
Abstract
High-chromium ferritic steels are current the only viable candidates for cheap interconnect materials for application in high-temperature solid oxide fuel and electrolyzer cells (HT-SOFCs/SOECs). The durability and operating characteristics of interconnects manufactured using these materials may be improved significantly by applying a protective-conducting MoCo2O4 coating and depositing an intermediate layer consisting of nanoparticles of Gd2O3—a reactive element oxide—on the surface of the steel substrate. The study demonstrated that the conditions of the thermal treatment of this layered system determine the efficacy of the applied modification with the reactive element. The persistence of this effect was tested over 7000 hours of quasi-isothermal oxidation in air at 800 °C.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The ability to produce electrical energy and store it without adversely impacting the environment is one of the main challenges in view of climate change caused by greenhouse gas emissions. Some of the most relevant solutions in this regard include solid oxide fuel cells (SOFCs), which use hydrogen as fuel to produce clean electrical energy, and solid oxide electrolyzer cells (SOECs), which make it possible to store surplus electrical energy in the form of hydrogen fuel obtained via the electrolytic decomposition of water. Improving the operating characteristics and reducing the cost of producing the components required for the construction of both types of devices have thus been the aims of intensive research in recent years.[1] This is especially true with regard to one crucial component—the interconnect.
Planar interconnects provide a simple way to join single SOFCs into stacks with operating voltage and power output that are proportional to the number of such cells. An interconnect with this geometry has the shape of plate that has a thickness of several mm; there are channels that allow gas reagents to be supplied to the cathode and anode on both sides of this plate.[2] Interconnects can be manufactured from metal alloys or ceramics, and the type of applied material has a significant influence on the operating characteristics of the component.[3,4] One particularly important factor that determines if a certain material is viable for the construction of interconnects is its price.[1] All of the above-described concerns are also true in the case of SOECs, since these devices are for all intents and purposes almost identical to SOFCs.
Interconnects serve a number of roles in a fuel or electrolyzer cell stack, and the material applied therefore needs to exhibit certain properties. An essential requirement is that the thermal expansion coefficient (TEC) of an interconnect material needs to be similar in value to the TECs of the ceramic components of the cell.[5] This criterion restricts the of available metal alloy choices to two types—high-chromium ferritic steels[6,7,8] and oxide dispersion strengthened alloys with a chromium matrix, the latter of which are incomparably more expensive to produce.[9,10] Another purpose of the interconnect is to ensure that the entire stack is sufficiently rigid and gas-tightness.[3,4]
Interconnects must also provide sufficiently good electrical contact between the neighboring cells over the entire lifetime of the device.[11] Interconnects made from high-chromium ferritic steels are characterized by gradually increasing resistance associated with the growing thickness of the high-temperature corrosion product formed on their surface.[12,13] The norm which had been specified for the maximum area-specific resistance (ASR) for a single interconnect over its entire operating time is 0.1 Ω cm2.[7,10,14] It is worth emphasizing that it is not the wear of the interconnect due to corrosion-related degradation that is the ultimate measure of its usefulness; the main factor is the resistance value which the oxide product of corrosion reaches over time. The influence of wear can be effectively mitigated by increasing the interconnect’s thickness as appropriate. It is also possible to reduce the rate at which oxide scales grow on steel interconnects by reducing the operating temperature of the fuel cell.[15] This brings with it a number of additional advantages and has therefore been one of the main research strategies in recent studies on SOFCs. The adoption of new materials and design solutions has made it possible to reduce fuel cell operating temperatures to the level of ca. 600 °C.[16] A new term was hence coined and applied in the subject literature, namely intermediate-temperature solid oxide fuel cell (IT-SOFC). Unlike electronic conductivity, the ionic conductivity of an interconnect should be as close to zero as possible.
The last requirement that interconnect materials are expected to meet is chemical inertness in relation to other materials in the fuel cell. In the case of high-chromium ferritic steels, this condition has proven rather difficult to satisfy, since Cr2O3—the main component of scales formed on such steels—readily reacts with oxygen and water vapor, forming volatile oxyhydroxides.[17] This process has particularly high potential to inflict damage on the cathode side, since reactions between volatile chromium compounds and cathode material, which contains oxide with a perovskite structure, such as (La,Sr)MnO4, lead to the formation of (Cr,Mn)3O4 spinel, which has inferior catalytic properties compared to the perovskite; this is referred to as cathode poisoning.[18,19] To prevent this from happening, interconnects are coated with protective-conducting oxide layers with a thickness of several dozen micrometers, which effectively separate the afore-mentioned reagents from one another.[20] Studies on such protective-conducing coatings initially investigated oxide materials with a perovskite structure, such as LaCrO3,[11] La1−xSrxCrO3[21,22] or La1−xCaxCrO3.[21] However, there is currently a preference for spinel oxide materials, e.g.,: MnxCo3−xO4,[23,24,25,26,27,28] Mn1+xCr2−xO4,[28] Co2−xMn1−xMex, where: Me = Cu, Ni, Fe.[29,30] The MnCo2O4 sinters that belong to this group are considered some of the most effective in this regard. It should be added that a protective-conducting coating on the surface of a high-chromium ferritic steel also reduces the rate of its oxidation and subsequently the rate at which the ASR of the interconnect increases.[27]
An alternative which to a large extent eliminates the formation of volatile chromium oxides, was the development of a high-chromium ferritic steel with the trade name Crofer 22 APU, the chemical composition of which was designed in such a way as to form a dual-layer scale composed of a Cr2O3 inner layer and an MnCr2O4 outer one.[31,32] This was achieved via a 0.8 wt pct addition of manganese to the alloy. The role of the outer layer is to reduce the rate of volatile chromium compound formation.[33] The Crofer 22 APU also includes up to 0.2 wt pct of lanthanum, which is a reactive element that improves the adhesion of the Cr2O3 scale to the alloy and reduces the rate of its formation—this is known as the reactive element effect (REE).[34,35,36,37,38] The choice of lanthanum over the more commonly applied yttrium was not without reason. Yttrium has a lower solubility in the α-Fe phase and unlike lanthanum it has a tendency to form intermetallic phases with iron (e.g., Fe17Y2); as a result, the concentration of these elements in the solid solution is lower. The solubility of lanthanum in ferrite is ca. five times higher, which is why it is a more effective reactive element in this context.[38] The addition of a small amount of titanium in the Crofer 22 APU is aimed at increasing the electronic conductivity of Cr2O3 via doping with Ti4+ cations.[39]
To recap, a metallic material applied for the construction of interconnects should meet four basic criteria—it should have a TEC similar to those of the remaining fuel cell components, be characterized by high structural stability that ensures that its mechanical properties are retained at high temperatures, exhibit a low ASR that increases at a very slow rate, and be chemically inert in relation to other fuel cell components. High-chromium ferritic steels are currently the only affordable metal alloys that can be applied as interconnect materials, but for the obvious reasons they do not meet every single one of the listed requirements.
It is therefore imperative that the search for new ways in which the utility of steel interconnects can be improved continues. The solutions that are the most desirable in this regard are those that do not entail a considerable increase in production cost. One example is the solution proposed in the present study, which involves a modified process used to manufacture a layered system composed of a Crofer 22 APU steel substrate, a protective-conducting MnCo2O4 spinel coating and an intermediate layer in the form of nanoparticles of the Gd2O3 reactive element oxide; in this case, the modification is the introduction of an additional oxidation stage after the nanoparticle deposition stage.
Practice has shown that the deposition of nanoparticles of a reactive element oxide (Y2O3, Sm2O3, Gd2O3, …) on the surface of the Crofer 22 APU leads to an increase in the oxidation resistance of both uncoated steel[37,38,40,41,42] and layered systems[43] with Crofer 22 APU as the substrate and an MnCo2O4 protective-conducting coating in which these nanoparticles form an intermediate layer. It should, however, be emphasized that the formation of such a coating from the MnCo2O4 spinel is a complex process and involves two stages: 2 hours of annealing in an Ar + 10 pct H2 reducing atmosphere at 900 °C and 4 hours of re-oxidation in air at the same temperature. Such a process may therefore adversely impact the effectiveness of the afore-mentioned oxide nanoparticles that should serve as an additional source of the reactive element other than lanthanum. To dispel these doubts, an additional stage involving 1 hour of oxidation in air at 800 °C was introduced after the deposition of the oxide nanoparticles onto the surface of the steel. The rationale was that the forming layer of Cr2O3 should retain contact with the nanoparticles and ensure that an additional amount of the reactive element incorporates into this oxide before the deposition of the MnCo2O4 coating.
To implement this, a set of 5 samples for comparative thermogravimetric studies was prepared. The samples included unmodified Crofer 22 APU acting as the reference material and four different layered systems based on the Crofer 22 APU: one for which only the gadolinium oxide nanoparticles had been deposited, one with an MnCo2O4 coating, and two systems with the spinel coating and a Gd2O3 nanoparticle intermediate layer and with or without the afore-mentioned additional oxidation stage. To determine the differences in the corrosion process, all samples underwent 7000 hours quasi-isothermal oxidation tests in air at 800 °C. After the oxidation experiment, the structure and morphology of the samples was examined, and their electrical resistance was measured to determine for which of them ASR did not exceed the 0.1 Ω cm2 threshold value.
2 Experimental
2.1 Sample Preparation
The chemical composition of the Crofer 22 APU ferritic stainless steel (ThyssenKrupp VDM GmbH, Germany) is shown in Table I.
A set of five samples was prepared for comparative studies. All samples were cut from a rolled sheet of steel with a uniform thickness of 0.3 mm. The samples for quasi-isothermal studies had dimensions of 20 × 10 mm, and were cut with high precision using a guillotine. The surface area of the samples was determined taking into account their edges and the inner surface of the cylinder made after drilling a 1 mm hole to allow each sample to be suspended. The surface of the steel was polished using SiC abrasive paper with grit ranging from 1000 to 2000, and then degreased in an acetone solution.
Sample 1 was unmodified Crofer 22 APU and served as a reference material. Samples 2 and 3 were prepared by depositing either Gd2O3 nanoparticles or an MnCo2O4 spinel coating on the surface of the Crofer 22 APU steel, respectively. Samples 4 and 5 had the same composition—Crofer 22 APU as the substrate, an MnCo2O4 spinel layer as the coating and Gd2O3 nanoparticles as an intermediate layer; however, sample 5 underwent an additional hour of oxidation in air at 800 °C between the deposition of the nanoparticles and coating the steel with the spinel. Table II shows the designations and preparation details for each sample.
The Gd2O3 layer was deposited via dip-coating in a 0.01 M solution of gadolinium nitrate in ethanol. The solution was prepared from two analytical-grade reagents: Gd(NO3)3·6H2O (Sigma-Aldrich) and C2H5OH (POCH Poland S.A). The procedure through which the nitrate layer was deposited on the steel surface involved three dipping the samples in the solution three times for 10 seconds each. After the first and second dip the samples were annealed in air at 400 °C for 15 minutes, whereas after the third dip the annealing lasted 30 minutes. During this thermal treatment, the gadolinium nitrate was decomposed into an oxide that was insoluble in ethanol. Without the thermal treatment, a certain amount of gadolinium nitrate would be likely to dissolve in the solution again upon re-immersion. As shown in previous studies, the oxide produced via the decomposition of nitrate takes the form of nanoparticles with a size of several dozen nanometers.[43]
The MnCo2O4 spinel coating was produced via electrophoretic deposition (EPD), using a suspension consisting of an MnCo2O4 spinel powder (fuelcellmaterials) and organic solvents mixed at a ratio of 50/50 (ethanol, 99.8 pct purity; isopropanol, 99.7 pct purity). Every 100 mL of the suspension contained 1 g of the MnCo2O4 powder and 0.005 g of iodine (Sigma-Aldrich, ≥ 99.8 pct purity) that played the role of a dispersant. Analysis of the grain size distribution in this powder showed two fractions with an average size of 118 and 1230 nm.[25] Prior to the EPD procedure, the suspension was homogenized for 15 minutes in an ultrasonic bath. The parameters of EPD were as follows: voltage—60 V, deposition time—30 seconds, distance between electrodes—10 mm. After deposition, the spinel layer was dried for 10 hours in air at 70 °C, and then underwent a two-stage thermal treatment at 900 °C. The first stage, which lasted 2 hours, was carried out in a reducing atmosphere (Ar + 10 pct H2 mixture), while an oxidizing atmosphere (air) was applied during the 4 hours second stage.
2.2 Research Methodology
Quasi-isothermal oxidation tests were performed in laboratory air at 800 °C over a time of 7000 hours. Mass change was determined using a Radwag XA 210 analytical balance with the sensitivity of 10−5 g. Time intervals between consecutive measurements ranged from 160 and 290 hours.
Electrical resistance was measured using a 2-probe 4-point method described in detail elsewhere.[45] The opposing sides of the samples were covered with a conducting Pt paste. The measurements were conducted at a constant current density of 0.1 A cm−2. The difference in potentials was measured by means of an HP 34401 digital multimeter with an error of 0.3 pct. The obtained data were applied to determine ASR values from Eq. [1]:
where R—electrical resistance [Ω] and A—surface area of Pt conducting layer [cm2].
Electrical resistance measurements were performed in air at temperatures ranging from 350 °C to 800 °C, starting at 800 °C and with 50 °C decrements until the final temperature was reached.
The Pt conducting layer was then removed from the sample surface by dissolving it in acetone using an ultrasonic washer. After embedding the samples in a Struers EpoFix epoxy resin in a vacuum, polished metallographic specimens were prepared using the Struers Tegramin-20.
The phase composition of the oxidized samples was determined using X-ray diffraction (XRD) performed with an X’Pert Pro device and CuKα radiation. Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) were used for microscopic observations and element composition analysis of the oxidation product; the applied apparatus was a Phenom XL scanning electron microscope (Thermo Fisher Scientific).
In order to determine the phase composition within the layered systems more precisely, the same cross-sections were investigated using Raman spectroscopy (confocal Raman imaging) with a WITec alpha 300 M+ spectrometer. These studies were performed with a laser excitation wavelength of 488 nm, and 100× ZEISS Epiplan-Neufluar objective with NA = 0.9 and 600 grating (spectral resolution of 3 cm−1). The WITec Control FIVE software was used to collect Raman data. The scanned areas were 15 × 20 µm, 8 × 25 µm, and 20 × 20 µm, with a lateral resolution equal to 0.5 µm. Different dimensions were chosen in order to cover all distinctive layers in the investigated systems. The integration time at a single point was equal to 2 seconds. The WITec Project FIVE 5.3 PLUS software was used to analyze the collected maps. The recorded spectra underwent preliminary mathematical processing—extraction of the desired range (110 to 1800 cm−1), baseline correction and cosmic spike removal (CRR filter). Such spectra were subsequently used to generate chemical distribution maps—an integration filter (with specific position and width on the spectrum) was applied to the characteristic spectral regions (e.g., ca. 555 cm−1 for the Cr2O3 band). As a result, a set of distribution images was obtained, with the corresponding spectra confirming the presence of predominant phases chosen manually.
3 Results and Discussion
3.1 Oxidation Kinetics Studies
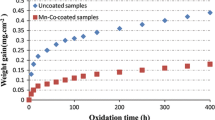
Curves representing sample change over 7000 hours of quasithermal oxidation in air at 800 °C are shown in Figure 1.

Curves representing the mass gain of the samples over 7000 hours of quasi-isothermal oxidation in air at 800 °C: (a) Crofer 22 APU, (b) Crofer 22 APU, Crofer 22 APU/Gd2O3 and Crofer 22 APU/MnCo2O4, (c) Crofer 22 APU, Crofer 22 APU/Gd2O3/MnCo2O4 and Crofer 22 APU/Gd2O3/HT/MnCo2O4. The curve for sample 5 on the figure is adapted from Ref. [53]
It should be noted that there is a certain risk inherent to formulating conclusions concerning the oxidation kinetics based on thermogravimetric curves recorded during a quasithermal oxidation process. This stems from the fact that each time the samples are removed from the furnace for weighing the metal-scale-coating system is subject to mechanical stress arising from differences in the thermal expansion coefficient of the metallic phase and the oxide phases present in the layered systems in question. In an extreme case, these stresses can cause fragments of the protective-conducting coating to spall together with the scale. Table III lists the parabolic oxidation rate constants (\(k_{p}^{^{\prime\prime}}\)) calculated based on the recorded mass change curves as well as total mass gain (Δm) measured upon the conclusion of the oxidation study.
The data presented in Table III shows that coating the Crofer 22 APU with a protective-conducting MnCr2O4 spinel coating and the deposition of a Gd2O3 nanoparticle layer serving as an additional reactive element source in the system have a similar effect as far as the reducing the amount of oxygen bound by the system. The mass gain observed for these two sample types was around half of the value observed for the unmodified steel. For the layered sample which had both the spinel coating and the Gd2O3 nanoparticle intermediate layer modifications and was also oxidized for an additional hour in air at 800 °C (Crofer22APU/Gd2O3/HT/MnCo2O4), a synergistic effect was observed—the amount of oxygen bound was reduced by ca. 75 pct compared to the unmodified steel, which translated to the parabolic oxidation rate constant (\(k_{p}^{^{\prime\prime}}\)) lower by more than an order of magnitude.
3.2 Microscopic Examination of Sample Cross-Sections
Figure 2(a) shows the cross-section of unmodified steel after the oxidation test (sample 1—Crofer 22 APU), while Figure 2(b) shows an EDS line scan of the area indicated in the SEM micrograph.

(a) Cross-section of sample 1 (Crofer22APU); (b) EDS spectra recorded for the indicated area
As reported by other authors, the Crofer 22 APU steel forms a dual-layer scale in a wide range of temperatures; the thicker inner layer of this scale is composed of Cr2O3, while the thinner outer scale consists of the MnCr2O4 spinel.[31,32] A scale with precisely this composition was found at the site for which the EDS analysis had been performed. The spectra recorded for Cr, Fe, Mn, O, and C suggest that the thickness of the chromium-containing inner layer was ca. 10 µm, whereas the thickness of the outer layer that also contained manganese was around 5 µm. The addition of carbon to the group of detected elements made it easier to pinpoint the location at which the surface of the scale ends and the resin used to create the metallographic specimen starts. A noticeable peak in the carbon spectrum, located within the outer layer, most likely originates from diamond grains that came from the polishing paste used to prepare the cross-section and had accumulated inside a single pore. After the 7000 hours quasithermal oxidation test, the scale retained contact with the metallic substrate, but its thickness was not uniform across the entire surface area of the sample. In the presented micrograph [Figure 2(a)] scale thickness ranged from ca. 8 to ca. 12 µm. The variation in scale thickness can be presumed to be associated with spallation that occurred at surface regions due to stresses arising while the sample was cooled to ambient temperature for weighing.
Figure 3(a) shows the cross-section of steel coated with nanoparticles of the reactive element oxide (sample 2—Crofer22APU/Gd2O3), and Figure 3(b) presents the EDS line scan spectra recorded for Cr, Fe Mn, Gd, O, and C.

(a) Cross-section of sample 2 (Crofer22APU/Gd2O3); (b) EDS spectra recorded for the indicated area
The predominant part of the surface area of this sample developed a very thin scale with a thickness of 2 to 3 µm. Despite having a much lower thickness than the scale that had grown on unmodified steel, this scale also had a dual-layer structure. The analysis of the recorded spectra indicates that the inner layer consisted solely of chromium, while the outer layer also contained manganese. On the surface of this scale, a very thin, locally discontinuous oxide layer containing gadolinium was found. At a small number of sites on the surface of the sample the scale had considerably higher thickness—comparable to what had been observed for the reference sample. This implies at these sites on the steel surface gadolinium did not induce the reactive element effect for reasons that are hard to establish. One possible cause is that after the deposition of Gd2O3 nanoparticles they were accidentally removed during one of the later preparation stages, and another explanation is that after the sample had been cooled to ambient temperature for weighing they lost contact with the substrate. In the area selected for investigation [Figure 3(a)] both scale parts—the thinner and thicker one—were in the immediate vicinity of one another.
The next figure [Figure 4(a)] shows the cross-section of another sample—the one with the spinel coating (sample 3—Crofer22APU/MnCo2O4)—as well as the EDS line scan results for Cr, Fe, Mn, Co, O, and C [Figure 4(b)].

(a) Cross-section of sample 3 (Crofer22APU/MnCo2O4); (b) EDS spectra recorded for the indicated area
At the site for which the EDS analysis had been performed (dotted line in the micrograph) scale thickness did not exceed 4 µm, and was thus two to three times thinner than the scale on the reference sample. As in the previous described cases, the Cr2O3 retained good contact with the metallic phase across the entire surface of the sample, which indicates very good adhesion to the steel substrate. It should be noted that on the steel with the MnCo2O4 coating the scale only had a single layer. It can be presumed that an MnCr2O4 spinel outer layer had not been formed because there was a concentration gradient of the manganese diffusing through the Cr2O3 scale and it dissolved in the spinel upon reaching the Cr2O3/MnCo2O4 interface.
The cross-section of the layered system for which additional thermal treatment (1 hour of oxidation in air at 800 °C) had been performed between the deposition of the Gd2O3 nanoparticles and the deposition of the MnCo2O4 coating (sample 5—Crofer22APU/Gd2O3/HT/MnCo2O4) is shown in Figure 5(a). Figure 5(b) shows the EDS line scan spectra recorded for Cr, Fe, Mn, Co, Gd, O, and C.

(a) Cross-section of sample 5 (Crofer22APU/Gd2O3/HT/MnCo2O4); (b) EDS spectra recorded for the indicated area
The scale formed on sample 5 is clearly around half as thick as the scale formed on sample 3, i.e., the sample with the spinel coating as the only modification. It can therefore be assumed that in this case there was synergy between the effects stemming from the presence of the spinel coating and the Gd2O3 nanoparticles. The EDS spectra recorded for chromium and manganese indicate that unlike the scale formed on sample 3 the scale on sample 5 again had a dual-layer structure. The oxide layer composed of Gd2O3 nanoparticles could have acted as a barrier preventing the dissolution of manganese in the MnCo2O4 coating. As in all previous cases, the Cr2O3 layer exhibited good adhesion to the steel surface across the entire sample area.
The microscopic image of the cross-section of sample 4 (Crofer22APU/Gd2O3/MnCo2O4), for which the MnCo2O4 coating had been deposited directly after the Gd2O3 nanoparticles—without any intermediate oxidation stage—did not differ significantly from the one for sample 5 and was therefore not included in the paper. Closed porosity within the MnCo2O4 coating was observed for samples 3, 4 and 5.
3.3 Phase Composition Study
An XRD phase composition analyses in the Bragg-Brentano geometry were conducted for all samples.
For sample 1 (unmodified steel), the following oxide phases were detected: Cr2O3, (Mn,Cr)3O4, Mn2O3 and the α-Fe phase originating from the steel substrate (Figure 6). The same phases were also detected for sample 2—the steel modified using Gd2O3 nanoparticles. The XRD patterns for samples 3 to 5, all of which had a spinel coating with a thickness of several dozen micrometers, indicated only the presence of the MnCo2O4 phase. Since the XRD pattern for sample 2 did not include reflections originating from oxide phases containing gadolinium, an attempt to identify such phases using confocal Raman imaging was undertaken. These investigations were conducted using this sample’s cross-section and the results are presented in Figure 7.

XRD patterns recorded for all samples after 7000 hours of quasi-isothermal oxidation in air at 800 °C. Diffractogram for the sample 5 on the figure is adapted from Ref. [53]

Confocal Raman imaging results obtained for the cross-section of sample 2 (Crofer22APU/Gd2O3) oxidized for 7000 hours in air at 800 °C: (a) confocal image; (b) overlay bitmap; (c) through (g) distribution images of (c) resin, (d) GdCr1−xMnxO3, (e) MnO2 + Mn2O3, (f) (Mn,Cr)3O4, (g) Cr2O3, and (h) combined distribution image obtained by integrating the most specific bands on the corresponding spectra indicated with the dotted line in (i)
Confocal Raman imaging was performed for two samples. Sample 2 was examined first—the analyzed region is marked in the confocal image using a rectangular red frame [Figures 7(a), (b)]. The Raman spectra (chosen manually) recorded for all phases are shown in Figure 7(i). Four different phases were observed (apart from carbon-based resin), forming four distinct layers. Starting from the bottom, the thickest layer consisting of Cr2O3 can be observed, the presence of which was confirmed based on the presence of four typical bands at around 304, 352, 556 (the most intense and characteristic), and 615 cm−1.[46] The next layer on top of the chromia one was determined to be (Mn,Cr)3O4 spinel based on the occurrence of Raman modes at ca. 515 and 680 cm−1 (the most intense and characteristic).[47] What is more, the bands located at 532, 576, and 638 (the most characteristic)[48,49] as well as at ca. 658 cm−1 (the most characteristic)[50] strongly suggest the formation of a mixture of MnO2 and Mn2O3, respectively, on top of the Mn–Cr spinel. Most importantly, given the fact that the spectrum recorded for the phase containing Gd included bands with Raman shift values of 216 cm−1 and 623 cm−1, it was assumed that this phase, formed on the top of the scale, was a GdCr1−xMnxO3 perovskite;[51,52] a similar assumption had been made in paper.[53]
The second investigated specimen was sample 5 (Figure 8). In this case, the results showed that the scale formed on this sample consisted of the same layers as in the case of sample 2 (Figure 7)—Cr2O3 with a top layer composed of Mn species (taking into consideration the intensity of the most characteristic bands, a major fraction of Mn–Cr spinel and two minor Mn oxide fractions) that were impossible to distinguish due to a very complicated spectrum contour. However, the distribution images of particular phases revealed that the Cr2O3 layer was thinner owing to the presence of the MnCo2O4 coating. In the case of protective layers no significant differences were observed for the Gd-containing phase other than additional bands at ca. 449 and 496 cm−1,[54] which can be associated with its greater thickness and thus stronger signal. Moreover, a thick, protective Mn–Co spinel layer with a high compactness can clearly be seen on top of the whole system. Even though no typical bands within the 670 to 690 cm−1 range were determined, the actual presence of such spinel structure can be stated based on the occurrence of modes at around 176, 491, and 629 cm−1 and on previous reports.[55,56,57] It had already been discussed in Reference [53] that the main reason for the shifting and broadening of bands in this context is the octahedral coordination of most Mn cations within the spinel structure and a thermally induced Jahn-Teller effect. The sparse pores within this layer were filled with carbon-based resin (the most intense and characteristic band at 1589 cm−1).[54]

Confocal Raman imaging results obtained for the cross-section of sample 5 (Crofer22APU/Gd2O3/HT/MnCo2O4) oxidized for 7000 hours in air at 800 °C: (a) confocal image; (b) overlay bitmap; (c) through (g) distribution images of (c) MnCo2O4, (d) GdCr1−xMnxO3, (e) (Mn,Cr)3O4/MnO2 + Mn2O3, (f) Cr2O3, (g) resin and (h) combined distribution image obtained by integrating the most specific bands on the corresponding spectra indicated with the dotted line in (i)
3.4 ASR Measurements
After the 7000 hours quasi-isothermal oxidation test, area-specific resistance (ASR) was measured for selected sample in laboratory air at temperatures ranging from 350 °C to 800 °C. The samples included in these investigations were sample 1 (unmodified steel), sample 4 (Crofer22APU/Gd2O3/MnCo2O4) and sample 5 (Crofer22APU/Gd2O3/HT/MnCo2O4). An Arrhenius plot showing ASR as a function of temperature is shown in Figure 9.

Arrhenius plot representing the temperature dependence of ASR for selected samples. The curve for the sample 5 on the figure is adapted from Ref. [53]
The plot shows that the dependence of ASR on temperature was linear for all three samples, which indicates that the mechanism underlying charge transport in the scale at temperatures in the investigated range did not change and that it was a thermally activated process. If it is assumed that charge is transported mostly via the quasi-particles known as small polarons, then the dependence of ASR on temperatures can be represented as follows[7]:
where A—material constant [Ω cm−2 K−1], Ec—activation energy of electrical conductivity [kJ mol−1], k—Boltzmann constant [eV K−1], T—temperature (Kelvin scale).
In the case of sample 1 the ASR value measured at 800 °C was 0.147 Ω cm2, and therefore it exceed the 0.1 Ω cm2 threshold acceptable for interconnects by ca. 47 pct. The corresponding values for samples 4 and 5 were far below the acceptable value—reaching as low as 0.019 Ω cm2 for sample 5, which is five times lower than the threshold.
The electrical conductivity of Cr2O3 at 800 °C is ca. 0.008 S cm−1.[11] At the same temperature the (Mn,Cr)3O4 spinel exhibits conductivity that is 25 times higher—ca. 0.2 S cm−1,[58] and the conductivity of the MnCo2O4 spinel (~ 60 S cm−1[28,59]) is more than three orders of magnitude higher than that of the Cr2O3 scale. The main component that is responsible for the total electrical resistance of this type of layered system is therefore the layer with the lowest conductivity, namely the inner scale layer composed of Cr2O3.
3.5 Estimating the Maximum Operating Time of an Interconnect
As mentioned, the ASR measured for sample 5 at 800 °C after the conducted 7000 hours oxidation test was 0.019 Ω cm2 and was thus five times lower than the accepted threshold (0.1 Ω cm2). It is therefore worth considering after what time an interconnect material with the same composition as sample 5 will reach the threshold value. Although ASR is a parameter that can be used to evaluate an interconnect’s suitability in terms of its electrical properties, another criterion related to corrosion processes should also be taken into account. The growth of the inner layer composed of Cr2O3 is associated with a gradual drop in the concentration of chromium in the metallic phases. Thicker interconnects have larger chromium reservoirs. If, however, the growth of a scale consisting mostly of Cr2O3 causes the concentration of chromium in the Crofer 22 APU steel to fall from 22 wt pct to a threshold value of ca. 12 wt pct, catastrophic corrosion is observed, leading to a very rapid degradation of the material.[60] In such a case iron oxides form on the surface of the scale in place of the protective chromia, and these oxide layers grow at rates that are order of magnitude higher.
It is possible to estimate the time needed for an interconnect in a solid oxide fuel cell to reach either the threshold ASR value or the threshold chromium concentration; this can be done based on the mass change curve recorded during quasi-thermal oxidation in laboratory air, but it requires making a number of assumptions. First and foremost, the actual operating conditions of a fuel cell feature different gas media on each side of the interconnect. On the cathode side air with a certain humidity is present, while the anode side is exposed to a mixture of hydrogen and water vapor—the former of these two gases is the substrate, while the latter is the product of the oxidation reaction. The first assumption is thus that the differences in how the Crofer 22 APU undergoes corrosion in these two atmospheres are negligible, or that the corrosion of this steel in the H2/H2O medium proceeds at a slower rate than in air. A lower oxidation rate on the anode side had in fact been reported by some researchers, for example Huczkowski et al.,[61] who investigated the JS-3 ferritic stainless steel that has a similar chemical composition to the Crofer 22 APU.[62] The cyclic oxidation tests they conducted showed that catastrophic corrosion in an H2/H2O medium was seen after a higher number of cycles than in the case of an air atmosphere. If it were otherwise, making projections concerning operating time based on data obtained for an air atmosphere would not be reasonable.
The performed microscopic examination had shown that the scale formed on sample 5 has a dual-layer MnCr2O4/Cr2O3 structure that is typical to the Crofer 22 APU steel. The thickness ratio of the outer layer to the inner layer is nevertheless not constant over the entire duration of the oxidation process and in time the inner layer becomes thicker. The starting concentration of manganese in the Crofer 22 APU is ca. 0.8 wt pct, but with time this value decreases until there is no more manganese in the metallic phase. Huczkowski et al.[61] found no manganese in the interior of a 0.3 mm sample of the JS-3 ferritic stainless steel after 1000 hours of cyclic oxidation in air at 900 °C. It follows that the growth of the outer scale layer ceases after a certain time, when the manganese reservoir in the steel had been depleted. This state is reached sooner for thinner samples.
The electrical resistivity of Cr2O3 at SOFC operating temperatures is many times higher than that of the MnCr2O4 spinel layer, which is why the thickness of inner layer and not the outer layer that is the main factor that determines the ASR of the interconnect. If it also taken into account that the outer layer was thinner than the inner layer in all cases investigated in the present study, another assumption can be made, namely that disregarding the contribution of the outer layer to the ASR value will not skew it by any significant margin.
This can be easily demonstrated. Literature data indicate a rather large spread in the electrical resistivity values reported for chromia—from 33 to 1000 Ω cm.[63] This is predominantly due to the doping of chromia by alloying agents and the impurities present in alloys. A similar tendency can be observed for samples in the form of ceramic sinters. The resistivity of the MnCo2O4 spinel from the protective-conducting coating is composed is around 0.02 Ω cm at 800 °C.[28] At a thickness of 20 µm its contribution to ASR is thus at the level of 4 × 10−5 Ω cm2. This means that for sample 5 the contribution of the spinel coating to its ASR of 0.019 Ω cm2 does not exceed 0.2 pct. The resistivity of the LaCrO3 at 800 °C is ca. 9 Ω cm.[64] If it assumed that the resistivity of the manganese-doped gadolinium–chromium perovskite (GdCr1−xMnxO3)—which the gadolinium oxide deposited on the surface of the steel had transformed into—is close to that of the lanthanum–chromium perovskite, then in the case of sample 5 its contribution to the total ASR value given that it has a thickness of a fraction of a micrometer can be disregard, especially since this layer is not continuous across the entire sample surface. The contribution of the metallic part of the interconnect can likewise be ignored, since the resistivity of ferrite at 800 °C is only 1.2 × 10−5 Ω cm. If these assumptions are made, then the following simplified dependence of ASR on the thickness of the inner layer (\({x}_{{{\text{Cr}}}_{2}{{\text{O}}}_{3}}\)) can be made:
where \({\rho }_{{{\text{Cr}}}_{2}{{\text{O}}}_{3}}\)—electrical resistivity of Cr2O3 [Ω cm], \({x}_{{{\text{Cr}}}_{2}{{\text{O}}}_{3}}\)—thickness of the Cr2O3 layer after oxidation time t [cm].
The mass change curves recorded during the quasi-isothermal oxidation experiments yielded data on the total mass of oxygen per unit are that had been bound during the formation of the oxide product of corrosion and had dissolved in the metallic core of the sample. It should, however, be emphasized that the presence of the protective-conducting coating does not block the oxidation of chromium oxide to its volatile compounds (CrO3, CrO2(OH)2, …) entirely, only slowing its rate to a significant degree. The term adopted in the literature for this oxidation of the chromia scale and the subsequent formation of volatile chromium compounds is “chromium evaporation”. In the case of a layered system with a structure analogous to sample 5 an experimentally determined chromium evaporation rate constant for 1000 hours of oxidation at 800 °C was 1.25 × 10−9 mg cm−2 s−1.[65] For the Crofer 22 APU steel with no spinel coating, oxidized in the same conditions, it was higher by more than an order of magnitude and was equal to 5.05 × 10−8 mg cm−2 s−1.[65] These data were used to estimate the mass loss stemming from chromium evaporation for sample 5 and the unmodified reference steel sample, both of which had been oxidized at 800 °C for 7000 hours; the calculated values were 3.15 × 10−2 mg cm−2 for sample 5 and 1.27 mg cm−2 for the reference sample, which was equal to 2 and 19 pct of the above-cited experimentally determined values, respectively. This suggests that for longer oxidation times chromium evaporation should absolutely be taken into account when considering total chromium consumption during the oxidation process.
It is impossible to determine the growth kinetics of individual constituent layers of the scale based on the obtained experimental data. However, since once the manganese reservoir in the metallic core has been depleted only the inner layer will continue to grow, it can be assumed that the parabolic oxidation rate constant of the inner layer alone (\(k_{{p_{{{\text{Cr}}_{2} {\text{O}}_{3} }} }}^{^{\prime\prime}}\)) will not differ significantly from the parabolic oxidation rate constant of the scale as a whole (\(k_{p}^{^{\prime\prime}}\)), especially with regard to very long oxidation times. Assuming that \(k_{{p_{{{\text{Cr}}_{2} {\text{O}}_{3} }} }}^{^{\prime\prime}} \cong k_{p}^{^{\prime\prime}}\), the mass gain per unit area (Δm/A) squared will be a linear function of time and according to the Pilling-Bedworth equation will take the following form[66]:
The relation of \({\left(\frac{\Delta m}{A}\right)}_{{{\text{Cr}}}_{2}{{\text{O}}}_{3}}\) to scale thickness (\({x}_{{Cr}_{2}{O}_{3}}\)) can be expressed as follows:
where \({d}_{{{\text{Cr}}}_{2}{{\text{O}}}_{3}}\)—density of chromia [g cm−3], \({M}_{{{\text{O}}}_{2}}\)—molecular mass of oxygen [g mol−1].
The following simplified notation can be used:
From Eqs. [4] and [5] the following equation can be derived:
Transforming Eq. [7] made it possible to arrive at an expression that describes the dependence of the thickness of the inner layer on oxidation time:
After substituting \({x}_{{{\text{Cr}}}_{2}{{\text{O}}}_{3}}\) in Eq. [3] with the right side of Eq. [8], ASR can be expressed as a function of oxidation time:
ASR values for samples 1, 4 and 5 were calculated using formula [1] based on their resistance values measured using the 2-probe 4-point method after the completed oxidation experiments. By substituting the calculated ASR values into the left side of formula [9], the value of resistivity (\({\rho }_{{{\text{Cr}}}_{2}{{\text{O}}}_{3}}\)) can be determined. The three afore-mentioned samples had similar electrical resistivity values at the level of ca. 60 Ω cm. It should, however, be stressed that resistivity calculated in this way is not just the resistivity of the Cr2O3 inner layer, but the resistivity of the entire scale together with half of the steel substrate. Regardless, the contribution of the Cr2O3 layer to the system is the highest. By applying formula [9] and substituting the \({\rho }_{{{\text{Cr}}}_{2}{{\text{O}}}_{3}}\) values determined as described, ASR can be calculated as a function of time. The plot showing the projected values of ASR(t) for samples 4 and 5 and times of over 7000 hours are presented in Figure 10.

Projected operating times of interconnect materials with compositions corresponding to samples 4 and 5
In the case of the reference sample, only the ASR value after the completed oxidation experiment is shown, since this value had already exceeded the accepted ASR threshold (0.1 Ω cm2). On the other hand, the ASR of samples 4 and 5 only exceeds this threshold after 120,000 and 182,000 hours, respectively. For samples 1 and 5 and assuming a thickness of 0.3 mm, the estimated oxidation time after which the Cr content would drop to the threshold value of 12 wt pct was 24,000 and 179,000 hours, respectively. Since the scale forms on both sides of the sample, these calculations were performed for a volume corresponding to half of each sample’s thickness (0.015 cm3), using the parabolic oxidation rate constants (\(k_{p}^{^{\prime\prime}}\)) determined in the present study (Table III).
The depletion of chromium due to its evaporation was also taken into account—chromium evaporation rate constants determined for similar samples in another study were used for this purpose.[65] The performed calculations showed that the time needed for catastrophic corrosion to occur for samples 1 and 5 is far longer than the time after which the ASR threshold would be reached for these two samples. It should, however, be stressed that the point at which catastrophic corrosion occurs is strongly dependent on the operating conditions of a SOFC. If the fuel cell is activated and deactivated frequently, as is the case in the automotive industry, it will last for a shorter time than if it operates at a constant temperature. Research carried out for the purpose of determining the catastrophic corrosion point for a given material involves cyclic temperature changes, with a single oxidation cycle lasting an hour or two. The time at which catastrophic corrosion occurs depends mostly on the number of thermal shocks that the system consisting of an oxide scale and a metallic substrate is subjected to. During the quasi-thermal oxidation test conducted in the present study, the samples were only removed from the furnace 28 times. Had the experiment involved cyclic oxidation and 2 hours cycles, this number would be equal to 3500.
4 Discussion
The oxidation kinetics curves (Figure 1) for samples 2 (Crofer22APU/Gd2O3) and 3 (Crofer22APU/MnCo2O4) confirm that both surface modification methods—the deposition of active element oxide nanoparticles or a protective-conducting spinel coating—reduce the oxidation rate of the Crofer 22 APU. In the case of the first method, the lower growth rate of the scale is caused by the more effective action of active elements from two sources—lanthanum is an alloying addition, and gadolinium is present in the oxide nanoparticles deposited directly on the surface of the unoxidized steel. In the second case, the lower rate at which mass is gained can be attributed to the lower partial pressure at which the steel if oxidized, which is equal to the partial pressure of the MnCo2O4 spinel. If the two modifications are applied concurrently, then both effects are observed, and this was reflected in the mass change curves for samples 4 (Crofer22APU/Gd2O3/MnCo2O4) and 5 (Crofer22APU/Gd2O3/HT/MnCo2O4). The mass change rate for sample 5 was lower than for sample 4, indicating that preliminary oxidation between the deposition of Gd2O3 nanoparticles and the MnCo2O4 spinel coating improves the resistance of the layered system to corrosion further. The expectation that the deposition of the coating material via electrophoresis and the subsequent thermal treatment may reduce the effectiveness of the Gd2O3 nanoparticles therefore proved to be fully justified.
The performed microscopic observations of the polished metallographic specimens of the investigated samples revealed numerous pores within the protective-conducting coating. The formation of a porous structure should be considered typical of ceramic materials obtained via free sintering, i.e., without the application of high pressures. The closed porosity in the coating, which was the only type observed in this case, does not imply loss of continuity. The partial pressure of oxygen underneath a coating with this type of porosity is equal to the partial pressure of spinel, as would be the case for a dense coating. This means that the growth of a scale on the surface of steel with a spinel coating proceeds at a lower gradient of the chemical potential of the oxidizing agent than when no coating is applied. This gradient is the main factor responsible for the transport processes within the scale, and its lower value translates to lower corrosion rates. In this particular case it does not matter if the predominant reagent transport mechanism within the oxidation product is the inward diffusion of oxygen across grain boundaries in Cr2O3 or the inward diffusion of oxygen via point defects in the anion sublattice of this oxide, as suggested by some research results published in recent years.[34,67] In both cases the presence of a continuous spinel layer on the steel surface will reduce the rate at which the studied steel undergoes corrosion.
As far as the other factor is concerned, namely the presence of the nanoparticles of an active element oxide on the surface of the steel, the only conclusions that can be drawn are somewhat speculative in nature, since the underlying mechanisms was not investigated in the present study. Due to its complexity, detailed studies of the REE require the application of atomic-resolution transmission electron microscopy; the use of this method entails very high costs and is only justified in the case of basic research involving simplified model systems, and in studies on the application potential of complex systems such as those investigated in the present study. Nevertheless, the available body of knowledge on the REE and our own experience in this regard leads to the assumption that during the preliminary oxidation of the steel coated with Gd2O3 nanoparticles (800 °C, air, 1 hour), gadolinium starts to segregate at the grain boundaries in the chromia that is forming a continuous layer on the steel surface. In this case this process is not disrupted by other factors such as the electrophoretic deposition of the spinel and its sintering in a reducing and oxidizing atmosphere. The segregation of active elements at grain boundaries has been documented by numerous researchers[34,43,68,69] over the course of the last 40 years and it is inextricably associated with the REE, which significantly reduces the growth rate of the Cr2O3 scale and at the same time improves its adhesion to the metallic substrate. Even when admitting that the knowledge on the mechanisms underlying the REE is incomplete, the view that a more intense segregation of active elements at grain boundaries in chromia should not be questioned.
Microscopic observations combined with phase composition analyses via Raman spectroscopy revealed that the gadolinium oxide nanoparticles were located on the surface of the Cr2O3 layer. If these nanoparticles are treated as an inert marker, the conclusion can be drawn that the predominant reagent transport mechanism in the Cr2O3 scale is the inward diffusion of oxygen. In this case, the marker experiment cannot be used to determine if this diffusion occurred via grain boundaries in chromia or via point defects in the anion sublattice of this oxide.
ASR measurements for selected layered systems and the Crofer 22 APU steel serving as the reference sample were conducted over the temperature range of 350 °C to 800 °C (Figure 3). The conclusions drawn based on these measurements are consistent with the observations based on the mass change curves recorded for these samples. The lowest ASR value was found for sample 5 (Crofer22APU/Gd2O3/HT/MnCo2O4) and it was also for this sample that the lowest mass gain had been observed. The general conclusion based on these is results is therefore that preliminary oxidation after the deposition of the Gd2O3 nanoparticles is advantageous with regard to the transport processes in the layered system, resulting in the reduced thickness of the layer consisting of the oxide product of oxidation formed between the spinel coating and the steel substrate.
The mass change curve for sample 5 (Crofer22APU/Gd2O3/HT/MnCo2O4) was used to calculate the parabolic oxidation rate constant—it was determined that \(k_{p}^{^{\prime\prime}}\) = 1.19 × 10−14 g2 cm−4 s−1. It was estimated that given this \(k_{p}^{^{\prime\prime}}\) value an interconnect fabricated from this material would exceed the threshold ASR value of 0.1 Ω cm2 only after an oxidation time of around 182,000 hours (Figure 10). Although reducing the operating temperature of a SOFC also reduces the corrosion rate of steel interconnects, it is also associated with increased ASR, since the electronic conductivity of chromia decreases with temperature. The performed ASR measurements indicate that for sample 5 this parameter reaches the threshold value at ca. 600 °C. However, it should be stressed that if the oxidation process involved a temperature of 600 °C, then the time required for the corrosion product in this layered system to reach the same thickness as after 7000 hours of oxidation at 800 °C would be much longer. To obtain reliable information on the electrical parameters of an interconnect operating at 600 °C, it would be necessary to conduct another quasi-thermal oxidation experiment featuring such conditions.
Finally, it should be emphasized that for substrates other than the Crofer 22 APU the ASR reduction achieved by means of the described modification may be smaller. This is especially true for steel types with a high impurity content. Muelenberg et al.[70] showed that during the oxidation of a layered system obtained by pressing two metallic foils (Ni/FeCrAl) in an Ar/H2H2O atmosphere, areas consisting of oxides with high electrical resistance formed underneath the nickel surface. These areas formed along grain boundaries in nickel as a result of the internal oxidation of metallic impurities originating from the FeCrAl foil. Grain boundaries are two-dimensional structural defects and the diffusion of both the alloying components and the metallic impurities present in the alloy proceeds at a faster rate along them. This is also true of the oxygen dissolving in the metallic phase. As a result of this process, metal oxides with high affinity for oxygen may precipitate at grain boundaries. Typical impurities in high-temperature ferritic stainless steels include silicon and aluminium. The SiO2 and Al2O3 precipitates forming at grain boundaries may bind with one another and form areas that significantly hinder the flow of current. In extreme cases such precipitates may even form continuous insulating layers across the entire surface of the interconnect, preventing the flow of current altogether.
5 Conclusions
Five types of samples were investigated. Unmodified Crofer 22 APU served as the reference sample, while the remaining four samples were layered systems—one modified only via the deposition of Gd2O3 nanoparticles, one modified only with a protective-conducting MnCo2O4 coating, one with both types of modifications, and one with both types of modifications and an additional pre-oxidation stage (800 °C, air, 1 hour) between the deposition of the nanoparticles and the spinel coating. A comparative study was conducted by oxidizing all samples for 7000 hours in air at 800 °C, under quasi-thermal conditions. It was determined that the layered system for which the pre-oxidation stage had been applied exhibited the lowest rate of corrosion and also the lowest ASR value. This finding has major implications for practical applications, since it provides a relatively simple way to prolong the operating time of a layered interconnect applied in a planar SOFC.
References
S. Zarabi Golkhatmi, M.I. Asghar, and P.D. Lund: Renew. Sustain. Energy Rev., 2022, vol. 161, p. 112339.
A.D.N. Kamkeng and M. Wang: Chem. Eng. J., 2022, vol. 429, p. 132158.
T.A. Morris, E.A. Barringer, S.C. Kung, and R.W. McKain: MRS Bull., 2005, vol. 30, pp. 596–600.
K. Kendall and M. Kendall: High-Temperature Solid Oxide Fuel Cells for the 21st Century: Fundamentals, Design and Applications, 2nd ed. Elsevier Ltd, Amsterdam, 2016.
C.M. Hsu, A.C. Yeh, W.J. Shong, and C.K. Liu: J. Alloys Compd., 2016, vol. 656, pp. 903–11.
M.J. Reddy, T.E. Chausson, J.E. Svensson, and J. Froitzheim: Int. J. Hydrogen Energy, 2023, vol. 48, pp. 12893–2904.
Z. Yang, K.S. Weil, D.M. Paxton, and J.W. Stevenson: J. Electrochem. Soc., 2003, vol. 150, p. A1188.
T. Brylewski, T. Maruyama, M. Nanko, and K. Przybylski: J. Therm. Anal. Calorim., 1999, vol. 55, pp. 681–90.
J.C.W. Mah, A. Muchtar, M.R. Somalu, and M.J. Ghazali: Int. J. Hydrogen Energy, 2017, vol. 42, pp. 9219–29.
W.J. Quadakkers, J. Piron-Abellan, V. Shemet, and L. Singheiser: Mater. High Temp., 2003, vol. 20, pp. 115–27.
W.Z. Zhu and S.C. Deevi: Mater. Sci. Eng. A, 2003, vol. 348, pp. 227–43.
W.Z. Zhu and S.C. Deevi: Mater. Res. Bull., 2003, vol. 38, pp. 957–72.
J.W. Fergus: Mater. Sci. Eng. A, 2005, vol. 397, pp. 271–83.
J. Molenda, J. Kupecki, R. Baron, M. Blesznowski, G. Brus, T. Brylewski, M. Bucko, J. Chmielowiec, K. Cwieka, M. Gazda, A. Gil, P. Jasinski, Z. Jaworski, J. Karczewski, M. Kawalec, R. Kluczowski, M. Krauz, F. Krok, B. Lukasik, M. Malys, A. Mazur, A. Mielewczyk-Gryn, J. Milewski, S. Molin, G. Mordarski, M. Mosialek, K. Motylinski, E.N. Naumovich, P. Nowak, G. Pasciak, P. Pianko-Oprych, D. Pomykalska, M. Rekas, A. Sciazko, K. Swierczek, J. Szmyd, S. Wachowski, T. Wejrzanowski, W. Wrobel, K. Zagorski, W. Zajac, and A. Zurawska: Int. J. Hydrogen Energy, 2017, vol. 42, pp. 4366–4403.
Z. Gao, L.V. Mogni, E.C. Miller, J.G. Railsback, and S.A. Barnett: Energy Environ. Sci., 2016, vol. 9, pp. 1602–44.
K.H. Tan, H.A. Rahman, and H. Taib: Int. J. Hydrogen Energy, 2019, vol. 44, pp. 30591–605.
K. Hilpert, D. Das, M. Miller, D.H. Peck, and R. Weiß: J. Electrochem. Soc., 1996, vol. 143, pp. 3642–47.
E. Konysheva, H. Penkalla, E. Wessel, J. Mertens, U. Seeling, L. Singheiser, and K. Hilpert: J. Electrochem. Soc., 2006, vol. 153, p. A765.
M.H.S. Bidabadi, T. Siripongsakul, T. Thublaor, P. Wiman, and S. Chandra-ambhorn: Surf. Coat. Technol., 2022, vol. 434, p. 128176.
H. Zhu, J. Zhang, and W. Cao: Int. J. Hydrogen Energy, 2023, https://doi.org/10.1016/j.ijhydene.2023.09.242.
J.W. Fergus: Solid State Ion., 2004, vol. 171, pp. 1–15.
T. Brylewski, J. Dabek, K. Przybylski, J. Morgiel, and M. Rekas: J. Power Sources, 2012, vol. 208, pp. 86–95.
F. Smeacetto, A. De Miranda, S. Cabanas Polo, S. Molin, D. Boccaccini, M. Salvo, and A.R. Boccaccini: J. Power Sources, 2015, vol. 280, pp. 379–86.
S. Molin, A.G. Sabato, M. Bindi, P. Leone, G. Cempura, M. Salvo, S. Cabanas Polo, A.R. Boccaccini, and F. Smeacetto: J. Eur. Ceram. Soc., 2017, vol. 37, pp. 4781–91.
M. Bobruk, S. Molin, M. Chen, T. Brylewski, and P.V. Hendriksen: Mater. Lett., 2018, vol. 213, pp. 394–98.
S. Molin, A.G. Sabato, H. Javed, G. Cempura, A.R. Boccaccini, and F. Smeacetto: Mater. Lett., 2018, vol. 218, pp. 329–33.
B. Talic, V. Venkatachalam, P.V. Hendriksen, and R. Kiebach: J. Alloys Compd., 2020, https://doi.org/10.1016/j.jallcom.2019.153229.
X. Chen, P.Y. Hou, C.P. Jacobson, S.J. Visco, and L.C. De Jonghe: Solid State Ion., 2005, vol. 176, pp. 425–33.
T. Brylewski, A. Kruk, M. Bobruk, A. Adamczyk, J. Partyka, and P. Rutkowski: J. Power. Sources, 2016, vol. 333, pp. 145–55.
E. Zanchi, B. Talic, A.G. Sabato, S. Molin, A.R. Boccaccini, and F. Smeacetto: J. Eur. Ceram. Soc., 2019, vol. 39, pp. 3768–77.
Z. Yang, J.S. Hardy, M.S. Walker, G. Xia, S.P. Simner, and J.W. Stevenson: J. Electrochem. Soc., 2004, vol. 151, p. A1825.
M. Naik, K. Santhy, and B. Rajasekaran: Thin Solid Films, 2023, vol. 782, p. 140043.
W. Zhang, S. Wu, L. Chen, J. Liu, L. Jia, D. Yan, and L. Jian: J. Electrochem. Soc., 2023, vol. 170, p. 024509.
A. Gil, O. Kryshtal, T. Brylewski, and A. Czyrska-Filemonowicz: Surf. Coat. Technol., 2019, vol. 368, pp. 232–42.
A. Mosavi and H. Ebrahimifar: Int. J. Hydrogen Energy, 2020, vol. 45, pp. 3145–62.
K. Przybylski and G.J. Yurek: Mater. Sci. Forum, 1991, vol. 43, pp. 1–74.
S. Fontana, R. Amendola, S. Chevalier, P. Piccardo, G. Caboche, M. Viviani, R. Molins, and M. Sennour: J. Power. Sources, 2007, vol. 171, pp. 652–62.
A. Gil, J. Wyrwa, and T. Brylewski: Oxid. Met., 2016, vol. 85, pp. 151–69.
T.C. Kaspar, P.V. Sushko, M.E. Bowden, S.M. Heald, A. Papadogianni, C. Tschammer, O. Bierwagen, and S.A. Chambers: Phys. Rev. B, 2016, vol. 94, pp. 1–12.
B. Lemieszek, J. Ignaczak, B. Kamecki, J. Karczewski, M.B. Mogensen, S. Molin, and P. Jasiński: Int. J. Hydrogen Energy, 2022, https://doi.org/10.1016/j.ijhydene.2022.07.220.
D.E. Alman and P.D. Jablonski: Int. J. Hydrogen Energy, 2007, vol. 32, pp. 3743–53.
P.F. You, X. Zhang, H.L. Zhang, H.J. Liu, and C.L. Zeng: Int. J. Hydrogen Energy, 2018, vol. 43, pp. 7492–500.
T. Brylewski, S. Molin, M. Marczyński, Mazur, K. Domaradzki, O. Kryshtal, and A. Gil: Int. J. Hydrogen Energy, 2021, vol. 46, pp. 6775–91.
VDM ® Crofer 22 APU. Material Datasheet no. 4046, vol. 10, 2010, https://www.vdm-metals.com/fileadmin/user_upload/Downloads/Data_Sheets/Data_Sheet_VDM_Crofer_22_APU.pdf, (accessed 13 July 2021).
E. Zanchi, J. Ignaczak, B. Kamecki, P. Jasiński, S. Molin, A.R. Boccaccini, and F. Smeacetto: Materials (Basel), 2021, vol. 14, p. 3836.
J. Mougin, T. Le Bihan, and G. Lucazeau: J. Phys. Chem. Solids, 2001, vol. 62, pp. 553–63.
B.D. Hosterman: "Raman Spectroscopic Study of Solid Solution Spinel Oxides". UNLV Theses, Dissertations, Professional Papers, and Capstones, 2011, https://doi.org/10.34917/2476131.
C.M. Julien, M. Massot, and C. Poinsignon: Spectrochim. Acta A, 2004, vol. 60, pp. 689–700.
S. Bernardini, F. Bellatreccia, A. Casanova Municchia, G. Della Ventura, and A. Sodo: J. Raman Spectrosc., 2019, vol. 50, pp. 873–78.
F. Hong, B. Yue, N. Hirao, Z. Liu, and B. Chen: Sci. Rep., 2017, vol. 7, pp. 1–7.
Y. Romaguera-Barcelay, J. Agostinho Moreira, A. Almeida, P.B. Tavares, and J. Pérez De La Cruz: Thin Solid Films, 2014, vol. 564, pp. 419–25.
P. Tiwari, S. Kumar, and C. Rath: RSC Adv., 2019, vol. 9, pp. 39871–82.
Ł Mazur, J. Ignaczak, M. Bik, S. Molin, M. Sitarz, A. Gil, and T. Brylewski: Int. J. Hydrogen Energy, 2022, vol. 47, pp. 6295–311.
A.C. Ferrari and J. Roberston: Phys. Rev. B, 2000, vol. 61, p. 14095.
P.L. Meena, S. Pal, K. Sreenivas, and R.A. Kumar: Adv. Sci. Lett., 2015, vol. 21, pp. 2760–63.
S. Yuvaraj, A. Vignesh, S. Shanmugam, and R. Kalai Selvan: Int. J. Hydrogen Energy, 2016, vol. 41, pp. 15199–207.
P. Stelmachowski, A.H.A. Monteverde Videla, T. Jakubek, A. Kotarba, and S. Specchia: Electrocatalysis, 2018, vol. 9, pp. 762–69.
W. Qu, L. Jian, J.M. Hill, and D.G. Ivey: J. Power. Sources, 2006, vol. 153, pp. 114–24.
A. Petric and H. Ling: J. Am. Ceram. Soc., 2007, vol. 90, pp. 1515–20.
D. Koszelow, M. Makowska, F. Marone, J. Karczewski, P. Jasiński, and S. Molin: Corros. Sci., 2021, https://doi.org/10.1016/j.corsci.2021.109589.
P. Huczkowski, S. Ertl, J. Piron-Abellan, N. Christiansen, T. Höfler, V. Shemet, L. Singheiser, and W.J. Quadakkers: Mater. High Temp., 2008, vol. 22, pp. 253–62.
W.J.Q. Pawel Huczkowski: Effect of Geometry and Composition of Cr Steels on Oxide Scale Properties Relevant for Interconnector Applications in Solid Oxide Fuel Cells (SOFCs), 2007, vol. 65, pp. 1–159.
R.W. Jackson, J.P. Leonard, F.S. Pettit, and G.H. Meier: Solid State Ion., 2008, vol. 179, pp. 2111–20.
S. Gupta, M.K. Mahapatra, and P. Singh: Mater. Res. Bull., 2013, vol. 48, pp. 3262–67.
M.J. Reddy, B. Kamecki, B. Talic, E. Zanchi, F. Smeacetto, J.S. Hardy, J.P. Choi, L. Mazur, R. Vasßen, S.N. Basu, T. Brylewski, J.E. Svensson, and J. Froitzheim: J. Power Sources, 2023, https://doi.org/10.1016/j.jpowsour.2023.232831.
S. Mrowec: An Introduction to the Theory of Metal Oxidation, National Bureau of Standards and National Science Foundation, Washington, D.C., 1982.
Y. Parsa, L. Latu-Romain, Y. Wouters, S. Mathieu, T. Perez, and M. Vilasi: Corros. Sci., 2018, vol. 141, pp. 46–52.
K. Przybylski, A.J. Garratt-Reed, and G.J. Yurek: J. Electrochem. Soc., 1988, vol. 135, p. 509.
C.M. Cotell, G.J. Yurek, R.J. Hussey, D.F. Mitchell, and M.J. Graham: Oxid. Met., 1990, vol. 34, pp. 201–16.
W.A. Meulenberg, A. Gil, E. Wessel, H.P. Buchkremer, and D. Stöver: Oxid. Met., 2002, vol. 57, pp. 1–12.
Acknowledgments
Funding as part of the National Science Centre (NCN) Project No. 2021/41/B/ST8/02187 is Gratefully acknowledged (TB). This work was also supported from the subsidy of the Ministry of Education and Science for the AGH University of Kraków (Project No 16.16.160.557).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mazur, Ł., Gil, A., Kamecki, B. et al. On the Possibility of Improving the Oxidation Resistance of High-Chromium Ferritic Stainless Steel Using Reactive Element Oxide Nanoparticles. Metall Mater Trans A 55, 2555–2570 (2024). https://doi.org/10.1007/s11661-024-07424-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-024-07424-6