
Abstract
Platinum group metals (PGMs) are primarily used in automotive exhaust catalysts (autocatalysits). Spent autocatalysts are the most important secondary resource for PGMs. However, transporting autocatalyst scraps and recovering PGMs from the scraps are costly and time-consuming, owing to the low PGM content in spent autocatalysts. Thus, an effective PGM-concentration technology for the pretreatment of scrap prior to transport is required. This study develops a new pretreatment technique that is applied prior to the flotation concentration of PGMs in autocatalysts. This method utilizes electroless Cu plating followed by sulfurization. In the electroless Cu-plating process, which uses glyoxylic acid as a reducing agent, Cu is deposited on the PGM particles in the washcoat of the autocatalyst. During the sulfurization process, S vapor sulfurizes the deposited Cu into copper sulfide, which is hydrophobic. Prior to the experiments, thermodynamic considerations were made to predict the reactivity of Cu and the representative elements constituting the autocatalyst with S vapor, and the sulfurization conditions were designed. Sulfurization experiments were performed at 850 K (577 °C) in the presence of carbon (C), and the results show the successful conversion of only Cu to copper sulfide without sulfurizing the representative oxides (MgO, Al2O3, SiO2, CeO2, and ZrO2) present in the autocatalyst. Finally, in the flotation process, the copper sulfide-coated PGMs are separated from the ceramic components of the autocatalyst, which is hydrophilic; thus, it is concentrated in the froth. Flotation experiments utilizing a microbubble flotation method were successfully performed to recover the PGM concentrates. This innovative pretreatment technique is expected to reduce the cost and time required for the entire PGM recycling process.
Similar content being viewed by others

Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Platinum group metals (PGMs) are used in various industrial fields, owing to their excellent corrosion resistance, heat resistance, and unique catalytic properties. Among PGMs, platinum (Pt), palladium (Pd), and rhodium (Rh) are primarily used as catalyst metals for automotive exhaust catalysts (autocatalysts), which purify automobile exhaust gases. In 2020, the global demand for Pt, Pd, and Rh for autocatalysts accounted for 31, 85, and 93 pct of the total demand of each element, respectively.[1] The demand for PGMs as autocatalysts is expected to increase with the breakaway from the COVID-19 impact and tighter environmental regulations. Natural PGM resources are scarce and distributed in limited regions, such as South Africa and Russia. This results in a low annual production of PGMs and price fluctuations, which is associated with social conditions.[1] In addition, the concentrations of each PGM in natural ores are very low—below 10 mass ppm PGM.[2] Therefore, PGM smelting results in excessive energy consumption, long process time, and the generation of a large amount of waste. However, the total concentration of Pt, Pd, and Rh in autocatalysts is as much as 500–5000 ppm PGM,[3,4,5,6] which is hundreds to thousands of times their concentration in ores. Therefore, spent autocatalysts are important secondary PGM resources, and recycling PGMs from spent autocatalysts is imperative for reducing environmental loads and securing resource supply.
In the autocatalysts, a PGM-containing washcoat is loaded onto a substrate, which is typically made of ceramic materials, such as cordierite (2MgO·2Al2O3·5SiO2), and it has a honeycomb structure consisting of multiple channels (60–120 cells cm−2).[8,9] The main component of the washcoat is gamma-alumina (γ-Al2O3), on which nanometer-sized PGM particles are loaded.[3,7] The washcoat also contains a ceria (CeO2) or ceria–zirconia (CeO2–ZrO2) solid solution, which promotes the purification reaction of exhaust gases using the catalyst. Baria (BaO) and lanthana (La2O3), which provide high thermostability to γ-Al2O3, are added to the washcoat.[8,9]
Currently, PGMs are recycled from spent autocatalysts using two main methods: pyrometallurgical and hydrometallurgical processes. In the pyrometallurgical process, the PGMs in scraps are extracted and concentrated by alloying the PGMs with collectors, such as copper (Cu) and iron (Fe), which have strong chemical affinities with PGMs. Subsequently, the extracted PGMs are dissolved in an aqueous solution and then separated and purified using various techniques, such as solvent extraction, precipitation, and ion exchange. The pyrometallurgical process has a high processing speed and recovery rate. However, it requires a large-scale facility, which restricts location options, and consumes a large amount of energy. The Rose process is a typical pyrometallurgical process that uses Cu as the collector. In hydrometallurgical processes, the PGMs in scraps are extracted via direct dissolution using acids with strong oxidants. The hydrometallurgical processes can be operated at low energy costs and in small-scale facilities compared to the pyrometallurgical processes. However, the hydrometallurgical process has low PGM recovery rates and requires long processing time, and it generates a large amount of difficult-to-treat liquid waste containing heavy metals.[2,6,10] Furthermore, the PGM concentration in autocatalysts is low compared with that of the target elements contained in other general scrap for recycling. Hence, owing to the large amount of scrap per unit of PGM, transporting scraps from scrap-collection sites to refineries requires a long time and is costly, which is a considerable bottleneck in PGM recycling.[11] Therefore, an efficient method for high-speed PGM concentration in scrap prior to transport is required. Producing PGM-concentrated scraps with a small volume reduces transportation costs via sea or land and enables high-speed transportation via air. Thus, cost reduction and a shorter process lead time for the overall PGM recycling scheme can be achieved.
Many technologies have been developed to concentrate PGMs in spent autocatalysts via physical separation. Owada et al. studied mineral-processing techniques to physically separate and concentrate PGMs from spent autocatalysts. Magnetic separation was experimentally conducted for pulverized autocatalysts containing nickel (Ni) in the washcoat, and this technique demonstrated a positive relationship between the recovery of PGMs and Ni.[12,13] Notably, magnetic separation is only effective for old types of autocatalyst that contain Ni and are magnetic. By assuming that PGM particles and γ-Al2O3 in the washcoat are not separated during pulverization, Owada et al. also attempted flotation for pulverized autocatalysts to recover PGMs together with γ-Al2O3 by separating γ-Al2O3 from the cordierite substrate.[14,15] Owada et al. also experimentally showed that γ-Al2O3 and cordierite could be separated by flotation. However, the separation efficiencies and enrichment ratios of PGMs in their experiments using spent autocatalysts were low.
Several studies have reported PGM concentration methods that use size separation after grinding. Kim et al. reported that PGMs can be efficiently separated and concentrated from a cordierite substrate via size separation by coarse crushing spent autocatalysts, followed by fine grinding using a wet attritor.[16] Liu et al. reported that 76 pct of the washcoat was separated from the substrate via size separation after coarse crushing with a jaw crusher and roll crusher, followed by fine grinding with a dry attritor.[17] Liu et al. also concentrated PGMs using a heat-and-quench treatment, followed by selective grinding.[18,19] A heat-and-quench treatment can generate cracks at each interface, owing to the difference in thermal-expansion coefficients between various materials in the substrate and washcoat, which leads to the destruction and even separation of some parts of the washcoat. After the heat-and-quench treatment, the fine particles were enriched with PGMs via two-step grinding using a jaw crusher and roll crusher. Subsequently, PGM-rich fine particles were recovered using size separation.
Okabe et al. proposed a method that combined chemical treatment with physical separation. Okabe and Mitsui proposed new processes to concentrate PGMs in scrap via physical separation after various chemical reactions [20]. For example, PGMs were alloyed with Fe, and the obtained Fe–PGM alloys were recovered using magnetic separation. They also reported that alloying PGMs with Cu or lead (Pb), which causes a change in the volume of PGM particles and improves the detachability of PGMs from ceramics in scrap, is effective as a pretreatment for a separation process that combines sulfurization and flotation. They also suggested that alloying PGMs with Pb is an effective pretreatment for specific gravity separation, owing to the high specific gravity of Pb.[20]
Recently, Okabe et al. demonstrated a method that combined chemical treatment and magnetic separation. Taninouchi et al. developed a process in which PGMs in the washcoat are magnetized by depositing or alloying them with Ni or Fe through electroless plating or FeCl2 vapor treatment; the PGMs are then enriched by magnetic separation.[21,22,23,24] They performed electroless Ni and Fe plating on simulated and actual autocatalysts containing Pt, Pd, and Rh, followed by magnetic separation.[21,22] Electroless Ni and Fe plating processes were conducted in plating baths that contained sodium hypophosphite (NaPO2H2·H2O) and sodium borohydride (NaBH4) as the reducing agents, respectively. In the experiments with the simulated autocatalysts, almost all PGMs were recovered by both the Ni and Fe plating methods. The highest enrichment ratio was 6.5 times for Ni plating and 4.6 times for Fe plating. However, in the experiments that utilized unused actual autocatalysts, the highest recovery rate and enrichment ratio were approximately 80 pct and 2 times, respectively. Taninouchi et al. also reported a process for enriching PGMs via magnetic separation after the formation of Fe–PGM alloys using FeCl2 vapor.[23,24] Pt wire or a simulated autocatalysts were heated together with Fe powder and FeCl2 powder at 1200 K (927 °C), and the Pt wire or simulated autocatalysts were then magnetized. The treated simulated autocatalysts were pulverized and magnetically separated. The recovery rates and enrichment ratios of the process were greater than 96 pct and 3–5 times, respectively.
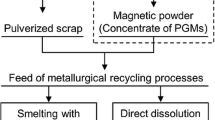
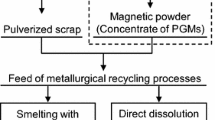
In this study, we developed a new PGM concentration process that combines electroless Cu plating, sulfurization, and flotation for autocatalysts.[25] Figure 1 shows the flowchart of the entire PGM recycling scheme that incorporates the proposed process. The flowchart of our novel PGM concentration method is shown in Figure 2. First, Cu is electrolessly plated onto the PGM particles in the washcoat of the pulverized autocatalysts. Next, the deposited Cu is sulfurized into hydrophobic copper sulfide using sulfur vapor. Finally, in the flotation process, the copper sulfide-coated PGMs are separated from the hydrophilic ceramic components of the autocatalyst and recovered in the froth. Cu was selected as the plating material because the flotation process for copper sulfide ores is well established in the mining industry. In addition, flotation has many adjustable parameters, such as various additives and pH, which make it advantageous for optimization compared with magnetic separation. Notably, the PGM concentrate produced by this method can be processed using existing Cu refineries. Okabe and Mitsui previously demonstrated the sulfurization of PGMs and Cu–PGM alloys with sulfur vapor; however, the sulfurization behavior of the oxides in the autocatalyst has not been experimentally confirmed.[20] When these oxides are converted to sulfides or sulfates via the sulfurization process, they float together with copper sulfide-containing PGMs or dissolve in the flotation solution during flotation, which may reduce the concentration efficiency of PGMs. There are no existing studies on the concentration behavior and recovery efficiency of PGMs in autocatalysts through the integrated processes of electroless Cu plating, sulfurization, and flotation.

Flowchart of the novel physical concentration method proposed in this study and common procedure for recovering PGMs from spent autocatalysts

Schematic of the PGMs concentration process from spent autocatalysts proposed in this study
In this study, we experimentally investigate the sulfurization reactivities of typical oxides (MgO, Al2O3, SiO2, CeO2, and ZrO2) in autocatalysts to demonstrate the selective sulfurization of Cu. We also evaluate the effectiveness of an integrated process that combines electroless Cu plating and sulfurization with flotation for simulated autocatalysts.
Theoretical Background
Electroless Copper Plating
Formaldehyde (HCHO) is widely used as a reducing agent for industrial electroless Cu plating. Formaldehyde is inexpensive and has a high oxidation rate; however, it has a strong odor and adverse effects on the human body and environment.[26,27] In this study, glyoxylic acid (CHOCOOH) was used as a reducing agent; it has a high Cu deposition rate, no irritating odor, and is easier to handle.[27,28,29,30,31,32,33] Cu is deposited on PGM particles in the washcoat of the autocatalyst, which have catalytic activity for glyoxylic acid; however, Cu is not deposited on ceramic components with no catalytic activity.
Sulfurization Using Sulfur Vapor
In our proposed process, after the electroless Cu plating of spent autocatalysts, Cu deposits on PGMs are converted to copper sulfide via sulfurization. Copper sulfide, which is hydrophobic, is separated from the hydrophilic oxide components in the spent autocatalyst via flotation. Hydrogen sulfide (H2S) is widely used as a sulfurizing agent in industrial sulfurization processes; however, H2S is a highly toxic and flammable gas. In this study, we investigated sulfurization using pure sulfur (S), which is less toxic and can be safely handled.
There have been many studies on sulfurization processes in the past[34,35,36,37]. In the following section, the sulfurization process for Cu using sulfur vapor is thermodynamically evaluated to determine the optimal conditions for the sulfurization of Cu. The reactivities of typical oxide components in autocatalysts with sulfur vapor are also evaluated to verify the feasibility of the selective sulfurization of Cu.
Reactivity of copper with sulfur vapor
Sulfur has a relatively low boiling point of 717.8 K (444.6 °C) at 1 atm[38,39], which suggests that the sulfurization of metal can be carried out utilizing sulfur vapor at low temperatures of approximately 673–873 K (400–600 °C). Sulfur has many allotropes and isotopes (Sx (g); (x = 1, 2, ··, 8, etc.)) as a gas species; however, the reference state of the vapor species for sulfur in this study is arbitrarily defined to be 0.5 S2 (g) according to the thermodynamic database, which is used for the thermodynamic assessment of sulfurization reaction in this study.[40] The calculated arbitrary boiling point, Tb.S, for the process S (l) = 0.5 S2 (g) is 882 K (609 °C)), which is considerably different from the normal boiling point of 717.8 K (444.6 °C) at 1 atm. If all allotropes and isotopes of sulfur are considered in the vaporization process, the normal boiling point (1 atm) is closely reproduced using the same thermodynamic database.[40]
Figure 3(a) shows the phase stability diagram (\(\log {p_{{{\rm{O}}_2}}} - \log {p_{{{\rm{S}}_2}}}\)) of the Cu–O–S system at an experimental temperature of 850 K (577 °C). Table I summarizes the thermodynamic data that was used to calculate the phase stability diagrams.[40,41] The stable phase of sulfur at 850 K (577 °C) is assumed to be S (l) [40]. S (g), S2 (g), S3 (g), S4 (g), S5 (g), S6 (g), S7 (g), and S8 (g) are formed. Furthermore, when oxides are installed in the system, SO (g), SO2 (g), SO3 (g), and S2O (g) are possibly formed. The 1 atm isobar lines considering O2 (g), S (g), S2 (g), S3 (g), S4 (g), S5 (g), S6 (g), S7 (g), S8 (g), SO (g), SO2 (g), SO3 (g), and S2O (g) are shown in Figure 3 as black dashed lines. The isobar indicates the partial pressure of S2 (g) with the presence of S (l) (aS(l) = 1) respecting to the partial pressure of O2 (g).

Phase stability diagrams: (a) Cu–O–S, (b) Mg–O–S, (c) Al–O–S, (d) Si–O–S, (e) Zr–O–S, and (f) Ce–O–S systems superposed with Cu–O–S and C–O–S systems at 850 K (577 °C). [40,41] Activity and partial pressure of each constituent is assumed as unity. The 1 atm isobar lines considering O2 (g), S (g), S2 (g), S3 (g), S4 (g), S5 (g), S6 (g), S7 (g), S8 (g), SO (g), SO2 (g), SO3 (g), and S2O (g) are shown as black dashed lines
As shown in Figure 3(a), the sulfurization of Cu by sulfur at 850 K (577 °C) produces copper (I) sulfide (Cu2S) under low-oxygen partial pressure and copper (II) sulfate (CuSO4) under high-oxygen partial pressure. During the flotation process, Cu2S floats in the flotation solution, whereas CuSO4 dissolves in the solution. In the latter case, it is expected that the nanometer-sized PGMs contained in CuSO4 will disperse or precipitate and cannot be recovered in the froth.
In this study, the oxygen partial pressure in the sulfurization process was set to be low by installing carbon (C) in the reaction system to convert Cu into Cu2S. Thermodynamic data indicates that the presence of excess C leads to the formation of carbon dioxide (CO2), carbon disulfide (CS2), and carbonyl sulfide (COS) gases. However, it has been reported that at 850 K (577 °C), the operating temperature in this study, CS2 formation is not observed with the reaction between S2 (g) and C (s) (C (s) + S2 (g) = CS2 (g)).[34,42] When CS2 and COS gases are not produced, C (s)/CO2 (g) equilibrium determines the partial pressure of oxygen.
Reactivity of oxides in autocatalyst with sulfur vapor
Cordierite is widely used as a substrate for autocatalysts. In a washcoat, γ-Al2O3 is typically used as a support for PGMs, and CeO2 or CeO2–ZrO2 solid solution is used as an additive to promote the catalytic reaction for PGMs. As shown in Figures 3(b) through (f), the M–O–S phase-stability diagrams for various elements in the substrate and the washcoat at 850 K (577 °C) were constructed to evaluate the possibility of sulfurization for each of the constituents (M = Mg, Al, Si, Zr, and Ce).
According to the M–O–S phase stability diagrams, when sulfurizing with sulfur vapor under a high-oxygen partial pressure, the stable phases of Mg, Al, and Ce are magnesium (II) sulfate (MgSO4), aluminum (III) sulfate (Al2(SO4)3), and cerium (III) sulfate (Ce2(SO4)3), respectively. These sulfates can dissolve in flotation solutions, owing to their high solubility in aqueous solutions. Their dissolution can change the pH of the flotation solutions and cause problems in the flotation collector, which may reduce the separation efficiency of the flotation process.
In contrast, when sulfurizing with sulfur vapor under a low-oxygen partial pressure under C (s)/CO2 (g) equilibrium, the stable phases of Mg, Ce, and Zr are magnesium (II) sulfide (MgS), cerium (III) sulfide (Ce2S3), and zirconium (IV) oxide (ZrO2), respectively. In this case, MgO and CeO2 in the autocatalyst are sulfurized with Cu during the sulfurization process and float with copper sulfide during flotation, which can reduce the PGM enrichment ratio.
Materials and Methods
Preparation of Simulated Autocatalyst
Simulated autocatalysts were prepared by coating a PGM-bearing washcoat onto an Al2O3 substrate.[21,43,44] First, a mixture of γ-Al2O3 powder (> 97.0 pct, Strem Chemicals Inc.), CeO2 powder (> 99.5 pct, Kanto Chemical Co., Inc.) or CeO2–ZrO2 solid solution powder (Ce0.5Zr0.5O2, > 99.0 pct, Sigma-Aldrich), Pt aqueous solution (Pt(NH3)2(NO2)2 (aq.), Tanaka Kikinzoku Kogyo K.K.), Pd aqueous solution (Pd(NH3)2(NO2)2 (aq.), Tanaka Kikinzoku Kogyo K.K.), and Rh aqueous solution (Rh(NO2)2 (aq.), Tanaka Kikinzoku Kogyo K.K.) was dried at 423 K (150 °C) for 12 hours and sintered at 673 K (400 °C) for 60 minutes to prepare the catalyst powder, in which Pt, Pd, and Rh were loaded on Al2O3 and CeO2 or Ce0.5Zr0.5O2. Next, the catalyst powder was mixed with boehmite powder (AlOOH-nH2O, Wako Pure Chemical Industries, Ltd.) and a 5 vol pct nitric acid solution (HNO3 (aq.), Kanto Chemical Co., Inc.) to form a slurry. The mixing weight ratio of boehmite powder, catalyst powder, and 5 vol pct HNO3 (aq.) was 1: 10: 15. The simulated autocatalyst was prepared by coating the surface of an Al2O3 plate (99.5 pct, thickness: 0.5 mm, Nikkato Co., Ltd.) with the slurry, drying the pasted Al2O3 plate thoroughly at 423 K (150 °C) for 12 hours, and sintering the dried plate for 180 minutes at 1073 K (800 °C) under a vacuum. The moisture and HNO3 in the slurry evaporated during the drying step, and boehmite was dehydrated to Al2O3 during the sintering step. Figure 4(c) shows a cross-sectional image obtained using scanning electron microscopy (SEM, JSM-6510LV, JEOL). A porous washcoat with a thickness of 150–200 μm was loaded onto the surface of the Al2O3 substrate. The compositions of the simulated autocatalysts were set to 0.15–0.20 mass pct for Pt, Pd, and Rh (Ccat,i_exp), 27 mass pct for CeO2, and 9–10 mass pct for CeO2–ZrO2 solid solution.

Photographs of (a) side and (b) top views of initial simulated autocatalyst, and (c) secondary electron image of the cross section of the simulated autocatalyst
Electroless Copper Plating
Table II shows the composition of the electroless Cu plating bath used in this study.[29] Copper (II) sulfate pentahydrate (CuSO4·5H2O, > 99.5 pct, Kanto Chemical Co., Inc.) was used as a copper ion source, 2,2′-bipyridyl (C10N2H8, > 99.5 pct, Kanto Chemical Co., Inc.) as a stabilizing agent, glyoxylic acid (C2H2O3·H2O, 98 pct, Sigma-Aldrich Co., LLC.) as a reducing agent, and ethylenediaminetetraacetic acid ([CH2N(CH2COOH)2]2; EDTA, > 99.0 pct, Kanto Chemical Co., Inc.) as a complexing agent.
The pH of the plating bath was adjusted to 12.5, using an 8 mol L−1 hydroxide solution (NaOH (aq.), Kanto Chemical Co., Inc.) at 298 K (25 °C). The plating bath was then heated to 333 K (60 °C) using a water bath, and electroless Cu plating was conducted by immersing the simulated autocatalysts in the plating bath for 90 minutes. Some of the simulated autocatalysts were pulverized with an agate pestle and mortar prior to Cu plating. Electroless Cu plating of the pulverized simulated autocatalysts was conducted under magnetic stirring. The microstructure and composition of the unpulverized simulated autocatalysts before and after electroless Cu plating were analyzed using SEM combined with energy-dispersive X-ray spectroscopy (EDS). Some samples were heated at 1073 K (800 °C) for 180 minutes under a vacuum after the electroless Cu plating to promote the alloying of Cu with PGMs.
Sulfurization
The reactivities of the oxide components in the autocatalysts with sulfur vapor under low-oxygen partial pressure were investigated. The sulfurization experiments were conducted using a mixture of MgO (> 98.0 pct, Wako Pure Chemical Industries, Ltd.), Al2O3 (94 pct, Wako Pure Chemical Industries, Ltd.), and SiO2 (99.9 pct, Kojundo Chemical Laboratory Co., Ltd.) powders (molar ratio, MgO : Al2O3 : SiO2 = 2: 2: 5) and a mixture of CeO2 (> 99.5 pct, Kanto Chemical Co., Inc.) and ZrO2 (> 99.0 pct, Kanto Chemical Co., Inc.) powders (molar ratio, CeO2 : ZrO2 = 1: 1). The particle size distributions of these oxide samples were measured using a particle size analyzer (SALD-7100, Shimazu) and shown in Figure 5.

The particle size distributions of these oxide samples used in the experiment: (a) CeO2, MgO, ZrO2, SiO2, and (b) Al2O3
Five grams of S powder (98.0 pct, Wako Pure Chemical Industries, Ltd.) was fed into the system to sulfurize all the samples. One gram of C powder (99.9 pct, Kojundo Chemical Laboratory Co., Ltd.) was used. Finally, Cu powder (~ 75 µm in diameter, 99.9 pct, Wako Pure Chemical Industries, Ltd.) was fed into the system (weight ratio, Cu : mixture of oxides = 1: 10). The same experiments were conducted on simulated autocatalysts after electroless Cu plating.
Figures 6(a) and (b) show the schematic of the experimental apparatus[20] and the temperature distribution in the reaction chamber inside the electric furnace, respectively. The sample, S powder, and C powder were placed in individual quartz crucibles, which were then placed in a quartz reaction chamber (inner diameter: 51 mm, length: 710 mm), followed by evacuation. The reaction chamber was then installed in an electric furnace at 850 K (577 °C) and heated for 60 minutes to proceed sulfurization reactions. The reaction chamber was then removed from the furnace and air-cooled to room temperature. The crystalline phases of the samples after sulfurization were identified using X-ray diffraction (XRD; D2PHASER, Bruker Corporation, Cu-Kα radiation).

(a) Schematic of the experimental apparatus for sulfurization[20] and (b) temperature distribution in the quartz tube at the experimental temperature of 850 K (577 °C)
Microbubble Flotation
A column-type microbubble flotation method, which can separate micrometer-sized particles with diameters of 10 μm or less, was employed in the experiment.[44] Figure 7 shows the photograph and schematic of the flotation apparatus used in this study. The flotation solution was prepared by adding a predetermined amount of collector and frother to ion-exchanged water. Potassium amylxanthate (CH3(CH2)4OCS2K, > 97.0 pct, Tokyo Chemical Industry Co., Ltd.) with 0.1 g L−1 was used as a collector for copper (II) sulfide (CuS), and 4-methyl-2-pentanol ((CH2)2CHCHCH2CH(OH)CH2, > 99.0 pct, Kanto Chemical Co., Inc.) with 0.03 mL L−1 was used as a frother. The pH of the flotation solution was maintained between 5.8 and 6.0 throughout the experiments. The flotation apparatus was constructed by attaching a gas-injection filter (pore size: 10–20 μm) to the bottom of a quartz tube (inner diameter: 30 mm; length: 900 mm). Nitrogen (N2) gas was injected through the gas-injection filter at a flow rate of 0.25 L min−1 to generate microbubbles. After the bubbles were stabilized for 10 minutes, a mixture of 20–30 mL of the flotation solution and simulated autocatalyst powder, which was subjected to Cu plating and sulfurization, was fed into the upper part of the flotation column. The particle size distribution of the simulated autocatalyst powder were shown in Figure 8. After 10 minutes, froth was collected using a silicon ladle, and tailing was collected via filtration. The obtained froth and tailings were then dried on a hot plate.

(a) Photograph and (b) schematic of the experimental apparatus for flotation

The particle size distribution of the simulated autocatalyst powder, which was subjected to Cu plating and sulfurization. It was fed into the upper part of the flotation column
Analysis of PGM Concentration
The PGM concentrations in the floating and settled samples obtained via flotation were analyzed using inductively coupled plasma atomic emission spectrometry (ICP-AES, SP3520UV, SII NanoTechnology Inc.). The sample solutions for ICP-AES were prepared using alkali fusion and tellurium precipitation techniques.[21,45] The sample powder was mixed with sodium peroxide (Na2O2) in an Al2O3 crucible (weight ratio, sample : Na2O2 = 1: 10). The crucible was heated for 20 minutes using a burner to fuse the samples. After cooling, 100 mL of ion-exchanged water and 50 mL of hydrochloric acid (HCl (aq.), Kanto Chemical Co., Inc.) were added to dissolve the sample. After the dissolution, the solution was heated on a hot plate for precipitation. The precipitate was dissolved in 1.93 mol L−1 HCl. The undissolved residue was removed via filtration through a glass filter (pore size: 5–10 μm) and glass fiber paper (pore size: ~ 0.7 μm). Approximately 100 mL of the filtration was mixed with 10 mL of 10 mg·mL−1 Te aqueous solution (H6TeO6, > 99.0 pct, Kanto Chemical Co., Inc.) and heated to 363 K (90 °C) in a water bath. Then, 30 mL of 20 mass pct SnCl2 aqueous solution (> 97.0 pct, Wako Pure Chemical Industries, Ltd.) was added to the filtration solution, and the mixture solution was kept at 363 K (90 °C) for 300 minutes to completely coprecipitate the PGMs with Te. The precipitate was collected via filtering through a glass filter and glass fiber paper. The black precipitate was dissolved in aqua regia (volume ratio HNO3 (aq.) : HCl (aq.) = 1: 3), and the solution was heated on a hot plate to evaporate the solution and gas components. The salt residue was dissolved in 1.93 mol L−1 HCl to prepare a sample solution for ICP-AES analysis. The PGM concentrations in the sample solutions were analyzed using an external standard calibration method. Calibration curves were prepared using blank, 10, 20, and 50 ppm of the Pt, Pd, and Rh standard solutions. The PGM standard solutions were adjusted by adding 1.00 PGM mg mL−1 of PGM solution (Wako Pure Chemical Industries, Ltd.) to 1.93 mol L−1 HCl. The analysis wavelengths for Pt, Pd, and Rh were 203.7118, 324.3632, and 343.5871 nm, respectively.
As shown in Table III, the analysis error of the PGM concentration in the simulated autocatalyst, Ei, was evaluated by comparing the calculated concentration of element i based on the amount of input materials (Ccat,i_prep) and the measured concentration of i based on ICP-AES (Ccat,i_icp). Ccat,i_prep, and Ei were calculated using Eqs. [1] and [2], respectively:
where Ccat. powder,i_prep is the calculated concentration of element i in the catalyst powder based on the amount of input materials, wcat is the weight of the simulated autocatalyst, and wsub is the weight of the Al2O3 substrate of the simulated autocatalyst. The expected PGM concentrations (Ccat. powder,i_prep) in the catalyst powders for the simulated autocatalysts were 2.00 mass pct PGM for sample Nos. 17–31 and 1.50 mass pct PGM for sample Nos. 35–52. The weight ratio of the catalyst powder to the washcoat is calculated to be 10:11, based on the amount of components in the slurry (boehmite: catalyst powder: 5 vol pct HNO3 (aq.) = 1: 10: 15) used as the intermediate for the simulated catalysts and the following two assumptions: (1) all HNO3 in the slurry evaporates during the drying step; (2) the weight change due to the dehydration of boehmite during the sintering step is negligible.
In this study, the initial PGM concentration (Ccat,i_est) of the simulated autocatalyst is the estimated value based on the following equation, assuming that Ei is constant for all simulated autocatalysts in the same lot:
Results and Discussion
Electroless Copper Plating of Simulated Autocatalyst
Figures 9(a) and (b) show the appearance of the unpulverized simulated autocatalyst (Sim_PGM/CeZrO4_38) before and after the electroless Cu plating, respectively. Figure 9(c) and (d) show the appearance of the pulverized simulated autocatalyst (Sim_PGM/CeZrO4_40) before and after the electroless Cu plating, respectively. Table IV shows the weights of the simulated autocatalyst samples before and after electroless Cu plating. After the Cu plating, the weight of the unpulverized autocatalyst increased by 0.20–1.56 pct, whereas the weight of the pulverized simulated autocatalyst increased by 1.80–3.66 pct.

Photographs of catalyst samples before and after electroless Cu plating: the unpulverized sample (Sim_PGM/CeZrO4_38) (a) before and (b) after plating; and the pulverized sample (Sim_PGM/CeZrO4_40) (c) before and (d) after plating
Figure 10 shows the SEM image and EDS analysis areas of the simulated autocatalyst (Sim_PGM/CeO2_22) processed with electroless Cu plating without pulverization. Table V presents the EDS analysis results for the numbered areas in Figure 10(a). Cu was deposited on the surface of the washcoat via electroless plating, and the Cu concentration decreased with depth from the surface of the washcoat. Figure 10(b) shows the EDS mapping of Cu on the cross section of the same sample. Cu was only deposited on the surface of the washcoat. These results suggest that the plating solution was not sufficiently supplied to the inside of the washcoat on the unpulverized simulated autocatalyst, thereby leading to the absence of Cu deposition inside the washcoat.

Secondary electron image of the cross section of the simulated autocatalyst (Sim_PGM/CeO2_22) (a) after elctroless Cu plating at 333 K (60 °C) for 90 min and (b) the composition map of Cu. EDS results for the numbered areas are shown in Table V
Sulfurization
Sulfurization of oxides
Table VI shows the weight changes of the samples during the sulfurization experiments. After the experiments, all the sulfur powder disappeared from the original position, and some of the sulfur vapor condensed on the low-temperature side of the reaction chamber. The weight change in C was extremely small, which suggests that the formation rate of CS2 gas was low under the current experimental conditions. This is consistent with what was reported in the literature [34,42].
Figure 11(a) shows the XRD pattern of the mixture of MgO, Al2O3, and SiO2 powders after the sulfurization experiment (Exp. no. M-S exp.6). Figure 11(b) shows the XRD pattern of the Cu powder that was processed with the mixture of MgO, Al2O3, and SiO2 powders. The results confirm that only Cu was sulfurized to Cu1.8S and CuS, whereas MgO, Al2O3, and SiO2 were not sulfurized.

XRD profiles of (a) the mixture of MgO, Al2O3, and SiO2 powder and (b) Cu powder after sulfurization at 850 K (577 °C) for 60 min (Exp. no. M-S exp.6)
Figure 12(a) shows the XRD pattern of the mixture of CeO2 and ZrO2 powders after the sulfurization experiment (Exp. no. M-S exp.7). Figure 12(b) shows the XRD pattern of the Cu powder that was processed with the mixture of CeO2 and ZrO2 powders. It was confirmed that only Cu was sulfurized to Cu1.8S and CuS, whereas CeO2 and ZrO2 were not sulfurized.

XRD profiles of (a) the mixture of CeO2 and ZrO2 powder and (b) Cu powder after sulfurization at 850 K (577 °C) for 60 min (Exp. no. M-S exp.7)
The CuS phase, which is not a stable species at 850 K (577 °C), was detected via XRD analysis. One possible reason for the detection of the CuS phase in the above experimental results is the CuS formation during the cooling step after sulfurization. Figure 13 shows the phase diagram of the Cu–S binary system.[46] Figure 13 indicates that in the S-rich (Xs ≥ 50 mol pct) region, Cu1.8S (s) and S (l) co-exist at 850 K (577 °C). CuS (s) can coexist with S (l) at temperatures below 780 K (507 °C). Therefore, it is considered that by including a large amount of sulfur, an S-rich atmosphere was formed during the sulfurization process, and Cu1.8S reacted with residual sulfur vapor to form CuS during the cooling step.

Calculated condensed T–XS phase diagram of the binary copper–sulfur system at a total pressure of 1 bar. Dg digenite (Cu1.8S), fcc face-centered cubic copper phase, hiCh high-temperature chalcocite (Cu2S), loCh low-temperature chalcocite (Cu2S), Cov covellite (CuS).[46]
The thermodynamic considerations indicate that MgO, and CeO2 may be sulfurized to MgS, and Ce2S3 during the sulfurization process, respectively. However, the experimental results showed that the typical oxide components of the autocatalyst were not sulfurized, and only Cu in the same system was sulfurized. One possible reason for these results is that, unlike the assumption in thermodynamic calculations, the partial pressures of oxygen, pO2, during the actual reaction does not decrease to the level of C (s)/CO2 (g) equilibrium; therefore, the equilibrium points in the phase stability diagrams shifted to the stable regions of MgO, CeO2, and ZrO2.
Sulfurization of simulated autocatalyst
Figures 14(a) and (b) show photographs of the simulated autocatalyst (Sim_PGM/CeO2_22) before and after the sulfurization experiment, respectively. After sulfurization, the color of the sample changed to blue-black. Figure 14(c-1) shows the XRD spectrum of the sample surface (Sim_PGM/CeO2_22) after electroless Cu plating and sulfurization (Exp. no. M-S exp.10). Figure 14(c-2) shows the XRD spectrum of the powdered sample (Sim_PGM/CeO2_22) pulverized using a ball mill after electroless Cu plating and sulfurization (Exp. no. M-S exp.10). Only Al2O3, CeO2, and CuS existed in the powdered sample, which indicates that CeO2, and Al2O3 in the simulated autocatalyst were not sulfurized in this sulfurization experiment, whereas the Cu deposit in the washcoat was sulfurized. No XRD peaks of the Cu1.8S phase were detected for the sulfurized simulated autocatalyst; however, peaks were detected for the sulfurized Cu powder. Honma reported that the deposition rate of Cu on a thin Cu film during electroless plating under the same conditions was 4 μm h−1.[29] In our experiments, the expected thickness of the Cu coating on the PGM particles after 90 minutes of electroless plating was approximately 6 μm, which is considerably small compared to the diameter of the Cu powder particles (~ 37.5 μm). Sulfurization of the thin Cu plating at 850 K (577 °C) can produce a thin Cu1.8S coating. It is predicted that during the cooling step after sulfurization, sulfur rapidly diffused in the thin Cu1.8S coating and converted all of the Cu1.8S into CuS.

Photographs of Cu-plated simulated autocatalyst (Sim_PGM/CeO2_22) (a) before and (b) after sulfurization at 850 K (577 °C) for 60 min (Exp. no. M-S exp.10). (c) XRD profiles of the samples after sulfurization at 850 K (577 °C) ((c-1): ‘Sim_PGM/CeO2_22’, (c-2): ‘Sim_PGM/CeO2_22’ pulverized with a ball mill.)
Figure 15 shows the SEM image and EDS analysis results of the cross-sections of the simulated autocatalyst (Sim_PGM/CeO2_22) after the sulfurization experiment (Exp. no. M-S exp.10). The EDS analysis results for the numbered areas in Figure 15(a) are listed in Table V. The sulfur concentration at the surface of the washcoat was high, and its concentration decreased with depth from the surface of the washcoat. The extent of the decrease in the sulfur concentration was smaller than that of the Cu concentration. As shown in Figure 15(b) and (c), the EDS mappings of Cu and S indicate that Cu was primarily deposited on the surface of the washcoat, whereas sulfur was distributed deep in the washcoat. No sulfides other than copper sulfides were identified in the XRD analysis; however, a small number of ceramic components were possibly sulfurized. Alternatively, the residual sulfur vapor or liquid S co-existing with copper sulfide at 850 K (577 °C) may have adhered to the fine pores in the washcoat as solid S during the cooling step.

(a) Secondary electron image of the cross section of simulated autocatalyst (Sim_PGM/CeO2_22) after electroless Cu plating and sulfurization at 850 K (577 °C) for 60 min (Exp. no. M-S exp.10) and the composition maps of (b) Cu and (c) S. EDS results for the numbered areas are shown in Table V
Flotation
Table VII shows the initial PGM concentrations (Ccat,i_est) in the simulated autocatalysts, weights of the separated samples in the flotation experiments, and PGM concentrations in each sample. The enrichment ratio (Fi) and recovery rate (Ri) for each experiment were evaluated using Eqs. [4] and [5], respectively, as summarized in Table VIII:
where Fi is the enrichment ratio for element i in the integrated process of electroless Cu plating, sulfurization, and flotation; Cfloat,i is the concentration of element i in the floating sample; Ccat,i_est is the concentration of element i in the initial simulated autocatalyst; Ri is the recovery rate of element i in the integrated process; and wfloat is the weight of the floating sample.
For the simulated autocatalysts that were not pulverized prior to electroless Cu plating, the enrichment ratios for PGMs in the current concentration process were between 2.1 and 7.4; however, the recovery rates were low, i.e., 5.5–8.8 pct. For the simulated autocatalysts that were pulverized prior to electroless Cu plating, the enrichment ratios for PGMs in the current concentration process were between 4.6 and 5.6, and the recovery rates were significantly high, i.e., 71–90 pct. Figure 16 shows photographs of the samples before and after the flotation experiment (Exp. no. Flot.exp.11), which exhibited the highest enrichment ratio and recovery rate. For the unpulverized simulated autocatalyst, the PGMs that existed on the surface of the washcoat were collected because Cu was electrolessly deposited only on the surface of the washcoat. The heat treatment after electroless Cu plating had no effect on the enrichment ratios and recovery rates. In this study, the flotation experiments were conducted using simple equipment and conditions. The optimization of the flotation process may result in a higher enrichment ratio.

Photographs of (a) sample before flotation. (b) froth and (c) tailing collected after flotation (Exp. no. Flot.exp.11)
The above experimental verifications confirm that we developed a new process for efficiently concentrating PGMs in autocatalysts. This process begins with electroless Cu plating, followed by sulfurization and flotation. This process utilizes harmless agents, such as glyoxylic acid as a reducing agent for electroless Cu plating and elemental sulfur as a sulfurizing agent. In principle, this process can be operated using small-scale facilities with a short processing time. Although process optimization and experimental verification on actual spent autocatalysts are required for industrialization, the results show that this process can improve the PGM grade in scrap at scrap-collection sites in the future. PGM-enriched scrap treated with the developed process can be transported more rapidly at a low cost compared to unprocessed scrap. This study contributes to a reduction in the cost and time associated with the PGM refining process, thereby leading to the promotion of the recycling of PGM-containing scrap.
Conclusion
This study develops an efficient concentration method for PGMs in spent autocatalysts, and a novel process comprised of electroless Cu plating and sulfurization was experimentally investigated as a pretreatment prior to flotation. It was demonstrated that the typical oxide components in the autocatalysts were not sulfurized under the current sulfurization conditions and that only Cu was selectively sulfurized. A series of experiments for the entire process, from the electroless Cu plating to flotation, enriched PGMs in the simulated autocatalysts, by up to 5.6 times and recovered 90 pct PGMs at the highest. The results were obtained using simulated samples prepared by the authors, and it is possible that the results would be different if real scrap were used, due to the effects of oil and other contamination. However, with that in mind, this new process exhibits considerable potential for the concentration of PGMs from spent autocatalysts.
References
J. Matthey: PGM Marker Report May 2020, 2020. http://www.platinum.matthey.com/services/market-research/pgm-market-reports
M.K. Jha, J. Lee, M. Kim, J. Jeong, B. Kim, and V. Kumar: Hydrometallurgy, 2013, vol. 133, pp. 23–32. https://doi.org/10.1016/j.hydromet.2012.11.012.
R. K. Mishra: in Pro. 17th Int.Precious Metals Conf. 1993, pp. 449–74.
F. Habashi (ed.): Handbook of Extractive Metallurgy, VCH Verlagsgesellschaft mbH, Weinheim, 1997, vol. III, pp. 1269–326.
F.K. Crundwell, M.S. Moats, V. Ramachandran, T.G. Robinson, and W.G. Davenport: Extractive Metallurgy of Nickel, Cobalt and Platinum-Group Metals, Elsevier, 2011.
H. Dong, J. Zhao, J. Chen, U. Wu, and B. Li: Int. J. Miner. Process., 2015, vol. 145, pp. 108–13. https://doi.org/10.1016/j.minpro.2015.06.009.
D.J. Aberasturi, R. Pinedo, I.R. Larramendi, J.I.R. Larramendi, and T. Rojo: Miner. Eng., 2011, vol. 24, pp. 505–13. https://doi.org/10.1016/j.mineng.2010.12.009.
R.J. Farrauto and R.M. Heck: Catal Today, 1999, vol. 51(3–4), pp. 351–60. https://doi.org/10.1016/S0920-5861(99)00024-3.
J. Kaspar, P. Fornasiero, and N. Hickey: Catal Today, 2003, vol. 77(4), pp. 419–49. https://doi.org/10.1016/S0920-5861(02)00384-X.
H.B. Trinh, J.C. Lee, Y.J. Suh, and J. Lee: J. Waste Manag., 2020, vol. 114, pp. 148–65. https://doi.org/10.1016/j.wasman.2020.06.030.
T.H. Okabe: Mater. Jpn., 2007, vol. 46(8), pp. 522–29. https://doi.org/10.2320/materia.46.522.
S. Owada, H. Seshimo, M. Miyashita, and K. Fujiwara: in Proc. 2nd Int. Symp. East Asian Resour. Recycl. Technol., 1993, pp. 69–77.
S. Owada, Y. Tsubuku, and H. Nakayama: in Proc. Spring Meeting, MMIJ, 1994, pp. 282–83. (in Japanese)
S. Owada, Y. Tsubuku, and T. Ayase: in Proc. Spring Meeting, MMIJ, 1995, p. 107. (in Japanese)
S. Owada, and K. Shinoda: in Proc. Spring Meeting, MMIJ, 2006, pp. 69–70. (in Japanese)
W. Kim, B. Kim, D. Choi, T. Oki, and S. Kim: J. Hazard. Mater., 2010, vol. 183, pp. 29–34. https://doi.org/10.1016/j.jhazmat.2010.06.038.
G. Liu, T. Ichinose, A. Tokumaru, and S. Owada: Mater. Trans., 2014, vol. 55(6), pp. 978–85. https://doi.org/10.2320/matertrans.M2014082.
G. Liu, T. Ichinose, and S. Owada: in Proc. Spring Meeting, MMIJ, 2012, pp. 206–207.
G. Liu, A. Tokumura, and S. Owada: Resources Process., 2013, vol. 60, pp. 28–35. https://doi.org/10.4144/rpsj.60.28.
T. H. Okabe and J. Mitsui: Japan Patent, No. JP2013-147702A, 2013, No. JP5946034, 2016. (in Japanese)
Y. Taninouchi, T. Watanabe, and T.H. Okabe: Mater. Trans., 2017, vol. 58(3), pp. 410–19. https://doi.org/10.2320/matertrans.M-M2017801.
Y. Taninouchi, T. Watanabe, and T.H. Okabe: Metall. Mater. Trans. B, 2017, vol. 48B, pp. 2027–36. https://doi.org/10.1007/s11663-017-0999-4.
Y. Taninouchi and T.H. Okabe: Mater. Trans., 2018, vol. 59(1), pp. 88–97. https://doi.org/10.2320/matertrans.M-M2017844.
Y. Taninouchi and T.H. Okabe: Metall. Mater. Trans. B, 2018, vol. 49B, pp. 1781–793. https://doi.org/10.1007/s11663-018-1269-9.
S. Kang, T. Ouchi, and Toru H. Okabe: in Proc. MMIJ, 2020. (in Japanese)
S. Shimizu: J. Surf. Finish. Soc. Jpn., 2007, vol. 58(2), pp. 81–6.
C.C. Sharma and J. Jung: J. Microelectron. Packag. Soc., 2016, vol. 23(4), pp. 1–6. https://doi.org/10.6117/kmeps.2016.23.4.001.
J. Darken: Trans. Inst. Metal. Finishi., 1991, vol. 69(2), pp. 66–9. https://doi.org/10.1080/00202967.1991.11870895.
H. Honma, M. Komatsu, and M. Hujinami: J. Surf. Finish. Soc. Jpn., 1991, vol. 42(9), pp. 913–17. https://doi.org/10.4139/sfj.42.913.
H. Honma and T. Kobayashi: J. Electrochem. Soc., 1994, vol. 141(3), pp. 730–33. https://doi.org/10.1149/1.2054800/meta.
H. Watanabe, K. Chiba, Y. Fukuda, and H. Kurokawa: J. Jpn. Inst. Interconnect. Packag. Electron. Circuits, 1995, vol. 10(2), pp. 118–22. https://doi.org/10.5104/jiep1995.10.118.
Y.Y. Shacham-Diamand: Electrochem. Solid-State Lett., 2020, vol. 3(6), pp. 279–82. https://doi.org/10.1149/1.1391124.
X. Xu and W. Sha: Appl. Surf. Sci., 2009, vol. 255, pp. 4259–266. https://doi.org/10.1016/j.apsusc.2008.11.015.
C. Stinn and A. Allanore: Nature, 2022, vol. 602, pp. 78–83.
J. Han, W. Liu, T. Zhang, K. Xue, W. Li, F. Jiao, and W. Qin: Sci. Rep., 2017, vol. 7, p. 42536.
J. Han, W. Liu, D. Wang, F.F. Jiao, and W. Qin: Metall. Mater. Trans. B., 2016, vol. 47B, pp. 344–54.
N. Chakraborti: Can. J. Chem. Eng., 1983, vol. 61, pp. 763–65.
B. Meyer: Elemental. Sulfur Chem. Rev., 1976, vol. 76(3), pp. 367–88.
T. Watanabe: Encyclopedia of the Elements, Asakura Publishing Co. Ltd, Shinjuku, 2007, p. 572.
I. Barin: Thermochemical Data of Pure Substances, 3rd ed. VCH Verlagsgesellschaft mbH, Weinheim, 1995.
O.K. Knacke and K. Hesselmann: Thermochemical Properties of Inorganic Substances, 2nd ed. Springer, Heidelberg, 1991.
H.N. Madon and R.F. Strickland-Constable: Ind. Eng. Chem., 1958, vol. 50(8), pp. 1189–192.
S. Yamamoto and M. Takaya: U.S. Patent, No. US 2002/0197192 A1, 2002.
S. Yamamoto and M. Takaya: Japan Patent, No. JP2003-53152A, 2003. (in Japanese)
M. Etoh and H. Tokumori: Bunseki Kagaku, 1985, vol. 35, pp. T39-T42. (in Japanese).
P. Waldner: Metall. Mater. Trans. B, 2020, vol. 51B, pp. 805–17. https://doi.org/10.1007/s11663-020-01796-x.
Acknowledgments
The authors are grateful to Professor Yu-ki Taninouchi (Kyushu University), Akinari Suzue (Sumitomo Metal Mining Co., Ltd), Dr. Gen Kamimura, Dr. Akihiro Iizuka, Mr. Kenta Akaishi, and Mr. Hiromu Hiramatsu (The University of Tokyo) for their valuable suggestions and comments. The authors would like to thank Professor Shunsuke Yagi (The University of Tokyo) for his help with the particle size distribution analysis. This work was financially supported by the Japan Society for the Promotion of Science (JSPS) through a Grant-in-Aid for Scientific Research (S) (KAKENHI Grant No. 26220910 and 19H05623) and a Grant-in-Aid for Scientific Research (B) (KAKENHHI Grant No. 20H02493).
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Funding
Open access funding provided by The University of Tokyo.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kang, S., Ouchi, T. & Okabe, T.H. Novel Concentration Process for Platinum Group Metals in Automotive Exhaust Catalyst Using Electroless Copper Plating, Sulfurization, and Flotation. Metall Mater Trans B 55, 32–52 (2024). https://doi.org/10.1007/s11663-023-02880-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-023-02880-8