
Abstract
Metallurgical fundamentals for vacuum refining of molten silicon and the behavior of different impurities in this process are studied. A novel mass transfer model for the removal of volatile impurities from silicon in vacuum induction refining is developed. The boundary conditions for vacuum refining system—the equilibrium partial pressures of the dissolved elements and their actual partial pressures under vacuum—are determined through thermodynamic and kinetic approaches. It is indicated that the vacuum removal kinetics of the impurities is different, and it is controlled by one, two, or all the three subsequent reaction mechanisms—mass transfer in a melt boundary layer, chemical evaporation on the melt surface, and mass transfer in the gas phase. Vacuum refining experimental results of this study and literature data are used to study the model validation. The model provides reliable results and shows correlation with the experimental data for many volatile elements. Kinetics of phosphorus removal, which is an important impurity in the production of solar grade silicon, is properly predicted by the model, and it is observed that phosphorus elimination from silicon is significantly increased with increasing process temperature.
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Metallurgical grade silicon (MG-Si) is commercially produced through the reduction of silicon oxide with carbon in submerged arc furnaces.[1] MG-Si is the initial material to produce pure silicon for electronics applications. The minimum required purity of silicon for photovoltaic applications and that for silicon wafers used in the semiconductor industry are, respectively, 6 N and 9 N. With regard to the recent rapid growth of photovoltaic industry, there is a great demand for solar grade silicon (SoG-Si) feedstock. Traditionally, the waste materials from the electronic industry have been used to produce the crystalline silicon for photovoltaic applications. The concurrent effect of the increase in the demand for solar silicon and of the costs associated with pure silicon scrap from electronic industry led the photovoltaic industry to focus its efforts on the development of new production processes dedicated to solar silicon.[2]
Two main routes for the industrial production of silicon for photovoltaic applications are under development: a chemical route and the metallurgical route. Ultra-pure silicon for electronic/photovoltaic applications has been produced through the established, well-known Siemens process referred to as a chemical route.[2–4] The main disadvantage of this process has been the high energy consumption, and hence new chemical processes with higher production yields have been developed such as the Fluidized Bed Reactor developed by REC Silicon,[5] the Vapor to Liquid Deposition developed by Tokuyama Corporation,[2] or the closed loop process adopted by Wacker Polysilicon.[6] The metallurgical route for the production of silicon for photovoltaic applications has been a later development than the chemical route. The main advantage as claimed by manufacturers who developed dedicated metallurgical refining routes concerns the low energy consumption.[2]
Metallurgical grade silicon contains around 99 wt pct silicon, and its impurities are generated from the raw materials. The major impurities in MG-Si with regard to the quantities are Fe, Ca, Al, and Ti, which are usually from hundreds to thousands ppmw. On the other hand, B and P are the most important impurities in view of the difficulties encountered in their removal with their concentrations usually being less than 100 ppmw. Directional solidification, which is always a key process step in SoG-Si production through metallurgical route, is a purification method to remove all impurities efficiently; except B and P. These latter two elements have large segregation coefficients during silicon solidification, which are 0.8 and 0.35, respectively. Hence, the developed and the under development refining techniques have struggled to remove B and P through other purification methods. Vacuum refining can be an alternative process step for silicon refining in the metallurgical route to remove particular impurities, which are more volatile compared with silicon, in particular P. The scope of the current study is to develop fundamental knowledge on vacuum refining of silicon and the behavior of the impurities in silicon in vacuum refining process.
Vacuum Refining Fundamentals
The difference in the vapor pressures of the liquid metal components at elevated temperatures is the basic principle of crude metal vacuum distillation. The vapor pressures of selected pure substances between 1673 K and 2273 K (1400 °C and 2000 °C) were calculated using the available vapor pressure data of pure substances[7], and the results are shown in Figure 1. It is seen that the vapor pressures of many elements which exist in MG-Si are higher than that of silicon, and they can be evaporated from the molten silicon. However, when substance i goes into solution with silicon solvent at temperature T, its vapor pressure value is decreased from p 0 i to p e i . If the vapor above the solution has ideal behavior, which is a fair approximation for metal-gas mixtures, then the ratio of these vapor pressures can present the activity of i in the solution[8]:

The changes in standard vapor pressure of pure substances with temperature. Symbol ■: melting point
Equation [1] is valid for monatomic form of the solute i in the gas phase. Similarly, the activity of silicon solvent is defined as
It must be emphasized that Eqs. [1] and [2] are principally valid for the equilibrium conditions between the gas and liquid phases with no temperature gradient. Dividing the equilibrium partial pressures of impurity and silicon obtained from Eqs. [1] and [2], the following ratio is obtained:
where
in which parameter β i is called separation coefficient of impurity i.[9] If β i is equal to unity, then the concentrations of impurity in silicon and gas phase are equal, and no separation occurs. When β i is smaller than unity, then the equilibrium concentration of the impurity in silicon is more than that in the gas phase, meaning that the impurity cannot be separated. In contrast, with β i values being greater than unity, the separation of impurity from liquid silicon is possible. Assuming Raoultian behavior for the silicon solvent, γ Si ≈ 1, Henrian behavior for the solute component (\( \gamma_{i} \approx \gamma_{i}^{0} \)) can be considered. Therefore, taking natural logarithm from both sides of Eq. [4] gives
The activity coefficients of many dissolved elements in silicon binary melts have been recently determined by Safarian and coworkers at the silicon melting point.[10] These activity data are reproduced in Table I, along with reported activities for solute elements B and Ti in the literature.[11,12] The activity coefficient for phosphorus in molten silicon was calculated from the reported activities for the low phosphorus concentrations by Zaitsev et al.[13] Activity of Na was calculated by FactSage thermodynamic software. Assuming regular solution behavior for dilute solute elements in molten silicon, their activity coefficients at temperatures greater than the silicon melting point of 1687 K (1414 °C) can be estimated. The calculated lnβ i values for solute elements in silicon for temperatures ranging from 1687 K to 2273 K (1414 °C to 2000 °C) are graphically shown in Figure 2. Obviously, the separation of the dissolved Ti, B, and Fe from liquid silicon is impossible, whereas the separation of the other dissolved elements studied in this article is thermodynamically possible. From thermodynamics point of view, phosphorous should be easy to remove, while copper is the hardest.

The relationship between the separation coefficients and temperature for the dissolved elements in molten silicon
Thermodynamics alone is not enough to evaluate the production efficiency of a process, and thermodynamically feasible processes may be too slow to proceed practically. Vacuum refining is a process under non-equilibrium conditions in which pressures less than the melt vapor pressure is maintained by a vacuum pump. According to Olette,[14] the ratio of the vacuum evaporation rates of the solute i and silicon solvent can be written as
where
where α i is known as the evaporation coefficient and, as long as it is grater than unity, then the removal of impurity i from silicon melt is possible. The differences in the magnitude of α i from one solute to another indicates relative rates of refining when monatomic evaporation controls rates of refining. Polyatomic evaporation can be treated in a similar manner by substituting the vapor pressure of polyatomic species in terms of known solute properties. As mentioned above for the dilute solute element i in liquid Si, γ i is nearly constant and equal to γ 0 i . Thus, the volatility criterion for impurity removal from Si can be written as
Based on the activity coefficients displayed in Table I and the standard vapor pressures of different elements, the corresponding α i -values were calculated at temperatures greater than the silicon melting point, and the results are illustrated in Figure 3. Comparing Figures 2 and 3, it is seen that the shapes of the α i and β i curves for each element are similar, while the order of the curves are different. This can be explained through considering Eqs. [4] and [8] in which α i is actually β i multiplied by (M Si/M i )1/2, and this indicates the effect of the atomic weight on the evaporation rate of the solute element. The positive lnα Fe value obtained as shown in Figure 3 indicates that when silicon-rich Si-Fe melts are vacuum treated, the evaporation rate of Fe is faster than Si, and thus the separation of Fe is kinetically possible—even though lnβ Fe values were negative. However, the separation of Fe from silicon by vacuum treatment is slow, and a high silicon loss is expected for eliminating small amount of Fe.

The relationship between the volatility coefficient α i and temperature for the dissolved elements in molten silicon
Mass Transfer Model for Vacuum Refining of Silicon
In vacuum refining process, a volatile solute element in the melt is transferred from the melt to the gas phase and then condensed far away from the melt surface. This phenomenon is schematically shown in Figure 4(a) in which the elimination of volatile solute element i from molten silicon occurs through the following steps:

Schematic of the mass transfer of volatile impurity i from silicon melt in vacuum refining (a), and the concentration and pressure changes in the vacuum refining system (b)
-
(a)
Bulk mass transport of the solute element in the melt to a melt boundary layer.
-
(b)
Mass transport across the melt boundary layer to the melt/gas interface.
-
(c)
Chemical evaporation of the solute i at the melt/gas interface.
-
(d)
Mass transport of the evaporated element in the gas phase.
-
(e)
Condensation of the gaseous element on the condenser surface.
Depending on the characteristics of vacuum refining technique and process conditions, one or more combinations of these steps can control the process rate. Mass transport of the solute element in the bulk of the melt, step (a) is not usually a rate-limiting step, in particular, when the melt is stirred as, e.g., being done in induction furnace. Gaseous species in vacuum distillation process are finally condensed on the condenser surface or taken out by the pumping system. As long as the minimum thermodynamic conditions for the condensation of the gaseous species are provided, the condensation from the gas phase proceeds rapidly. In practice, continuous pumping and the existence of relatively large condensation surfaces support this, and therefore, the process step (e) is not generally considered as the rate-limiting step. Therefore, the kinetics of impurity removal from molten silicon can be controlled by individual steps from (b) to (d), or combinations of two or all the three. The mass transfer of solute element i in every individual step is studied in this article with respect to vacuum induction refining.
Mass Transport Through the Melt Boundary Layer
The solute element mass transfer in the melt boundary layer occurs through diffusion and convection mechanisms. The latter mechanism is more dominant for the inductively stirred melts where the molar flux of element i can be expressed as:
A kinetic theory for the vacuum induction refining has been developed by Machlin.[15] In this theory a rigid flow model is applied to describe the behavior of the melt in the vicinity of reaction surfaces. The presented equation by Machlin for deep, inductively stirred melts, has been successfully used for the mass transfer calculations in the vacuum refining of molten metals such as steel,[16] copper[17,18] and recently phosphorus evaporation from silicon.[19,20] According to this equation, the mass transfer coefficient of element i through a silicon melt boundary layer can be written as:
where r m and v m are the melt radius and the melt surface velocity, respectively, and they are related to the furnace characteristics, whereas the diffusion coefficient D m,i varies for different solute elements in silicon. The diffusivity of solute elements in liquids can be estimated through the well-known Stocks-Einstein equation. Engh[21] modified this equation by introducing a dimensionless ratio of the masses of solute and solvent elements. According to Engh, the diffusion coefficient of solute element i in Si is expressed by Eq. [11]:
The calculated diffusion coefficients of selected solute elements in molten silicon at 1687 K (1414 °C) from the above equation are listed in Table II along with the related characteristics of the elements appearing in this equation. It is worth noting that the silicon viscosity μ Si was calculated using the presented silicon viscosity relationship with temperature by Sato et al.[22]:
The calculated diffusivities at silicon melting point by Eq. [11] for Sb, B, Ga, and P are consistent with the recent experimental measurements by Garandet[23] as seen in Table II. The calculated diffusivities for Al and In are smaller than the measured values, which is hard to be explained. It is also observed that the experimentally measured diffusivities are in a wider range than the theoretically calculated values. As there are no experimental data on the diffusivities of many elements, the estimation of the diffusivities by Eq. [12] can be considered to be reliable as observed for the majority of the above elements. The diffusivities of elements at higher temperatures than silicon's melting point were calculated, and the results are shown in Figure 5. As the experimentally determined diffusivities for the above mentioned elements are considered more accurate, the diffusivities for these elements at higher temperatures were calculated by Eq. [13] with reference to the diffusivities at silicon melting point T Sif , 1687 K (1414 °C). This equation is simply obtained considering the diffusivity relationship with viscosity and temperature in Eq. [12]:

Diffusivities of the dissolved elements in molten silicon
Using the calculated diffusivities of volatile elements, the corresponding mass transfer coefficients were calculated considering the previously characterized melt properties as r m = 0.032 m and v m = 0.056 m/s.[19] The calculated k m,i values at 1687 K, 1773 K, 1873 K, and 1973 K (1414 °C, 1500 °C, 1600 °C, and 1700 °C) are illustrated in Figure 6. It is observed that k m,i for different solute elements is slightly affected by temperature. Moreover, the mass transfer coefficients of elements in the inductively stirred silicon melt are in a range between 2 × 10−4 and 8 × 10−4 m/s.

Mass transfer coefficients of the dissolved elements through a melt surface boundary layer
Chemical Evaporation of the Impurities
The molar flux of solute element i in step (c) of the process in which the chemical evaporation takes place is expressed by Hertz-Knudsen equation.[16–18]
Both p s,e i and p s i in Eq. [14], which are illustrated schematically in Figure 4(b), affect the molar flux through the chemical evaporation. The parameter p s,e i depends on the solute concentration on the melt surface and also temperature, while p s i is in addition dependent on the vacuum chamber pressure. These two partial pressures for different solute elements in silicon are studied in the following. In addition, the mass transfer coefficients for the evaporation of solute elements are calculated for perfect vacuum and typical low pressure conditions.
Equilibrium partial pressures
Based on the presented activity coefficients for the dilute solute elements in liquid silicon (Table I), the equilibrium partial pressures of different solute elements (p e i ) in silicon at the silicon melting point 1687 K (1414 °C) were calculated and the results are shown in Figure 7. Phosphorus is evaporated in monatomic or diatomic forms depending on its concentration in the melt,[19] and therefore p e i for both conditions are presented in Figure 7, which have been calculated based on the thermodynamic work on Si-rich melts of Si-P alloys.[13] Regarding the importance of phosphorus in silicon, the measured phosphorus vapor pressures over Si-P melts by Zaitsev et al.[13] are also presented which show the consistency of the calculations. Figure 7 is practically important to achieve thermodynamic overview about the gas-liquid equilibrium for silicon rich binary melts. Considering Eq. [1], the equilibrium partial pressure of solute element i at temperature T can be calculated with assuming the regular solution model for the dilute solutions of silicon binary melts:

Equilibrium partial pressure of the dissolved volatile elements in liquid silicon
The magnitude of the changes of p e i by temperature is depending on the temperature dependence of p 0 i and also the activity coefficient of the solute in the melt. It was found that the effect of p 0 i is more domineering, and p e i increases with increasing temperature for the majority of the solutions with both positive and negative deviations from the ideal solution. Especially, minimal decreases in p eAg and p eZn with increasing temperature were observed, which is because of the dominance of the decreases in the activity coefficients of Ag and Zn with increasing temperature.[10] The effect of temperature change on p e i can be seen in Figure 8 for silicon-rich Si-P melts. This figure indicates also that the concentration range in which the monatomic evaporation of phosphorus takes place is increased with increasing temperature. The monatomic evaporation of phosphorus is dominant for concentrations below 8, 18, 27, and 36 ppmw at 1687 K, 1773 K, 1873 K, and 1973 K (1414 °C, 1500 °C, 1600 °C, and 1700 °C), respectively.

Equilibrium partial pressure of the dissolved phosphorus in molten silicon at different temperatures
The dependence of p s,e i on the surface concentration (C s i ) can be calculated with regard to the equilibrium partial pressure relationship with composition as:
As C s i is smaller than C i , the equilibrium vapor pressure at the melt surface (p s,e i ) is always smaller than p e i . However, for the rapidly stirred melts in which there is a negligible difference between the concentrations on the surface and the bulk of the melt, it is a fair approximation to assume p s,e i ≈ p e i .
Partial pressures in the gas phase
In order to establish a relationship between p s i and vacuum refining conditions an analytical approach is applied in this study. In principle, the partial pressure of the solute volatile element, p s i , is dependent on its concentration in the gas phase (X sg,i ), and the total pressure in the vacuum chamber (p t). If we consider no pressure gradient in the gas phase over the whole vacuum chamber, the partial pressure of the solute element over the melt surface, p s i , can be expressed as:
Regarding the continuous flux of both solute and solvent elements from the melt to the gas phase, X sg,i is equal to the ratio of the evaporation molar flux of the solute over the total evaporation flux for the solute and solvent elements. Thus, considering Hertz-Knudsen equation for solute i in Eq. [14] and also a similar expression for silicon, we obtain:
On the other hand, the vacuum chamber pressure is the sum of the partial pressures of the gas components:
Combining Eqs. [17], [18] and [19], a quadratic equation for p s i is obtained:
where
The solution of this equation is:
It is observed that p s i is depending on the atomic weight of the melt components, vacuum chamber pressure p t, solute concentration at the melt surface, and temperature. The latter two parameters are hidden in p s,e i and p s,eSi . The relationship between p s i and concentration at the melt surface, X s i , is illustrated for several elements in Figure 9 under typical vacuum chamber pressure of 0.02 Pa at 1687 K (1414 °C). It is observed that p s i is increased with increasing solute concentration and it is levelled off in a particular concentration. Figures 7 and 9 indicate that for a given solute concentration the corresponding partial pressure in the gas phase is much less than the equilibrium partial pressure. Figure 10 shows the effect of the vacuum chamber pressure on p sP and p sSi at the silicon melting point and pressures lower than the vapor pressure of low phosphorus Si-P melts (0.049 Pa). It is observed that the partial pressure of phosphorus in vacuum is proportional to the chamber pressure and it is changed with almost similar changes in the total vacuum pressure. The effect of temperature changes on the partial pressure of phosphorus are also illustrated in Figure 11 for typical constant pressure of 0.02 Pa. This figure shows that in a given pressure the partial pressure of the volatile impurity is not significantly changed by temperature changes. The curvature for phosphorus apperaing around X P = 0.00001 is due to the change from monatomic to diatomic evaporations of phosphorus. It is worth noting that the calculated pressure for diatomic phosphorus is obtained by Eq. [22] with considering M i as the molecular weight of p 2.

Partial pressures of different volatile elements from silicon melt in the binary gas mixtures under typical process conditions

Partial pressures of P and Si above Si-P melts under different vacuum chamber pressures at constant temperature

Partial pressure of phosphorus (P or P2) above Si-P melts at different temperatures and constant pressure
Impurity evaporation rate in perfect vacuum
Under perfect vacuum conditions (p t → 0), p s i in Eq. [14] can be considered as zero. For extremely stirred melts, we may also consider p s,e i ≈ p e i and C s i = C i in Eq. [16]. Considering η as unity, the maximum evaporation flux of element i through chemical evaporation is obtained by combining Eq. [14] with Eq. [16]:
The evaporation coefficient k maxc,i for different solute elements in silicon were calculated, and the results for 1687 K, 1773 K, 1873 K, and 1973 K (1414 °C, 1500 °C, 1600 °C, and 1700 °C) are illustrated in Figure 12. It is observed that k maxc,i varies in a wide range for different elements. It is also found that, by comparing k m,i and k c,i values in Figures 6 and 12, the mass transfer rates in the melts for Zn, Pb, Bi, Na, Mg, and Sb are mainly slower than their evaporation rates, while the chemical evaporation rates of P, Al, Ca, Mn, and Cu are mainly slower than their transfer rates in the melt boundary later. Almost equal k m,i and k c,i values are observed for some elements in particular temperatures such as Ca at 1973 K (1700 °C), Ag at 1773 K (1500 °C), and so on. Therefore, in perfect vacuum conditions in which process step (d) is not rate limiting, the process kinetics is controlled by steps (b) and (c) individually or through a combination of both of them.

Mass transfer coefficient of the dissolved elements through chemical evaporation reaction at the melt surface
Impurity evaporation rate in low pressures
Based on the above determined equilibrium partial pressures of the solute elements, and the corresponding determined partial pressures in the gas phase in Section III–B–2, the difference between the pressures in Eq. [14], \( p_{i}^{\text{s,e}} - p_{i}^{\text{s}} \), can be calculated. Figure 13 shows the changes of \( p_{i}^{\text{s,e}} - p_{i}^{\text{s}} \) against the corresponding molar fraction in the melt surface (X s i ) for different typical temperatures and pressures. It is seen that \( p_{i}^{\text{s,e}} - p_{i}^{\text{s}} \) is a linear function of composition with a proportional constant (λ i ). Considering the relationship between the molar fraction and concentration (\( X_{i}^{\text{s}} = \frac{{M_{\text{Si}} }}{{\rho_{\text{Si}} }}C_{i}^{\text{s}} \)), Eq. [14] becomes:

The relationship between the driving force for chemical evaporation of volatile solute elements in silicon and their concentrations under typical process conditions
where
The mass transfer coefficient k c,i for selected process conditions was calculated with regard to the corresponding λ i values. It was observed that calculated k c,i values under the vacuum refining conditions considered are very close to the k maxc,i values shown in Figure 12. The applied vacuum pressures here over the vapor pressure of the Si-i melt (p t /p m °) are 0.4, 0.52, 0.63, and 0.18 at 1687 K, 1773 K, 1873 K, and 1973 K (1414 °C, 1500 °C, 1600 °C, and 1700 °C), respectively. Although the applied pressures are not far below the melt vapor pressure, the mass transfers of the volatile elements occur close to the maximum evaporation rate. This is because the partial pressure of the volatile element in the gas phase is much less than its equilibrium partial pressure at the melt surface. Considering these results, we may conclude that k c,i ≈ k maxc,i for the evaporation of volatile elements from molten silicon.
Mass Transport in the Gas Phase
Mass transfers of the evaporated species above the melt occur by diffusion and convection mechanisms. The total mass flux of element i may be written as
where x is a coordinate perpendicularly away from the liquid surface.[16] It has been postulated that the diffusion mechanism is not significant in vacuum refining.[16–18] In the current study, the diffusion flux for the pure silicon gas (D g,Si) by the presented formula for the self-diffusivity equation in the gas phase[24] was calculated which provides a much smaller diffusion flux than the above obtained flux by Hertz-Knudsen equation. This result supports also that the diffusion mass transfer is negligible compared to the mass transfer by convection. Extending this result to the evaporated solute element and considering mass transfer by convection as the dominant mechanism, Eq. [27] becomes
The gas-phase mass transfer coefficients of the volatile elements from silicon are calculated in Section III–D.
Overall Mass Transport Coefficient
Partial pressure of element i on the condensation surface, p c i , can be fairly approximated as zero in the vacuum refining process. Considering negligible accumulation of the solute atoms in the melt, at the interface, and in the gas phase (\( \dot{n}_{{{\text{m}},i}} = \dot{n}_{{{\text{c}},i}} = \dot{n}_{{{\text{g}},i}} \)), the flux of solute element i through all the process steps (b) through (d) can be obtained:
where the total mass transfer coefficient k i is a function of the mass transfer coefficients in all the process steps:
where k g,i is the mass transfer coefficient of solute element i in the gas phase, and it is related to the gas velocity v g along x coordinate:
As the gas phase is mainly formed of the silicon atoms, v g can be considered as the gas velocity obtained for the evaporation of pure silicon. It has been shown by the authors previously[25] that the gas velocity above the pure liquid silicon can be expressed as
The gas velocity is increased with the decreasing chamber pressure, and at a critical chamber pressure, which is p t ≈ 0.2p 0Si , it reaches its maximum. At this pressure, the silicon vapor velocity reaches the velocity of sound.[25] Gas velocities v g for the selected conditions of 1687 K (1414 °C); 0.02 Pa, 1773 K (1500 °C); 0.1 Pa, 1873 K (1600 °C); 0.5 Pa, 1973 K (1700 °C); 0.5 Pa, are 414, 264, 172, and 988 m/s, respectively. The latter velocity is actually the velocity of sound under the vacuum refining conditions considered, which is smaller than that obtained from Eq. [32]. The corresponding k g,i values for the above selected process conditions are shown in Figure 14. Based on the comparion of the magnitudes of the obtained k c,i , k g,i values shown in Figures 12 and 14, it is concluded that the mass transfers of some elements such as Al, Ca, and P can be controlled by both chemical reaction and mass transfer in the gas phase. Moreover, the data presented in Figures 6, 12, and 14 show that the removal rate of volatile impurities from silicon can be controlled by an individual process step, a combination of two particular steps or all three. Obviously, the share of each process step depends on the applied vacuum induction refining conditions. Considering the effect of temperatures, pressures, as well as the magnitudes of the calculated mass transfer coefficients in all the process steps (b) through (d), the main rate-limiting steps can be determined, as summarized in Table III.

Gas-phase mass transfer coefficients of the dissolved volatile impurities in silicon under typical process conditions
The calculated total mass transfer coefficients (k i ) for the volatile elements under the above refining conditions are shown in Figure 15. It is observed that the kinetics of vacuum removal of some elements such as Zn, Pb, Bi, Na, Mg and Sb are not significantly affected by the pressure and temperature changes. This is because the kinetics of removal of these impurities is controlled mainly through the mass transfer in the melt, process step (b). In contrast, the kinetics of the removal of other volatile elements, such as Ca, Sn, Al, P, Mn, Cu, Ga, and Ag, are affected by the pressure and temperature changes because of the importance of the process steps (c) and (d) in their mass transfer.

Total mass transfer coefficients for the dissolved volatile elements in silicon under typical process conditions
Validation of the Mass Transfer Approach
The results obtained from vacuum refining experiments in this study and from the literature data are compared with the calculation results through the mass transfer approach developed for the current study.
Experimental Work
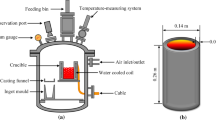
Silgrain silicon from Elkem AS was used for vacuum treatment. For every individual experiment around 300 g silicon was melted in a high quality graphite crucible which shows no silicon infiltration. The crucible dimensions were 64 mm inside diameter, 80 mm outside diameter and 150 mm. A drilled graphite rod was vertically fixed into the crucible wall for holding a thermocouple type C for temperature monitoring.
Vacuum refining experiments were carried out in a vacuum induction furnace. The crucible containing silicon was located in the middle of the induction coil. The experimental set-up has been previously described elsewhere.[19] For each individual run the furnace chamber was cleaned up three times through making the vacuum and flashing +99.99 pct Ar gas into the chamber. Then the sample was heated up rapidly from room temperature to 1873 K or 1973 K (1600 °C or 1700 °C) in vacuum. The heating rate from the completion of melting to these experimental temperatures was so fast to enable its completion within less than 1 minute. Vacuum pressure of 0.5 Pa was applied for both temperatures, and the refining was done for 1.0 and 2.5 hours at 1973 K (1700 °C). According the fundamental theories mentioned above, the applied pressure for vacuum refining must be lower than the melt vapor pressure. The selected pressure here is lower than the silicon vapor pressure, which is 0.79 and 2.82 Pa at 1873 K and 1973 K (1600 °C and 1700 °C), respectively. Regarding the theoretical discussions above, the rate of process is increased with decreasing pressure to critical chamber pressures as p t ≈ 0.16 and p t ≈ 0.56 at 1873 K and 1973 K (1600 °C and 1700 °C), respectively. Applying lower pressures than these critical pressures does not increase the process rate anymore. In order to see the effect of changing vacuum pressure on the process rate, the pressure must be changed in the ranges between 0.16 and 0.79 Pa at 1873 K (1600 °C), and between 0.56 and 2.82 Pa at 1973 K (1700 °C). In practice, however, it is very difficult to maintain a fixed low pressure in these narrow ranges. Therefore, the experiments were done only at a constant pressure (0.5 Pa), and temperature was considered as a main variable. The melt was casted in a water-cooled copper mold to solidify the treated silicon fast and minimize the segregation of the dissolved elements through solidification. Solidified silicon was then crushed down to a powder by a disk mill, and three samples of each silicon powder were thereafter analyzed by high-resolution Inductively Coupled Plasma Mass Spectrometry (ICP-MS).
The measured chemical compositions of silicon before and after vacuum treatment are listed in Table IV. It is observed that volatile elements such as Ca, Na, Mg, Al, P, and Mn have been removed through vacuum refining, while not considerable removal of B, Ti, and Fe is observed. These observations are in agreement with the theoretical predictions considering the volatility coefficients' values shown in Figure 3 in which small amount of Fe loss and no B removal are expected to occur. Assuming that the evaporation of the impurities takes place according to a first-order reaction, the mass transfer coefficient of the impurities can be calculated from the following relation.[19]:
The calculated evaporation mass transfer coefficients for the volatile elements are listed in Table V. It is observed that the application of higher temperature affects the kinetics of evaporation significantly, and the evaporation rate increases 10 times with only 100 degrees temperature rise. This result is in agreement with the presented theoretical approaches shown in Figure 15.
Model Correlation with Experimental Measurements
Figure 16 shows the measured mass transfer coefficients for P evaporation against the corresponding theoretical calculated values according to the outlined method in Section III. The presented experimental data in this figure has been all obtained through vacuum induction refining.[19,20,26–29] Obviously, there is a good correlation between the results, indicating the reliability of the current model for studying the kinetics of P removal, which is the main volatile impurity that has to be removed for SoG-Si production. The differences between the experimental works in Figure 16 can be related to the applied vacuum refining conditions and also the utilized raw materials. While examining the existing literature, it was found that the P removal rate is faster during MG-Si treatment than that during clean silicon melt treatment. This result may be due to the effect of the dissolved elements such as Fe on the increase in the activity coefficient of phosphorus.[30]

Plot of the experimentally determined k P values against the corresponding values obtained based on the current theoretical kinetic approach
Figures 17 and 18 show the measured k Al and k Ca values against their corresponding calculated values. It is observed that there is a fairly good correlation between theoretical and experimental results for Al, while the theoretical values for Ca evaporation are higher than the experimental ones. Similar results as in the case of Ca evaporation are also observed for Na and Mg evaporation (Figure 19). This may indicate that the evaporation kinetics of these metals from liquid silicon is in a similar manner lower than the theoretical. As similar behavior for these elements in liquid silicon is expected with regard to their location in the periodic table and their characteristics, we may argue that their evaporation coefficients (η) are lower than unity so that a few of the Ca, Na, and Mg atoms which reach the surface are actually evaporated. The evaporation of other transition metals such as Mn and Al and non-metals such as P may, however, take place in a different manner (η = 1).

Plot of the experimentally determined k Al values against the corresponding values obtained based on the current theoretical kinetic approach

Plot of the experimentally determined k Ca values against the corresponding values obtained based on the current theoretical kinetic approach

Plot of the experimentally determined k Na, k Mg, and k Mn values against the corresponding values obtained based on the current theoretical kinetic approach
The mass transfer model as presented in the current study provides marginally higher removal rates than the experimentally observed rates. This is due to the ideal situations considered in the development of the current kinetic model. For instance, in the current model, constant temperature throughout the system has been considered. However, temperature at the melt surface is in reality lower than the bulk melt, which has been always measured in other experimental studies[19,20,26–29] similar to this study. The endothermic evaporation reactions take place on the surface, and therefore, a lower temperature is maintained at the melt/gas interface.[31] On the other hand, the gas-phase temperature above the melt is always lower than that at the melt surface, which also causes slightly slower gas phase mass transfer.
Conclusions
Thermodynamics for silicon purification through vacuum refining process was theoretically studied. A kinetic approach was applied to calculate the mass transfer coefficients in different process steps. Expressions for calculating the partial pressures in the gas phase were derived, and it was indicated that the mass transfer coefficient for the chemical evaporation of volatile elements from silicon is close to that obtained from Hertz-Knudsen equation in perfect vacuum. It was found that the kinetics of vacuum refining of silicon can be controlled by mass transfer of the impurity in the melt, chemical evaporation at the melt surface, or mass transfer in the gas phase, or through combinations of two or all of the three steps. The vacuum removal rate of phosphorus, which is a main impurity in silicon in the production of solar grade silicon, is controlled by both chemical reaction and mass transfer in the gas phase. Moreover, the de-phosphorization rate is significantly increased with increasing temperature. The comparison of applied kinetic approach for this study with experimental data indicated that the model is reliable for kinetic study regarding removal of volatile elements from silicon.
Abbreviations
- a i :
-
Chemical activity of solute element i
- a Si :
-
Chemical activity of silicon
- A :
-
Melt surface area
- C i :
-
Concentration of element i in the melt
- \( C_{i}^{\text{s}} \) :
-
Concentration of element i at the melt surface
- \( C_{i}^{0} \) :
-
Initial concentration of element i in the melt
- \( C_{i}^{\rm t} \) :
-
Concentration of element i in the melt after vacuum treatment for t duration
- D g,i :
-
Diffusion coefficient of volatile element i in the gas phase
- D m,i :
-
Diffusion coefficient of solute i in the melt
- \( D_{{{\text{m}},i}}^{{{\text{T}}_{\text{f}}^{\text{Si}} }} \) :
-
Diffusion coefficient of solute i in the melt at silicon melting point
- k c,i :
-
Mass transfer coefficient of element i through chemical evaporation
- k maxc,i :
-
Maximum mass transfer coefficient of element i through chemical evaporation
- k g,i :
-
Mass transfer coefficient of element i in the gas phase
- k i :
-
Total mass transfer coefficient of element i
- \( k_{i}^{\exp } \) :
-
Experimentally determined mass transfer coefficient
- k m,i :
-
Mass transfer coefficient of element i in the melt boundary layer
- m 0 i and m 0Si :
-
Initial masses of solute i in Si solvent
- m i and m Si :
-
Remained masses of element i and Si solvent
- M i , M Si :
-
Molecular weight of element i and Si
- \( \dot{n}_{i} ,\dot{n}_{\text{Si}} \) :
-
Molar flux of element i and Si
- \( \dot{n}_{{{\text{c}},i}} ,\dot{n}_{\text{c,Si}} \) :
-
Molar flux of element i and Si through chemical evaporation reaction
- \( \dot{n}_{{{\text{g}},i}} \) :
-
Molar flux of element i in the gas phase
- \( \dot{n}_{{{\text{m}},i}} \) :
-
Molar flux of element i in the melt boundary layer
- p c i :
-
Partial pressure of volatile element i on the condenser
- p e i , p eSi :
-
Partial pressures of element i and Si at equilibrium
- \( p_{i}^{\text{s,e}} ,p_{\text{Si}}^{\text{s,e}} \) :
-
Equilibrium surface partial pressures of element i and Si
- \( p_{i}^{\text{s}} ,p_{\text{Si}}^{\text{s}} \) :
-
Surface partial pressures of element i and Si
- p 0 i , p 0Si :
-
Standard vapor pressures of pure substance i and Si
- p 0m :
-
Melt vapor pressure
- p t :
-
Vacuum chamber pressure
- r i :
-
Atomic radius of element i
- T :
-
Absolute temperature
- T fSi :
-
Melting point of silicon
- r m :
-
Melt radius
- R :
-
Universal gas constant
- t :
-
Refining time
- v m :
-
Melt surface velocity
- v g :
-
Gas phase velocity
- V :
-
Melt volume
- x :
-
Direction perpendicular on the melt surface
- X i :
-
Molar fraction of element i in the melt
- X s i :
-
Molar fraction of element i at the melt surface
- X sg,i :
-
Molar fraction of element i in the gas phase above the melt surface
- X Si :
-
Molar fraction of silicon in the melt
- α i :
-
Volatility coefficient of element i
- β i :
-
Separation coefficient of element i
- η:
-
Evaporation coefficient
- \( \varphi_{i}^{\text{s}} \) :
-
A function of \( M_{i} ,M_{\text{Si}} ,p_{i}^{\text{s,e}} ,p_{\text{Si}}^{\text{s,e}} ,p_{\text{t}} \)
- γ i , γ Si :
-
Activity coefficients of element i and Si
- γ 0 i :
-
Henrian activity coefficient of element i in Si
- \( \kappa_{\text{B}} \) :
-
Boltzman constant
- λ i :
-
Constant indicating the magnitude of (p e i − p s i ) for element i
- \( \mu_{\text{Si,T}} \) :
-
Viscosity of silicon at temperature T
- \( \rho_{\text{Si}} \) :
-
Density of molten silicon
References
A. Schei, J.K. Tuset, and H. Tveit: Production of High Silicon Alloys, Tapir Forlag, Trondheim, 1998.
A.F.B. Braga, P.R. Zampieri, J.M. Bacchin, and P.R. Mei: Sol. Energy Mater. Sol. Cells, 2008, vol. 92, pp. 418–24.
B.R. Bathey and M.C. Cretella: J. Mater. Sci., 1982, vol. 17, pp. 3077–96.
B.G. Gribov and K.V. Zinov: Inorg. Mater., 2003, vol. 39, pp. 653–62.
J. Safarian, G. Tranell, and M. Tangstad: Energy Procedia, 2012, vol. 20, pp. 88–97.
E. Schindlbeck: Alternative Energies Conference, Paris, 2007.
O. Kubaschewski and C.B. Alcock, Metallurgical Thermo-Chemistry, 5th ed., Pergamon Press Ltd., New York, 1979.
D.R. Gaskell: Introduction to the Thermodynamics of Materials, 5th ed., Chap. 10, Taylor & Francis Group, New York, 2008.
R. Liu: Trans. NFSoc., 1995, vol. 5, 39–42.
J. Safarian, L. Kolbeinsen, and M. Tangstad: J. Mater. Sci., 2012, vol. 47(14), 5561–80.
T. Yoshikawa and K. Morita: Mater. Trans., 2005, vol. 46, pp. 1335–40.
T. Miki, K. Morita, and N. Sano: Metall. Mater. Trans. B, 1997, vol. 28B, pp. 861–67.
A.I. Zaitsev, A.D. Litvina, and N.E. Shelkova: High Temp., 2001, vol. 39, pp. 227–32.
M. Olette: in Physical Chemistry of Process Metallurgy, Part 2, G.R. St-Pierre, ed., Interscience, New York, NY, 1961, pp. 1065–87.
E.S. Machlin: Trans. TMS-AIME, 1960, vol. 218, pp. 314–26.
R. Harris and W.G. Davenport: Metall. Trans. B, 1982, vol. 13B, pp. 581–88.
R. Harris: Metall. Trans. B, 1984, vol. 15B, pp. 251–57.
E. Ozberk and R.I.L. Guthrie: Metall. Trans. B, 1986, vol. 17B, pp. 87–103.
J. Safarian and M. Tangstad: High Temp. Mater. Proc., 2012, vol. 31, pp. 73–81.
S.S. Zheng, W.H. Chen, J. Cai, J.T. Li, C. Chen C, and X.T. Luo: Metall. Mater. Trans. B, 2010, vol. 41B, pp. 1268–73.
T.A. Engh: Principles of Metal Refining, Oxford University Press, Oxford, 1992, p. 116.
Y. Sato, Y. Kameda, T. Nagasawa, T. Sakamoto, S. Moriguchi, T. Yamamura, and Y.J. Waseda: Cryst. Growth, 2003, vol. 249, pp. 404–15.
J.P. Garandet: Int. J. Thermophys., 2007, vol. 28, pp. 1285–303.
R.B. Bird, W.E. Stewart, and E.N. Lightfoot: Transport Phenomena, 2nd ed.; John Wiley & Sons, Inc., New York, 2002.
J. Safarian and T.A. Engh: Unpublished research, 2012.
K. Suzuki, K. Sakaguchi, T. Nakagiri, and N. Sano: J. Jpn. Inst. Met., 1990, vol. 54, pp. 161–67.
N. Yuge, K. Hanazawa, K. Nishikawa, and H. Terashima: J. Jpn. Inst. Met., 1997, vol. 61, pp. 1086–93.
J. Safarian and M. Tangstad: 26th European PV Conference and Exhibition, Hamburg, Germany, 5–8 Sept. 2011, pp. 1810–13.
S.S. Zheng, T.A. Engh, M. Tangstad, and X.T. Luo: Metall. Mater. Trans. A, 2011, vol. 42A, pp. 2214–25.
S. Ueda, K. Morita, and N. Sano: Metall. Mater. Trans. B, 1997, vol. 28B, pp. 1151–55.
H.W. St. Clair and M.J. Spenlove: Trans. AIME, 1951, vol. 191, pp. 1192–97.
Acknowledgments
The authors acknowledge the funding provided through the BASIC project (191285/V30) program by the Norwegian Research Council. The authors acknowledge silicon material support from ELKEM AS.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted: April 12, 2012.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Safarian, J., Tangstad, M. Vacuum Refining of Molten Silicon. Metall Mater Trans B 43, 1427–1445 (2012). https://doi.org/10.1007/s11663-012-9728-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-012-9728-1