takeuchi, first-principles calculations of the growth of insb on GaSb(110). Phys. Rev. B 61: 15581. https://doi.org/10.1103/PhysRevB.61.15581.
C. Raisin, A. Rocher, G. Landa, R. Carles and L. Lassabatere. 1991. GaSb/GaAs heteroepitaxy characterized as a stress-free system. Appl. Surf. Sci. 50: 434. https://doi.org/10.1016/0169-4332(91)90213-4.
N. Tandon, L.R. Ram-Mohan and J.D. Albrecht. 2018. Electron-phonon scattering in alas and its response to hydrostatic pressure. J. Electron. Mater.https://doi.org/10.1007/s11664-018-6651-5.
N. Tandon, L.R. Ram-Mohan and J.D. Albrecht. 2019. Effect of hydrostatic pressure on the electron-phonon scattering in GaAs. J. Appl. Phys. 126: 015701. https://doi.org/10.1063/1.5094416.
N. Tandon, J.D. Albrecht and L.R. Ram-Mohan. 2015. Electron-phonon coupling and associated scattering rates in diamond. Diam. Relat. Mater. 56: 1.
N. Tandon, J.D. Albrecht and L.R. Ram-Mohan. 2015. Electron-phonon interaction and scattering in Si and Ge: implications for phonon engineering. J. Appl. Phys.https://doi.org/10.1063/1.4927530.
S. Poncé, F. Macheda, E.R. Margine, N. Marzari, N. Bonini and F. Giustino. 2021. First-principles predictions of hall and drift mobilities in semiconductors. Phys. Rev. Res. 3: 043022. https://doi.org/10.1103/PhysRevResearch.3.043022.
P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G.L. Chiarotti, M. Cococcioni, I. Dabo, A.D. Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A.P. Seitsonen, A. Smogunov, P. Umari and R.M. Wentzcovitch. 2009. Quantum espresso: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21: 395502. https://doi.org/10.1088/0953-8984/21/39/395502.
J.P. Perdewand, and A. Zunger. 1981. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 23: 5048.
C. Hartwigsen, S. Goedecker and J. Hutter. 1998. Relativistic separable dual-space gaussian pseudopotentials from h to rn. Phys. Rev. B 58: 3641.
S. Baroni, S.D. Gironcoli, A.D. Corso and P. Giannozzi. 2001. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73: 515.
A.A. Mostofi, J.R. Yates, G. Pizzi, Y.-S. Lee, I. Souza, D. Vanderbilt and N. Marzari. 2014. An updated version of Wannier90: a tool for obtaining maximally-localised Wannier functions. Comput. Phys. Commun. 185: 2309. https://doi.org/10.1016/j.cpc.2014.05.003.
J. Noffsinger, F. Giustino, B.D. Malone, C.H. Park, S.G. Louie and M.L. Cohen. 2010. Epw: a program for calculating the electron-phonon coupling using maximally localized Wannier functions. Comput. Phys. Commun. 181: 2140.
S. Poncé, E. Margine, C. Verdi and F. Giustino. 2016. Epw: electron-phonon coupling, transport and superconducting properties using maximally localized Wannier functions. Comput. Phys. Commun. 209: 116. https://doi.org/10.1016/j.cpc.2016.07.028.
M. Wuand, and C. Chen. 1992. Photoluminescence of high-quality GaSb grown from Ga- and Sb-rich solutions by liquid-phase epitaxy. J. Appl. Phys. 72: 4275. https://doi.org/10.1063/1.352216.
O. Madelung, 2004. Semiconductors: Data Handbook, 3rd edn. Berlin: Springer.
M. Bernardi, D. Vigil-Fowler, C.S. Ong, J.B. Neaton and S.G. Louie. 2015. Ab initio study of hot electrons in GaAs. Proc. Natl. Acad. Sci. 112: 5291. https://doi.org/10.1073/pnas.1419446112.
J.L. Birman, M. Lax and R. Loudon. 1966. Intervalley-scattering selection rules in III–V semiconductors. Phys. Rev. 145: 620. https://doi.org/10.1103/PhysRev.145.620.
K. Tanimura, H. Tanimura and J. Kanasaki. 2022. Ultrafast dynamics of photoinjected electrons at the nonthermal regime in the intra-\(\Gamma \)-valley relaxation in InP studied by time- and angle-resolved photoemission spectroscopy. Phys. Rev. B 106: 125204. https://doi.org/10.1103/PhysRevB.106.125204.
J.A. Kash, J.C. Tsang and J.M. Hvam. 1985. Subpicosecond time-resolved Raman spectroscopy of lo phonons in GaAs. Phys. Rev. Lett. 54: 2151. https://doi.org/10.1103/PhysRevLett.54.2151.
J. Sjakste, K. Tanimura, G. Barbarino, L. Perfetti and N. Vast. 2018. Hot electron relaxation dynamics in semiconductors: assessing the strength of the electron-phonon coupling from the theoretical and experimental viewpoints. J. Phys. Condens. Matter 30: 353001. https://doi.org/10.1088/1361-648x/aad487.
R. Sen, N. Vast and J. Sjakste. 2022. Hot electron relaxation and energy loss rate in silicon: temperature dependence and main scattering channels. Appl. Phys. Lett. 120: 082101. https://doi.org/10.1063/5.0082727.
Acknowledgments
This work was supported by the Air Force Research Laboratory and by the HPC facilities from the DoD.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tandon, N., Albrecht, J.D. & Badescu, S.C. Electron–Phonon Coupling and Carrier Relaxation Times in Gallium Antimonide Under Strain. J. Electron. Mater. 53, 1161–1168 (2024). https://doi.org/10.1007/s11664-023-10877-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11664-023-10877-x