
Abstract
In a study on parts of the system Fe2O3-CaO-Al2O3-MgO, the previously unknown compound Ca3MgAl6Fe10O28 or FCAM-I (iso-structural with SFCA-I) has been synthesized. The two principal aims of our investigations have been (i) analysis of the stability field of the new phase as a function of T and fO2 and (ii) determination of its crystal structure. Two experimental series in air and under controlled oxygen fugacity via the hematite-magnetite buffer were conducted. Pure polycrystalline FCAM-I has been obtained at 1463.15 K (1190 °C) in air. While increasing the temperature from 1573.15 K to 1673.15 K (1300 °C to 1400 °C), the FCAM-I phase breaks down forming a variety of new compounds depending on T and fO2. Ca3MgAl6Fe10O28 has a triclinic crystal structure (space group P \( \overline{1} \)). Basic crystallographic data are as follows: a = 10.2980(4) Å, b = 10.4677(4) Å, c = 11.6399(4) Å, α = 94.363(3)°, β = 111.498(3)°, γ = 109.744(3)°, V = 1069.81(7) Å3, Z = 2.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
SFCA is an acronym that was coined by Hancart et al.[1] rather generally for silico-ferrites of calcium and aluminum in order to describe the most important bonding constituents forming during the production of industrial iron ore sinters. Later, in a sequence of papers[2,3,4,5] Mumme and co-workers used this term more specifically for the description of several crystallographically different but structurally closely related phases whose chemical composition can be given as M14+6nO20+8n (where M = Si, Fe, Al, Ca, Mg, and n = 0 to 1). From a structural point of view, these compounds form a homologous series and the members with n = 0 and n = 1 were designated SFCA and SFCA-I, respectively. Furthermore, a SFCA-II phase has been mentioned representing an intermediate of SFCA and SFCA-I.[6] The stability regions of these compounds are only partially well characterized using in-situ synchrotron X-ray diffraction. The formation temperature[6] of SFCA-I via the reaction of SFCA + Fe2O3 was determined at 1392.15 K (1119 °C) by pO2 = 5 × 10−3 atm. Webster et al.[6] postulated that the differences in the temperature of formation can directly be related to variations in the oxygen fugacity and/or heating rate.
SFCA and SFCA-I show complex solid solution series involving a substitution mechanism based on the exchange of 2(Fe3+, Al3+) ↔ (Ca2+, Fe2+) + Si4+,[7] in which the increase of Al2O3 concentration has a positive effect of the stability field of SFCA-I at lower temperatures.[6] Furthermore, an increasing replacement of Fe3+ by Al3+ with increasing temperature has been observed. Depending on the chemical system, the two phases SFAC and SFCA-I can coexist in which SFCA-I is potentially more stable at higher temperatures. SFAC decomposes around 1743.15 K (1470 °C) completely and is progressively unstable with decreasing Al2O3 content.[7] Mumme[5] proposed that the three homologous SFCA structure-types have a much more limited temperature range than the so far suggested 973.15 K to 1743.15 K (700 °C to 1470 °C).
Triggered by previous studies on SFCA-related materials, this investigation focused on the influence of Mg incorporation in a silicon-free SFCA-I analogue. Adapting the nomenclature suggested by Mumme, the Mg-containing compounds will be labeled “FCAM-I.” According to Kalenga and Garbers-Craig,[8] the physico-chemical properties of iron ore sinters depend noticeably on the MgO content, which is due to the addition of Mg-containing fluxes such as dolomite or fused MgO during the sintering process. The magnesium oxide concentration, for example, is positively correlated with the abrasion and tumble indices as well as the coke rate. The present study focuses on the structural investigations on FCAM-I or Ca3MgAl6Fe10O28 (corresponding to the Si-free version of SFCA-I with an MgO content of up to 2.8 mol pct). Moreover, based on the quaternary system Fe2O3-Al2O3-CaO-MgO, a systematic examination of the T-fO2 stability of this FCAM-I phase with respect to the coexisting phase assemblages was performed.
Experimental Methods
Synthesis of FCAM-I
Synthesis experiments of silicon-free SFCA-I with a magnesium substitution of one atom per formula unit (apfu) were based on the molar ratio CaO:MgO:Al2O3:Fe2O3 = 3:1:3:5, which corresponds to a composition of Ca3MgAl6Fe10O28. Starting materials for the solid; state reactions were dried Fe2O3 (>99 pct), γ-Al2O3 (99,997 pct), MgO (99,998 pct), and CaCO3 (>99 pct) which were checked for phase purity using XRPD. Subsequently, the separate powders were mixed and homogenized under ethanol in a ball mill for 45 minutes.
Approximately 0.5 g of the powdered mixture was dried again and pressed into pellets with a diameter of 13 mm. In a first set of experiments, the pellets were sintered in air in the temperature range between 1473.15 K and 1673.15 K (1200 °C and 1400 °C) using an open Pt crucible placed in a chamber furnace. The samples were heated from room temperature up to the respective maximum temperature with a heating rate of 1.5 K/minutes. 20 K below the final temperature, the heating ramp was reduced to 0.5 K/minutes in order to avoid overheating of the sample. The maximum temperature was held for 24 hours. Afterwards, the samples were rapidly quenched to room temperature by placing the bottom of the Pt crucible into a water bath.
The second series of experiments was conducted using a platinum double-capsule geometry (Figure 1) controlling the oxygen fugacity via the hematite-magnetite buffer. The assembly consists of an outer Pt capsule with a length of 25 mm and an outer diameter of 5 mm containing the inner Pt capsule surrounded by the Fe2O3/Fe3O4 buffer. The container was filled only up to two-thirds of the sample volume. The inner capsule contains the pressed starting mixture which was previously heated to 1073.15 K (800 °C) for 12 hours for decarbonization. The temperature conditions as well as the heating ramps were the same as in the previous “in-air” experiments. In order to monitor temperature conditions as precisely as possible, a second external Pt-Pt90Rh10 thermocouple was placed on top of the sample.

Outline of the platinum double-capsule geometry
After the experiments, the buffer assemblage was analyzed by X-ray powder diffraction. The ratio between hematite and magnetite was calculated by quantitative phase analysis based on the Rietveld method using the software suite TOPAS 4.2.[9] Between 1473.15 K and 1683.15 K (1200 °C and 1410 °C), hematite (H) and magnetite (M) coexist in the experiments. The resulting oxygen fugacity was calculated by the model Reaction (A)
and the following thermodynamic relations (B)
at standard condition (1 atm) by using the thermodynamic data from Robie and Hemingway.[10]
At 1473.15 K (1200 °C), the hematite/magnetite ratio corresponds to 99:1 (HM). Above these temperatures, magnetite starts to form and the hematite-magnetite buffer is in equilibrium. The calculated \( \log_{10} P_{{{\text{O}}_{2} }} \) increases from −3.67 at 1473.15 K (1200 °C) to −1.48 at 1683.15 K (1410 °C). Exceeding 1683.15 K, hematite disappears and the experiments move into the stability field of magnetite (0.0 pct H to 100.0 pct M). Consequently, \( P_{{{\text{O}}_{2} }} \) drops down to \( \log_{10} P_{{{\text{O}}_{2} }} \) < −1.13 at 1723.15 K (1450 °C).
Single crystals of FCAM-I have been obtained somewhat serendipitously during previous experiments aiming on the preparation of Mg-containing C2F with a composition Ca1.9Mg0.1Fe2O5 in open corundum crucibles in air using the same starting materials as for the solid-state reactions. Two grams of the stoichiometrically weighed powders were homogenized in an agate mortar, transferred into an open 50 ml corundum crucible, and placed in a resistance-heated chamber furnace. Heating experiments were performed in air. The sample container was fired from 373.15 K (100 °C) with a rate of 200 K/hour to 1623.15 K (1350 °C), subsequently cooled with 1 K/hour to 1573.15 K (1300 °C), and finally quenched to ambient conditions. First visual inspection of the synthesis product revealed that the sample was highly crystalline. Furthermore, it was obvious that (i) the crystals had grown from a melt phase and (ii) reactions between the educts and the Al2O3 component of the crucible had occurred. The simultaneous presence of optically transparent and almost opaque crystals pointed to the existence of a complex phase mixture. The colors of the crystals ranged from honey-colored through reddish-brown to dark-brown or almost black. Concerning the sizes, the dark-brown compound was especially conspicuous: the lath-shaped crystals reached maximum dimensions of up to 1 mm and proved to be FCAM-I. Furthermore, single crystals of Ca(Al,Fe)O4 and Ca(Al,Fe)4O7 could be detected.
Subsequently, the resulting FCAM-I samples were analyzed by X-ray powder diffractometry, single-crystal X-ray diffraction, and EMPA.
X-ray Powder Diffraction
The BRUKER-AXS D8-Advance powder diffractometer used (Bragg–Brentano geometry with θ–θ coupling) is equipped with parallel beam optics and an energy-dispersive Si(Li) semiconducting Sol-X detector yielding CuKα1 and CuKα2 radiation. The settings for the recorded powder diffraction data were 40 kV and 40 mA and a LYNXEYETM silicon strip detector. Data were collected at ambient temperature in the 2θ range between 3 and 100 deg with a counting time of 10 seconds/step and a step size of 0.02 deg 2θ. For phase identification, LeBail fits using TOPAS 4.2[9] and the current version of the PDF-4+[11] database were used. The peak profiles in the fits were calculated using the fundamental parameter approach.[12] The background was simulated by Chebyshev polynomials of the 5th order.
Electron Microprobe Analysis
In order to obtain the compositions and to study phase relations of the experiments, part of the synthesized material was embedded in epoxy resin and polished with diamond paste (10, 5, and 1 μm grain size). Electron microprobe analysis in wavelength dispersive mode was performed using a JEOL JXA SUPERPROBE 8100. Measurement conditions were 15 kV acceleration voltage and 10 nA beam current. The measurement time for the peaks was 20 and 10 seconds for the background. A total number between 1 and 3 spots in various regions of the crystals were analyzed. The standard reference materials used for analysis of Ca, Al, Fe, and Mg were diopside (Ca), corundum (Al), hematite (Fe), magnetite (Fe), and magnesia (Mg). Intensities obtained were corrected for electron scattering, absorption, and fluorescence radiation (so-called ZAF correction). Finally, the Fe2+/Fetot ratio was calculated from the total iron content based on the crystal-chemical formula obtained from EMPA measurements and charge balance considerations.
Single-Crystal X-ray Diffraction
The basic crystallographic data at ambient temperature (293.15 K (20 °C)) of a sample retrieved from the single-crystal growth experiment were obtained by single-crystal diffraction using an Oxford Diffraction Gemini R Ultra single-crystal diffractometer. Therefore, a reddish fragment (210 × 100 × 90 µm) of a single crystal was mounted on top of a glass fiber. For the structure determination of the triclinic compound, a total of 583 frames using ω-scans with 1 deg scan width per frame covering a hemisphere of reciprocal space were collected.
The integration and data reduction were performed using the CrysAlis Pro software package[13] including corrections for Lorentz and polarization effects. Furthermore, an analytical absorption correction using accurately measured external faces has been employed. X-ray scattering factors together with real and imaginary coefficients for anomalous dispersion were taken from the International Tables for Crystallography, Vol. C.[14] The structure was solved in space group P \( \overline{1} \) using direct methods via the program SIR2004.[15] The final least-squares refinement using anisotropic displacement parameters for all atoms was performed with the program SHELX-97[16] embedded in the WinGX software suite.[17] The calculations converged to a residual of R1 = 0.0258 for all reflections with I > 2σ(I) using 450 parameters (Table I).
Since (i) the assignment of more than two chemical species to a specific position is mathematically undetermined and (ii) Mg and Al have almost identical scattering factors for X-rays, for site occupancy analysis on the different octahedral and tetrahedral sites the following procedure has been selected: Initially, the magnesium content was completely neglected and the total scattering power for each site was obtained by allowing for a potential Fe ↔ Al substitution under the assumption. For two of the three calcium positions in the asymmetric unit site, occupancy refinements indicated full occupation within 2 standard uncertainties. However, for the remaining “Ca-position” M2 the total scattering power was significantly less than 20 electrons pointing to a more complex cation distribution. A detailed analysis of the site populations, i.e., the assignment of different chemical species (Fe, Al, Mg, Ca) to each position was performed a posteriori using the program OccQP[18] where the full information from chemical analysis, refined total scattering powers for the positions, and bond distances is considered to find an optimal distribution. The mathematical algorithm is a quadratic numerical approach based on the least-squares method and a weighted linear system. The strength of the program is the minimization of the residual between formal and apparent valences for an acceptable crystal-chemically solution.[18]
Analysis of the atomic coordinates has proven the crystal structure of FCAM-I to be iso-structural with SFCA-I. For sake of clarity, labeling of the atoms in the asymmetric unit was therefore performed according to the already published crystal structure of SFCA-I.[4] The refined atomic coordinates, equivalent isotropic and anisotropic displacement parameters, as well as selected interatomic distances are listed in Tables II, III, and IV. Figures showing structural details were prepared using the program ATOMS6.4.[19]
TGA: DTA Analysis
Approximately 14 mg of the starting mixture was studied in an open platinum crucible by thermogravimetric (TGA) and differential thermal (DTA) analysis. The crucible was made out of a Pt tube with a diameter of 3 mm which was placed in an open corundum crucible (100 ml). The experiment was performed in a Setaram Setsys Evolution 2400 thermal analyzer. The device is coupled with a mass spectrometer OmniStar GSD-301 02 (Pfeifer Vacuum, Asslar, Germany). The sample holder is equipped with Pt/Pt90-Rh10 thermocouples. Temperature was increased with a ramp of 5 K per minute from room temperature to 1773.15 K (1500 °C). The experiments were done in air, in a helium atmosphere (99.999 pct), and in an oxygen atmosphere (99.995 pct) using a gas flow of 20 ml per hour.
Results
Crystal Structure
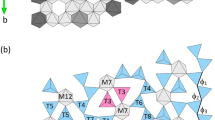
The crystal structure of FCAM-I is iso-structural with SFCA-I, whose structural features have been previously determined by Mumme et al.[4] Basic crystallographic data on FCAM-I are as follows: triclinic symmetry, space group P \( \overline{1} \), a = 10.2940(3) Å, b = 10.4393(3) Å, c = 11.5977(6) Å, α = 94.342(6) deg, β = 111.611(6) deg, γ = 109.617(3) deg, V = 1069.81(7) Å3, Z = 2. The structure can be built up from an alternating sequence of two different types of layers running parallel to (20-1) (see Figure 2(a)).

(a) Projection of the whole crystal structure of FCAM-I parallel to (20-1). Fundamental layers I (b) and II (c) are given in projections perpendicular to (20-1). For details, see text
Layer-type I contains MO6 octahedra as well as TO4 tetrahedra (T: Al, Fe, Mg), whereas layer II is exclusively based on MO6 octahedra (M: Ca, Fe, Al, Mg). Views of both sheets in projections perpendicular to the mean planes of the layers are given in Figures 2(b) and (c). Within layer I, twelve octahedral wide bands parallel to [010] can be identified. Linkage between the octahedra is provided by edge-sharing. The tetrahedra and octahedra of layer II (Figure 2(b)) share common corners. In more detail, this sheet can be subdivided into (i) vierer single chains of tetrahedra and (ii) ribbons containing octahedra and tetrahedra in the ratio 1:2. Again, both one-dimensional elements are running parallel to the b-axis. As can be seen from Figure 2(a), the “free” vertices of all tetrahedra in a specific chain point into the same direction, i.e., the sequence of directedness is either up or down. The fundamental units of the mixed-polyhedral bands are MT2O12 clusters, where the MO6 moieties, share trans-vertices with the adjacent two tetrahedral groups. It is this group that Mumme et al.[4] referred to as “winged octahedra.” One of these two tetrahedra resides above the hexagonally shaped vacancy of the neighboring octahedral sheet.
Notably, there is no direct connection between adjacent bands within the same octahedral layer. The ribbons are linked by (i) the three basal oxygens of the tetrahedral chains located directly above and below, respectively, and (ii) the octahedra of the MT2O12 clusters (by sharing common edges).
As mentioned above, the assignment of different chemical species to individual sites was performed by the program OccQP[18] using the chemical composition obtained by EMPA (corresponding to Ca2.90Mg0.95Fe10.11Al5.99O28) and the results of the structure refinement as input. Based on a quadratic approach, the program is able to assign occupancies at multiply occupied atom positions (Figure 3) in a structure.[18] Accordingly, nine of the twelve octahedral sites are preferentially occupied by Fe3+, (M4-M12), while calcium is predominantly located on the remaining three positions (M1 to M3). Moreover, the results indicate that the pure calcium positions M1 and M3 are located at the outer rims of the octahedral bands (see Figure 2(c)). The M2 site, in turn, occupying the center of the ribbons contains a larger amount of calcium cations but also magnesium and iron ions. Furthermore, it is the only position where the presence of a significant quantity of vacancies was suggested. Notably, in SFCA-I this site has been assigned to be fully occupied with Ca.
Most of the symmetrically independent TO4 groups show a clear preference for a specific cation type. Al3+ ions, for example, are preferentially located on the four tetrahedra (T4, T6, T7, T8) comprising the tetrahedral chains. Two of the tetrahedra, T3 and T5 belonging to the winged octahedra show a definite enrichment in iron, whereas the cation distribution on the remaining two tetrahedral sites (T1,T2) is more flexible. It is worth noting that the suggested cation distribution shows no clear preference of magnesium for a specific site. The single magnesium atom per formula unit is structurally distributed among several sites including tetrahedral and octahedral coordination environments. However, no magnesium incorporation could be detected on the tetrahedra forming the chains (Figure 3).

Optimized site populations obtained from the application of the program OccQP (Color figure online)
Based on results of the single-crystal structure analysis, the powder pattern of the polycrystalline material was studied by a LeBail fit[20] (see Figure 4) indicating that the materials obtained from the solid-state reactions at 1473.15 K (1200 °C) and the single-crystal growth experiments are equivalent. Moreover, no obvious unexplained peaks remained, strongly suggesting that FCAM-I has been synthesized in phase pure form.

Comparison between the observed X-ray powder diffraction pattern (blue) of the 1473.15 K (1200 °C) experiment and a LeBail fit (red) corresponding to the unit cell parameters obtained from the single crystals of FCAM-I. Tick marks for the Bragg peaks are also indicated. The gray line represents the difference curve between observed and calculated step intensities (Color figure online)
Stability Relations of FCAM-I
XRPD Measurements (Experiments in Air)
The phase evolution as a function of temperature was followed by a series of powder diffraction patterns, which were collected ex situ after quenching the samples from the respective maximum reaction temperature (see Figure 5). According to these measurements, FCAM-I seems to be stable up to a temperature of about 1573.15 K (1300 °C). Differences in relative intensities and peak positions between the patterns acquired for the samples prepared at 1473.15 K and 1573.15 K (1200 °C and 1300 °C) are not very pronounced and may be due to slightly different cation distributions on the various sites. The diffractogram for the 1673.15 K (1400 °C) sample, however, reveals drastic changes involving shifts of reflections as well as appearing of new and disappearing of already existing peaks. Three regions (at about 8, 35, and 57 deg 2θ) where these effects are especially obvious have been highlighted in Figure 5. Notably, the new isolated low-angle peaks at about 6.6 and 9.5 deg 2θ cannot be explained with unit cell metric of FCAM-I. There are at least two possible explanations for these observations: (i) a disintegration of FCAM-I into an assemblage of new phases or (ii) a phase transition of FCAM-I into a new high-temperature polymorph. At slightly higher temperatures (1698.15 K (1425 °C)), the reaction seems to be complete, i.e., the characteristic reflections of FCAM-I are barely visible.

Powder XRD patterns of samples from selected synthesis experiments 1473.15 K, 1573.15 K, 1673.15 K, and 1698.15 K (1200 °C, 1300 °C, 1400 °C, and 1425 °C) showing a shift of selected peak positions and a decrease in their intensities with increasing temperature. This compares well with the EMPA data and the change in the chemical composition of FCAM-I. At 1673.15 K and 1698.15 K (1400 °C and 1425 °C), the XRD diagrams at low 2θ angles confirm the presence of a newly formed phase
TGA: DTA Results
In addition, the thermal behavior of FCAM-I was investigated using TGA analysis under a controlled gas flow of air. Notably, after decarbonization of the starting composition between 873.15 K and 1043.15 K (600 °C and 770 °C), no clear exothermic peak is visible (Figure 6). However, the curve increases to a maximum at 1466.15 K (1193 °C). In contrast, the experiment in He atmosphere reveals a slightly more pronounced exothermic maximum in nearly the same temperature region (1441.15 K (1168 °C)). Under oxygen atmosphere, the exothermic maximum is shifted to slightly higher temperatures and can be located at 1485.15 K (1212 °C).

Differential thermal and thermogravimetric analysis of the FCAM-I starting mixture heated by 5 K/min in air, He, and O2 atmosphere
After 1466.15 K (1193 °C), the synthesis in air provides strong evidence for the stable occurrence of FCAM-I since the heat flow shows a constant negative or more precisely an endothermic gradient with a significant change in the slope at 1588.15 K (1315 °C). Contemporaneously above 1573.15 K (1300 °C), the TGA curve shows a gradual loss of mass. By heating the sample above 1685.15 K (1412 °C), a small exothermic maximum followed by melting accompanied with a significant weight loss at 1713.15 K (1440 °C) can be observed. In analogy, the curve measured in He gas has a similar shape with a substantial shift of the melting point to lower temperatures down to 1646.15 K (1373 °C). There is no significant exothermic maximum during the melting process in the O2 atmosphere, but it can be determined by the DTA curve during cooling. The mass loss due to the reduction of Fe2O3 to FeO and into pure iron occurs at lower temperatures of 1310.15 K (1037 °C).
The TG-DTA results in controlled O2 atmosphere clearly show similarities with the experiments in air. The formation of melt at 1727.15 K (1454 °C) occurs later and simultaneously the loss of mass accompanying the reaction of hematite to magnetite is negligible when compared with the other two experiments.
The synthesis experiments in the furnaces clearly correspond to the TG-DTA experiments in air. FCAM-I was not observed in the synthesis experiments below 1473.15 K (1200 °C). The synthesis experiments also disclosed that iron(III)oxide starts to be reduced to iron(II)oxide at temperatures between 1573.15 K and 1673.15 K (1300 °C and 1400 °C). Concurrently, at 1573.15 K (1300 °C) the FCAM-I phase breaks down forming a complex assemblage of new phases. Finally, the FCAM-I phase disappears completely at temperatures above 1673.15 K (1400 °C).
EMPA Results
Electron microprobe analysis clearly supports the previous results. Evaluation of the samples using backscatter electron (BSE) images yields the typical polycrystalline granular microstructure in the 1473.15 K (1200 °C) experiment (Figure 7(d)). After 24 hours, the crystallite size is 10 to 20 µm. The size increases from 10 µm in the 1473.15 K (1200 °C) experiments to about 100 µm in the highest temperature experiments performed at 1673.15 K (1400 °C). Exceeding 1673.15 K (1400 °C) melt dominates in the samples and the FCAM-I phase disintegrates completely. Chemical analysis confirms a homogenous composition of FCAM-I phases below 1573.15 K (1300 °C) which corresponds with the stoichiometric end-member composition. Increasing temperatures lead to a reduction of Fe3 to Fe2+ in FCAM-I (Figure 8; Table V). This is also accompanied by a decrease in the calcium content in FCAM-I and the formation of new calcium-rich Fe-Al-oxide phases. These new phases are solid solutions between calcium aluminates and calcium ferrites. During this reaction, the aluminum contents increase (Figure 9). The calculated Fe2+ concentrations of FCAM-I increase from 0 atoms per formula unit (apfu) up to 0.59 apfu. The concentration of calcium decreases considerably from 3.08 apfu down to 1.79 apfu (FCAM-I → FCAM-Ilow). Consequently, an increase in magnesium in the FCAM-I phase occurs. At 1573.15 K (1300 °C), FCAM-I starts to break down and reaction textures occur (Figure 7(e)), which can be explained with the following model Reaction (1).

BSE images of the experimental series with samples prepared at 1573.15 K, 1623.15 K, and 1673.15 K (1300 °C, 1350 °C, and 1400 °C) using the platinum double-capsule technique with a controlled oxygen fugacity via the hematite-magnetite buffer (a and c). (a) Illustrates the newly formed SFCA-I in a matrix of unreacted starting material. The disintegration of the polycrystalline FCAM-I phases starts at 1623.15 K (1350 °C) (b) and the formation of MA0.375F0.625 at temperatures above 1673.15 K (1400 °C) (c). For comparison, the BSE image of the 1473.15 K, 1573.15 K, and 1673.15 K (1200 °C, 1300 °C, and 1400 °C) experiments in air are shown in d through f. The first BSE image shows fine-grained, polycrystalline FCAM-I phases synthesized at 1473.15 K (1200 °C) in air (d). Disintegration of FCAM-I with a typical crystallite size of 10 to 20 µm starts at 1573.15 K (1300 °C). Simultaneously significant crystal growth of FCAM-I can be observed. The newly formed phases are shown in (e). The last BSE image (f) shows the 1673.15 K (1400 °C) experiment. The main phases in the sample are quenched materials, which are mostly solid solutions of C2A0.25F0.75 and CA0.75F1.25. Also clearly visible is the disintegration of FCAM-I and the new formation of MA0.375F0.625

Plot of Fe2+ content [apfu] in FCAM-I vs temperatures

The Fe2O3-CaO-Al2O3 (FCA) triangle (a) illustrates the starting composition and the chemical variation of FCAM-I phases as a function of temperature.[21] Also plotted (b) are the different quench phases observed in the samples which have been heated above 1573.15 K (1300 °C)
For comparison, the formation of FCAM-I in the platinum double-capsule technique with a controlled oxygen fugacity via the hematite-magnetite buffer begins at higher temperatures. Hence, the stability field of FCAM-I shifts to significantly higher temperatures. In the first experiment at 1473.15 K (1200 °C), no significant reaction occurs in the starting mixture. The BSE image (Figure 7(a)) and the EMPA analysis of the 1573.15 K (1300 °C) experiment show the formation of FCAM-I in a finely granulated matrix of the initial starting mixture. FCAM-I forms crystals with a size of ca. 100 µm. In contrast to the experiments in air, the FCAM-I phase is stable up to 1573.15 K (1300 °C). After heating the sample by another 50 K, the first breakdown textures of FCAM-I can be observed (Figure 7(b)). Equivalent to the experiments in air increasing the temperature leads to a decrease of the calcium content of FCAM-I. However, the positive shift of the aluminum content starts only after this heating step. At temperatures above 1673.15 K (1400 °C), the FCAM-I phase shows a significant increase of Al concentration from 6.08 to 6.92 apfu. The calculated Fe2+ content of the FCAM-I phase in this series is around 0.40 and 0.56 apfu.
The first quenched materials in the intergranular space have a chemical composition in the system CF-CA with the actual composition of Ca1.10Fe1.32Al0.65O4 (CA0.25F0.75). Between FCAM-I and the intergranular spaces are small quenched phases which exhibit a typical needle-like habitus. The chemical analyses correspond to the system of the so-called CF3-CA3 phases with the exact composition of Ca1.05Al1.95Fe4.00O10 or CAF2. In both phases, the calculated Fe2+ concentration is zero. The phases CA0.25F0.75 and CAF2 were detected at 1573.15 K (1300 °C) in air and in the platinum capsule experiment at 1623.15 K (1350 °C) (Figures 7(b) and (e)).
The synthesis in air proves that with increasing temperature the phase in the intergranular space changes the composition from Ca1.10Fe1.32Al0.65O4 to Ca1.97Al0.58Fe1.52O5 corresponding to a solid solution between the phases C2F and C2A with calculated Fe2+ contents of 0.05 apfu. The formation of iron (II) shows a change in the fO2 environment from oxidizing conditions at 1573.15 K (1300 °C) to slightly reducing settings at 1673.15 K (1400 °C) as indicated by model Reaction (2). Nevertheless, EMPA of the 1673.15 K (1400 °C) experiments confirm the presence of the CF2-CA2 phase with a composition of Mg0.01Ca0.94Al1.25Fe2.80O7 or simplified CaAlFe3O7 (CA0.75F1.25). The phase shows a similar needle-like habitus like the CAF2 phase.

The textures in the BSE image of Figure 7(f) clearly demonstrate the breakdown of FCAM-I to CAF2 and CA0.75F1.25. The calculated Fe2+ content in the CA0.75F1.25 phase is about 0.11 apfu.
The two phases C2A0.25F0.75 and CA0.75F1.25 could not be detected in the experimental series under controlled oxygen fugacity. At temperatures exceeding 1673.15 K (1400 °C) but prior to melting, the complete breakdown of FCAM-I to a newly formed phase (MA0.375F0.625) occurs (Figure 7(f)) as indicated by model Reactions (3) and (4). The calculated chemical composition based on 8 oxygens and 6 cations corresponds to \( {\text{Mg}}_{0. 5 1} {\text{Ca}}_{0.0 4} {\text{Fe}}_{0. 4 5}^{ 2+ } {\text{Fe}}_{1.16}^{3 + } {\text{Al}}_{0. 8 4} {\text{O}}_{ 4} {\text{or }}\left( {{\text{Mg}},{\text{Fe}}^{ 2+ } } \right)\left( {{\text{Al}},{\text{Fe}}^{ 3+ } } \right)_{ 2} {\text{O}}_{ 4} \). The stability field of this new spinel-type magnesium-aluminum-ferrite shows no correlation with oxygen fugacity (Figures 7(c) and (f)). In the double-capsule experiment as well as in the synthesis in air, MA0.375F0.625 can be detected at temperatures above 1673.15 K (1400 °C). It is chemically very homogenous and exhibits only a very small standard deviation of ±0.01 apfu in magnesium, aluminum, and iron. The calculated Fe2+ content is about 0.90 apfu.
Discussion
FCAM-I can be synthesized at temperatures above 1463.15 K (1190 °C) in air. This phase breaks down (Figure 7) in two steps: at 1583.15 K (1310 °C) due to the release of Ca, Fe, Al (FCAM-I → FCAM-Ilow) and the formation of new calcium-aluminum-ferrites (CA0.25F0.75 and CAF2). In the second step, at temperatures above 1683.15 K (1410 °C) FCAM-I disappears completely by forming magnesium-aluminum-ferrites (MF-MA) and subsequently melt. The growth of CAF2 and MA0.375F0.25 is predominantly a function of increasing temperatures. In contrast, increasing the oxygen fugacity leads to a positive correlation with the stability field of FCAM-I and hence the fO2 concentration has a significant influence on the nature of the newly formed components. Reducing conditions (decreasing O2 fugacity) favor the reduction of Fe3+ to Fe2+ and the formation of the phases C2F-C2A (C2A0.25F0.75) as well as CF2-CA2 (CA0.75F1.25).
The EMP analyses in conjunction with the textures observed in the BSE images clearly demonstrate that the calcium content in FCAM-I decreases with increasing temperature and simultaneously new calcium-rich phases appear. This suggests that FCMA-I still remains stable up to higher temperatures by simply adjusting its composition with respect to Ca and Al. Bond distance calculations indicate that the M2O6 polyhedron located in the center of the fundamental bands of layer-type I (Figure 2(a)) is significantly smaller (Table IV) compared to the octahedral sites around M1 and M3 (〈M-O〉 ~2.23 vs ~2.41 Å, respectively). On the other hand, site occupancy analysis indicates that the M2O6 octahedron shows a mixed cation population, whereas the octahedra M1 and M3 are almost exclusively occupied by calcium cations, indicating that the incorporation of calcium on M2 is the limiting factor for the incorporation of Ca2+ into the FCAM-I structure. Hence at higher temperatures, FCAM-I has presumably an ideal total calcium content of 2 calcium apfu that favors the stability of the structure (see Table VI).
Comparing the investigations using TG-DTA data acquired under different atmospheres demonstrates that the stability field of FCAM-I is mostly controlled by the concentration of Fe2+. Compared to the experiments in air, higher oxygen fugacity expands the stability field of FCAM-I of only about 30 K to higher temperatures Table VI.
Under atmospheric conditions in which the formation of Fe2+ is favored, the breakdown of FCAM-I shifts to a significantly lower temperature from 1683.15 K (1410 °C) down to 1622.15 (1349 °C) in He atmosphere—and concurrently the stability field of FCAM-I decreases noticeably.
It appears that with the increasing Fe2+ concentration in FCAM-I the charge balance can be maintained by the release of Ca2+. This observation is based on the increase of Fe2+ (≫0.6 apfu) and a simultaneous release of calcium from the M2O6 octahedra. Additional deficit of Ca on the octahedra around M1 and M3 (≪1.8 apfu) then ultimately leads to the breakdown of the FCAM-I structure.
References
J. Hancart, V. Leroy, A. Bragard: CNRM Metall, 1967, Report, pp. 3–7.
W. Mumme: N. Jb. Miner. Mh., 1988, pp. 359–66.
J. Hamilton, B. Hoskins, W. Mumme, W. Borbidge: Neues Jb. Miner-Abh., 1989, 161(1) 1-26.
W. Mumme, J. Clout, R. Gable: Neues Jb. Miner-Abh., 1998, pp. 93–117
W. Mumme: Neues Jb. Miner-Abh., 2003, 178 3 307-335.
N.A. Webster, M.I. Pownceby, I.C. Madsen, J.A. Kimpton: Metallurgical and Materials Transactions B, 2012, 43(6) 1344-1357
T.R. Patrick, M.I. Pownceby: Metallurgical and Materials Transactions B, 2002, 33(1) 79-89
M. Kalenga, A.M. Garbers-Craig: Journal of the South African Institute of Mining & Metallurgy, 2010, 110(8) 447
Bruker-AXS: TOPAS version 4.2—software for powder diffraction data, 2009, Karlsruhe, Germany
R.A. Robie, B.S. Hemingway: US Geol. Surv—Info. Ser., 1995, 1-5, pp. 192–93.
ICDD PDF-4+ Version 4: 1302 International Centre for Diffraction Data Powder Diffraction File, Newtown Square Pennsylvania, 2013
R.W. Cheary, A. Coelho: Journal of Applied Crystallogr., 1992, 25(2) 109-121
Program CrysAlis: Agilent Technologies, Yarnton, England, 2014
E. Prince, A.J.C. Wilson: Int. Tables Crystallogr., 2004, vol. C.
M.C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G.L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori, R. Spagna: Journal of Applied Crystallography, 2005, 38(2) 381-388
G.M. Sheldrick: Acta Crystallographica Section A, 2008, 64(1) 112-122.
L.J. Farrugia: Journal of Applied Crystallography, 1999, 32(4) 837-838
S.E. Wright, J.A. Foley, J.M. Hughes, S.E. Wright, J.A. Foley, J.M. Hughes: American Mineralogist, 2000, 85(3-4) 524-531
E. Dowty: ATOMS6. 4—Shape Software, Kingsport, USA, 2011
A. Le Bail: Powder Diffraction, 2005, 20(04) 316-326.
R. Dayal, F. Glasser: Science of Ceramics, Academic Press, London, 1967, pp. 191–214
Acknowledgments
Open access funding provided by University of Innsbruck and Medical University of Innsbruck. The corresponding author thanks the Association to Promote Scientific Education of South Tyroleans at the University of Innsbruck for the financial support. We thank Martina Tribus and Daniela Schmidmair for the support with the EMP and XRPD analyses.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted November 28, 2016.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zöll, K., Manninger, T., Kahlenberg, V. et al. Investigations on the Crystal Structure and the Stability Field of FCAM-I (Ca3MgAl6Fe10O28), an Iso-structure to SFCA-I. Metall Mater Trans B 48, 2207–2221 (2017). https://doi.org/10.1007/s11663-017-0988-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-017-0988-7