
Abstract
In this study, the deformation-induced α-martensite formation in AISI 304L steel was investigated in the temperature range between 75 °C and − 196 °C in the light of the temperature-dependence of hydrogen embrittlement phenomena. For this purpose, tensile tests with in-situ and ex-situ magnetic measurement of the α-martensite volume content as a function of plastic strain were carried out. In addition, a theoretical assessment of the temperature-dependence of the austenite stability was undertaken, evaluating chemical and non-chemical driving force contributions to the martensitic γ → α transformation as proposed by Ghosh and Olson. The experimental results clearly show an increase in the α-martensite volume content and a shifting of the phase transformation to lower strain levels upon reducing the temperature to − 75 °C. A further reduction of the temperature to − 196 °C revealed to have no significant impact on the α-martensite formation. The theoretical assessments indicate a similar temperature-dependence of the austenite stability as observed experimentally and suggest contributions of the thermal friction work at the fcc/bcc interface to be responsible for a constant austenite stability in low-temperature regimes. Additional investigations of deformed microstructures showed that element segregation effects on the local austenite stability are pronounced around room temperature, but become less relevant at low temperatures.
Similar content being viewed by others

1 Introduction
Austenitic CrNi-type steels belonging to the AISI 300-series have been used for decades in a wide variety of applications where corrosion resistance and formability are of crucial importance. More recently, specific properties of this alloy group, such as the resistance to hydrogen embrittlement, have moved into the focus of scientific interest.[1,2,3,4] The reason for this is that hydrogen-related technologies become more and more important in connection with the use of renewable energies. Material selection for hydrogen technologies must take into account that most metallic construction materials experience so-called hydrogen embrittlement, a deterioration of their ductility and fracture toughness when exposed to hydrogen.[5] Austenitic CrNi-type steels, especially high alloyed grades e.g., AISI 316L, are generally known to have a high resistance to hydrogen embrittlement, compared to other metallic construction materials. However, the resistance of austenitic steels to hydrogen embrittlement depends sensitively on the stability of the austenitic microstructure. Transformations of the austenitic fcc lattice to bcc martensite (γ → α) can drastically increase the susceptibility to hydrogen embrittlement.[6,7] This is based on an inherently high susceptibility of the α-martensite for hydrogen-induced decohesion and also a high diffusivity of hydrogen in the α-martensite, which enables a rapid accumulation of diffusible hydrogen atoms in critical sites, like stress fields in front of notches and crack tips.[6,7,8,9]
Martensitic γ → α phase transformations can be induced thermally by sufficient supercooling below the equilibrium temperature of the γ and α phase, T0, or through a deformation at temperatures < T0.[10] Usually, the martensite start temperature (Ms) of CrNi-type steels, which marks the temperature that has to be surpassed for thermal α-martensite formation, is sufficiently low so that exclusively thermally induced phase transformations are not relevant in most technical applications. But since the thermodynamic driving force for a γ → α transformation grows with increasing supercooling below T0, even small plastic deformations can be sufficient at low temperatures to provide an additional driving force to initiate a phase transformation.[11] While for most applications the presence of deformation-induced α-martensite is tolerable, when combined with hydrogen, even small amounts of α-martensite can lead to a catastrophic degradation of the ductility. In particular, this can be critical considering cryogenic hydrogen storage applications. Several studies indicate that α-martensite formation shifts to lower plastic strains with decreasing temperature and that the amount of deformation-induced α-martensite per increment of plastic strain tends to increase.[12,13,14,15,16,17] Since α-martensite facilitates hydrogen embrittlement, this results in an increase of hydrogen embrittlement with decreasing temperature, culminating in an embrittlement maximum at around − 50 °C to − 70 °C .[18,19] Towards lower temperatures, a reduction of hydrogen embrittlement, i.e., an increase in ductility, can be observed, commonly assumed to be related to the decreasing mobility of diffusible hydrogen atoms in the metal lattice and their resulting inability to follow the dislocation motion.[9,20] To what extent the kinetics of the deformation-induced α-martensite formation at temperatures around and below − 50 °C affect the characteristics of the embrittlement maximum has not been clarified yet. Although literature data on the deformation-induced α-martensite formation in this temperature regime, especially regarding α-martensite evolution during straining, is very limited, there is some evidence for a decrease of the deformation-induced γ → α transformation intensity in CrNi-type steel at low temperatures.[21,22] Other authors report that cooling from 223 K to 77 K does not further de-stabilize the austenitic phase.[23] Such observations have not been linked to hydrogen embrittlement effects yet.
An experimental assessment of the temperature-dependence of the austenite stability is usually performed by combining slow strain rate tensile tests at different temperatures with in-situ measurements of the bcc phase fraction using magnetic or X-ray measuring methods.[12,24] Especially in the sub-zero temperature range, the equipment for in-situ monitoring of the α-martensite content reaches its limits, which is probably the reason for the limited data available. For a theoretical assessment of the austenite stability there is a variety of empirical approaches, such as empirical equations for Ms- or Md-temperatures.[25,26,27,28] However, such approaches are not suited to assess the austenite stability at an arbitrary temperature. A thermodynamic approach for the evaluation of the austenite stability incorporates the difference of the molar Gibbs energies of the fcc phase \({G}_{\gamma }^{m}\) and the bcc phase \({G}_{\alpha }^{m}\).[10] \({G}_{\gamma }^{m}\) and \({G}_{\alpha }^{m}\) can easily be calculated for any temperature using Calphad software. The magnitude of the calculated chemical driving force \(\Delta {g}_{\mathrm{chem}}\) is often used to evaluate the austenite stability.[29,30,31,32,33] This is certainly reasonable for comparing different alloys at a given temperature. However, a consideration of the austenite stability over a temperature range must take into account that the γ → α transformation is counteracted by various energy contributions, e.g., interfacial energies and elastic strain energies, that impact the α-martensite formation with their individual temperature dependencies.[34] Ghosh and Olson[35] presented an approach, in which energy contributions counteracting a phase transformation are quantified and balanced with contributions that promote a phase transformation. The latter include the aforementioned chemical driving force \(\Delta {g}_{\mathrm{chem}}\) as well as the corresponding mechanical and magnetic contributions. Besides the calculation of an Ms-temperature, the approach allows a calculation of a net driving force over a temperature range, which can be used to quantify the transformation tendency at an arbitrary temperature.
In this work, the austenite stability of AISI 304L type steel is investigated in the temperature range between 75 °C and − 196 °C experimentally as well as theoretically. The experimental part covers slow strain rate tensile tests with a combination of in-situ and ex-situ quantification of strain-dependent α-martensite volume contents. Furthermore, local, segregation-related variations of the austenite stability are taken into account through investigations of the microstructure. The consequences of micro-segregations are mostly neglected when discussing austenite stability, but it was shown that local variations of the austenite stability can impact hydrogen embrittlement of austenitic steels.[36,37] The theoretical considerations cover an analysis of the temperature-dependence of the austenite stability on the basis of the aforementioned model by Ghosh and Olson.[35] From the results, a deeper understanding of the deformation-induced α-martensite formation in the temperature range relevant to hydrogen embrittlement is obtained, that aids to clarify the reasons for the peculiar temperature-dependence of hydrogen embrittlement.
2 Materials and Methods
2.1 Material and Sample Production
The experimental investigations presented in this work were conducted with an AISI 304L type austenitic stainless steel. The chemical composition was measured by optical emission spectrometry as listed in Table I. The steel was produced by continuous casting in an industrial scale and subsequently processed by electroslag-remelting (ESR) to improve the chemical homogeneity. The ESR-ingot with a diameter of 160 mm was hot forged to a Ø50 mm bar shape in multiple steps. From the bar, cylindrical tensile samples with a gauge length of 30 mm and a diameter of 5 mm were machined, parallel to the forging direction, by wet turning. The machined samples were heat treated in an industrial vacuum furnace for 30 minutes at 1050 °C and quenched with argon gas. This final solution-annealing after machining removes deformation-induced α-martensite, that possibly arises in surface-near regions of the samples during machining.[38]
2.2 Tensile Testing
Tensile tests in the temperature range from − 100 °C to 75 °C were performed with a universal testing machine (ZwickRoell GmbH, Germany), equipped with a temperature chamber and a sensor arm extensometer. All tensile tests were performed with a slow strain rate of 5.5 × 10−5 s−1 in order to minimize effects of quasi-adiabatic heating, that might impact strain-induced α-martensite formation.[39] From the tensile tests, the 0.2 pct offset yield strength Rp0.2, the tensile strength Rm, the uniform elongation Ag and the elongation at rupture A were obtained.
2.3 Martensite Formation
The formation of α-martensite during tensile deformation was monitored by means of magnetic induction measurements using a Feritscope® FMP30 device (Helmut Fischer GmbH, Germany) in a series of purpose-adjusted tensile tests. As the stress state of the tested material influences the material’s magnetic properties significantly, magnetic induction measurements were performed at loads lower than 100 N.[40] At testing temperatures between − 25 °C and 50 °C the loading process was therefore interrupted at increments of 5 pct engineering strain with an unload/load cycle. The resulting stress–strain profile is exemplarily illustrated in Figure 1. Since the Feritscope® is not designed for use at temperatures below − 25 °C and even opening the temperature chamber for a short time will lead to severe ice formation, for temperature regimes below − 25 °C, the tensile specimens were repeatedly deformed to certain engineering strains, then unloaded and warmed up to room temperature to perform the magnet inductive measurements. Following the same procedure, additional tensile tests to monitor the α-martensite formation at − 196 °C were performed in a liquid nitrogen bath. In this case, a clip on extensometer was used to monitor the strain.

Engineering stress–strain scheme of the in-situ magnetic measurements of α-martensite volume contents with loading and unloading cycles. The measurements were conducted in the unloaded state, as indicated by the red circles (Color figure online)
The values measured with the Feritscope® represent ferrite equivalent numbers that were corrected for the curved sample surface according to the manufacturers’ specifications. Furthermore a conversion of the ferrite equivalent numbers into a volume content of α-martensite was performed, as ferrite and α-martensite differ in their magnetic response.[41] The conversion was made with Eq. [1] as proposed in Reference 42:
where KB1 and KB2 are constants with the values − 22 and 102, FE represents the ferrite equivalent number and x0 = 23 and dx = 15 are constants as well. The constants were determined in Reference 42 by fitting experimental data from Feritsciope® measurements and X-ray diffraction measurements of the α-martensite volume contents.
2.4 Metallography and Microscopy
To investigate microstructural aspects of deformation-induced α-martensite formation at different testing temperatures, the microstructures of selected tensile specimens were examined via electron backscatter diffraction (EBSD) and energy dispersive X-ray spectrometry (EDS) after tensile testing. To that end, longitudinal sections were prepared metallographically by cutting and embedding, followed by mechanical grinding and polishing down to 1 µm. In order to remove deformation artifacts in the sample surfaces that may interfere with the EBSD-measurements, the samples were subjected to a final electrolytic polishing procedure.
The measurements were performed using a FEG-SEM Mira 3 (Tescan, Czech Republic) equipped with Oxford Instruments EDS and EBSD units and a 70 deg tilted specimen holder. The SEM was operated with an acceleration voltage of 20 kV and a working distance of 17 mm. EDS and EBSD maps were recorded with a step size of 0.3 µm. Quantification of the EDS data and analysis of the EBSD data were performed in the software AZtec.
2.5 Driving Force Calculations
The model adapted for the theoretical evaluation of the austenite stability is based on considerations by Olson and Cohen[43] regarding a heterogeneous martensitic nucleation, for which Ghosh and Olson[35] formulated a critical condition, incorporating a driving force for a barrierless nucleation:
with the driving force per unit volume \(\Delta {g}_{\mathrm{chem}}\), the elastic strain energy gel, the semicoherent interfacial energy σ, and the athermal and thermal components of the interfacial frictional work wµ and wth. n represents the thickness of the critical nucleus and is measured in units of interplanar spacing d of closest packed planes in the austenitic parent phase.[35] For the calculations in this paper, n = 18 was assumed, as reported in Reference 34. For d, a constant value of 2.0737 Å, measured for a similar alloy in Reference 44, was used. Thermal influences on d were neglected. The elastic strain energy gel (Eq. [3]) and the interfacial energy σ (Eq. [4]) can be calculated as products of the constants Kel and Kσ and the composition- and temperature-dependent shear modulus of the austenite \(\mu \left({X}_{j},T\right)\), respectively. The proportionality constants Kel and Kσ were adapted from Reference 35 as 9.4 × 10−4 and 1.8026 × 10−12 m.
The athermal friction work for the bcc/fcc martensitic interface wµ (Eq. [5]) is calculated as a product of the mole fraction of each solute element Xj, a respective coefficient Kµ and \(\mu \left({X}_{j}, T\right)\).[35]
For a multicomponent solid solution as treated here, the athermal friction work is calculated with the following superposition law:
where i = Al, C, N, Cr, Mn, Mo, Nb, Si, Ti, V, and W and j = Cu, Ni.[35] The respective coefficients Kµ that were used for the calculations can be looked up in Reference 35. The composition- and temperature-dependent shear modulus \(\mu \left({X}_{j},T\right)\) (Eq. [8]), required for the calculation of the above quantities, considers the influence of the solute elements Xj on the shear modulus µ of the austenite with a volumetric contribution \({\left(\frac{\mathrm{d}\mu }{{\mathrm{d}x}_{j}}\right)}_{\mathrm{vol}}\) and an electronic contribution \({\left(\frac{\mathrm{d}\mu }{{\mathrm{d}x}_{j}}\right)}_{\mathrm{elec}}\) that are summarized as \({\left(\frac{\mathrm{d}\mu }{{\mathrm{d}x}_{j}}\right)}_{\mathrm{tot}}\).[45] The values for those contributions were used as specified by Ghosh and Olson in Reference 45.
The thermal component of the friction work wth (Eq. 9) incorporates the temperature T, the composition-dependent frictional work W0 as well as the character of the dislocation–obstacle interaction during the phase transformation through the exponents p and q. For the case of martensitic transformation, where interfacial dislocations interact with the strain field of solute atoms, considered here, applies p = 0.5 and q = 1.5.[46] The calculation of W0 (Eq. [10]) is based on the same superposition law that was used for the athermal friction work with \({W}_{0}^{\mathrm{Fe}}\) representing W0 of pure iron and i = C, N; j = Cr, Mn, Mo, Nb, Si, Ti, V, and k = Al, Cu, Ni, W.[46] The coefficients \({K}_{0}^{i,j,k}\) as well as \({W}_{0}^{\mathrm{Fe}}\) were adapted from Reference 46.
The quantities calculated with aid of Eqs. [3] through [10] can be summarized as non-chemical driving forces or mechanical driving forces gmech. Assuming that there are no external magnetic fields and mechanical loads contributing further driving forces, gmech can be summed up with the chemical driving force \(\Delta {g}_{\mathrm{chem}}\) to form a net driving force gnet (Eq. [11]).[46]
\(\Delta {g}_{\mathrm{chem}}\) was calculated as the difference between the molar gibbs energies of the fcc and the bcc phase using the software Thermo-Calc version 2019a with the database TCFe10. For the conversion of the molar Gibbs energy difference to a driving force per unit volume, the molar volume of the fcc phase were calculated as well.
3 Results and Discussion
3.1 Mechanical Properties and α-Martensite Formation
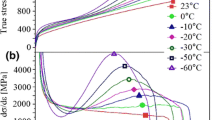
The testing temperature significantly influences the stress strain behavior of the examined alloy, as can be seen in Figure 2 and Table II. With decreasing temperatures, Rp0.2 and Rm increase continuously. The increase in Rp0.2 is mainly attributed to a temperature-related increase of the shear modulus, which delays the onset of the dislocation movement.[47] The alteration of Rm is due to an increase of work-hardening, that causes a change in the shape of the stress–strain curve from parabolic to sigmoidal with a reduction of the temperature.[17] The uniform elongation and elongation to fracture first increase between 75 °C and 25 °C and then decrease until − 100 °C as a result of the work-hardening behavior that is presumably dominated by α-martensite formation.

Engineering stress–strain curves of the steel AISI 304L at different testing temperatures (Color figure online)
Figure 3 presents the α-martensite volume contents measured in the interrupted tensile tests at temperatures between 75 °C and − 196 °C. Since no α-martensite was detected in the undeformed clamping region of the specimen tested at − 196 °C, sheer thermal α-martensite formation in the temperature range examined is ruled out, so that all measured values represent deformation-induced α-martensite. The first occurrence of α-martensite was observed at a testing temperature of 50 °C. This means that Md, which is the upper temperature limit for deformation-induced α-martensite formation, lies between 50 °C and 75 °C.[25] Below 50 °C, the total α-martensite contents drastically increase until 0 °C. The above-described initial increase and subsequent decrease in maximum strain values can be well explained by the occurrence of α-martensite. At temperatures above Md, the deformation is assumed to take place predominantly through dislocation slip, resulting in a minor work-hardening tendency. At lower temperatures, the increase in elongation at fracture occurs with the formation of α-martensite and is usually explained by the fact that, in the region of incipient necking of the specimen, there is a local increase in plastic strain causing local α-martensite formation and strengthening. As a consequence, the deformation is shifted to other regions of the specimen, that react similarly, delaying necking and fracture.[48] However, the early onset of α-martensite formation at low strains leads to an exhaustion of the work-hardening capacity before the onset of necking, so that the elongation to fracture is again reduced at lower temperatures.[49] At temperatures below 0 °C, the curves in Figure 2 follows approximately sigmoidal shapes with a temperature-independent saturation content of α-martensite between 80 and 90 vol pct, which has already been reported by other authors.[50,51,52] The transition from parabolic curve shapes above 0 °C to sigmoidal curve shapes below 0 °C is attributed to an increase of the work-hardening rate in early stages of the plastic deformation. The work-hardening rate is, in turn, dependent on the formation rate of α-martensite. In the temperature range between 0 °C and − 75 °C, the α-martensite formation is basically shifted to lower strains, which indicates lower additional mechanical driving forces to be required to trigger the phase transformation, i.e., the austenite stability declines. From − 75 °C to − 196 °C, the α-martensite formation behavior remains almost unchanged, which suggests that the austenite stability is not subjected to significant changes in this wide temperature range. This observation is in agreement with the above-mentioned reports of other authors, who stated an absence of changes in the austenite stability at low temperatures in other alloys.[21,23] However, the increase in austenite stability, that Hauser et al.[22] found in a Fe–19Cr–3Mn–4Ni–0.15C–0.17N austenitic steel, was not observed here. The named authors observed a sudden change in the amount of deformation-induced α-martensite below the Néel temperature TN in a high-interstitial CrNiMn austenitic stainless steel. This was attributed to the change of the magnetic state of the austenite from paramagnetic to antiferromagnetic, which causes changes in properties like shear modulus, Young’s modulus and thermal expansion, that in turn influence the phase transformation.[22,53,54,55] According to literature data, TN of the steel investigated in this paper can be assumed to lie somewhere below − 220 °C.[54,56] Despite some uncertainty about the exact location of TN, it is reasonable to assume that at temperatures around − 100 °C no influences from a change of the magnetic state on the austenite stability can be expected.

Measured α-martensite volume contents of AISI 304L tensile specimens at different testing temperatures (Color figure online)
3.2 Driving Force Calculations
The calculations of the driving force for a martensitic γ → α transformation of the investigated steel AISI 304L were performed as described in Section II–E with the chemical composition listed in Table I and in a temperature range from − 200 °C to 600 °C. Figure 4 illustrates the temperature-dependence of each component contributing to the non-chemical driving force gmech. All graphs present positive values throughout the whole temperature range, which clarifies that these contributions counteract the phase transformation. wth, gel and the interfacial energy term \(\frac{2\sigma }{nd}\) show a slight increase towards lower temperatures. The strongest temperature-dependence is found for wth with a parabolic curve that has its vertex at 227 °C.

Calculated contributions to the non-chemical driving force gmech as a function of the temperature (Color figure online)
The resulting sum curve gmech, illustrated in Figure 5, consequently ascends in the direction of lower temperatures with an increasing slope. The curve of the chemical driving force Δgchem declines almost linearly and crosses the x-axis at 364 °C, marking the T0 temperature. Thus, below 364 °C, there exists an increasing chemical driving force for the γ → α phase transformation that competes with gmech, which counteracts it. The sum of the non-chemical driving force and the chemical driving force can be interpreted as a net driving force gnet. This net driving force can serve as a general indicator for the stability of the austenite, as gnet must reach negative values for the transformation to occur.[43] Since the gnet curve does not intersect with the x-axis, no spontaneous phase transformation is to be expected during cooling and no Ms-temperature exists for the steel. This is in accordance with the experimental results, where no thermal α-martensite formation was found after to cooling to − 196 °C. For deformation-induced α-martensite formation, the difference between gnet and the x-axis must be provided as a mechanical driving force to initiate the transformation. The amount of driving force provided by a plastic deformation can be calculated as shown in Reference 32 or 57. The progression of the gnet curve is dominated by the slope of gchem at higher temperatures and therefore declines in an approximately linear manner towards lower temperatures. From approximately 100 °C, gmech increasingly influences the slope of gnet, which causes an eventual flattening of the curve and the occurrence of a local minimum at − 120 °C. This local minimum has a slightly asymmetric shape in that the slope to the right of the minimum (towards higher temperatures) is initially smaller than to the left of the minimum (towards lower temperatures) as can be seen in the first derivative of the net driving force curve \(\frac{\mathrm{d}{g}_{\mathrm{net}}}{\mathrm{d}T}\) illustrated in Figure 6. In Figure 6, also a temperature range of roughly − 70 °C to − 150 °C can be identified in which \(\frac{\mathrm{d}{g}_{\mathrm{net}}}{\mathrm{d}T}\) takes small values, which indicates a rather flat shape of gnet. In terms of austenite stability, this flat section of the driving force curve means a weak temperature-dependence. This agrees with the absence of a significant temperature-dependence of the α-martensite formation, that was observed experimentally below − 75 °C and also provides the explanation for the experimentally observed tendency. The pronounced increase of the net driving force curve left of the local minimum can, however, not be correlated with the experimental results, since the amount, as well as the strain dependence of the deformation-induced α-martensite produced at − 196 °C does not indicate any increase in austenite stability at this temperature. The reason for the discrepancy between experiment and calculation apparently lies in the contribution of the thermal friction work wth, which is clearly responsible for the increase in gnet below − 120 °C. Although the temperature-dependence of wth seems to correctly induce the flattening of gnet with a reduction of the temperature, Eq. [9] apparently overestimates the wth towards liquid nitrogen temperature. A similar observation was made in Reference 58 and attributed to a highly approximate character of the calculation method for wth. However, a revision of the temperature-dependence of wth is outside the scope of this work. Ghosh and Olson themselves specify an error of ± 40 K for Ms calculations using their model.[46] This somewhat mitigates the discrepancies observed here between calculations and experiments.

Calculated non-chemical driving force gmech, chemical driving force gchem and net driving force gnet as a function of the temperature (Color figure online)

First derivative of the net driving force curve gnet illustrated in Fig. 5
3.3 Microstructure Investigations
The EDS and EBSD investigations of the microstructure after tensile testing at different temperatures revealed some peculiarities regarding the distribution of α-martensite in the microstructure. Figure 7 illustrates these characteristics with inverse pole figure (IPF) maps showing the grain orientations parallel to the tensile axis, phase distribution maps and the local element distribution of Ni after tensile testing at 0 °C (a through c) and − 196 °C (d through f), respectively. After tensile deformation to a plastic strain of 73 pct at 0 °C, the IPF map [Figure 7(a)] exhibits a heavily deformed grain structure with grains elongated parallel to the tensile direction. The corresponding phase distribution map (b) shows that most of the microstructure has been transformed to α-martensite. The α-martensite is however not uniformly distributed and is rather arranged in a band-like structure with the remaining austenitic regions. The distribution of Ni, illustrated in (c) allows a correlation of the phase distribution with segregation bands that extend parallel to the forging direction. Regions with locally reduced Ni content are predominantly martensitic, while regions with increased Ni content remain austenitic. The distribution of the element Ni is shown here as a representative illustration of the segregation structures to which basically all alloying elements are subject of. An extensive analysis of element segregation effects in AISI 304L steels can be found in our previous publications.[33,36]] The apparent correlations between segregation structures and the spatial distribution of phase transformations found here have been observed previously and are attributed to segregation-induced local differences in austenite stability.[29,59,60,61,62] Considering the sample deformed at − 196 °C [Figures 7(d) through (f)], it becomes clear that the above-described band-like structure is not present in the phase distribution map. The latter is somewhat contrary to expectations, as pronounced segregation structures are present in the probed area of this tensile specimen as well. A possible explanation for the different dependence of deformation-induced α-martensite formation on local chemical composition at different temperatures might be found in different nucleation mechanisms. Generally, nucleation sites for deformation-induced α-martensite are assumed to be intersections of slip bands, ε-martensite plates and deformation twins.[63,64,65] The formation of these defects is closely linked to the stacking fault energy (SFE), which itself is subject to a strong temperature-dependence. The reduction in SFE with decreasing temperatures tends to favor the formation of ε-martensite as compared to twinning.[66] Figure 8 shows, for instance, the presence of ε-martensite as a transition phase between austenite and α-martensite. (a) shows the local phase distribution in an EBSD phase map of the specimen tested at − 196 °C at 20 pct plastic strain and (b) presents a detail of ε-martensite zones in the austenite-phase domains. This map is to be compared with the one shown in Figure 8(e): after 50 pct deformation at − 196 °C there are little to no ε-martensite areas observable. A resulting change in the dominating nucleation mechanism could possibly be a cause for a differing spatial distribution of α-martensite after deformation at very low temperatures. However, the exact determination of nucleation mechanisms is beyond the scope of this paper.

EBSD and EDS maps recorded in the microstructures of AISI 304L after tensile deformation at 0 °C (a through c) and − 196 °C (d through f). (a) + (d): IPF maps showing grain orientations ║ tensile direction, (b) + (e): phase distribution maps, (c) + (f): element distribution maps of Ni (Color figure online)

Presence of ε-martensite as a transition phase in the specimen tested at − 196 °C at 20 pct plastic strain. (a) EBSD phase map, (b) detail (Color figure online)
In order to clarify the direct influence of the observed segregation effects on the austenite stability, calculations of the net driving force were performed with the data obtained from the EDS measurements in the regions shown in Figure 7. For this purpose, gnet was calculated with the mean chemical compositions of both regions and with the respective segregation-related scattering. Table III shows the compositions employed. The calculation results shown in Figure 9 provide very similar results for both samples and regions. In both cases, the consideration of local chemical variations leads to the formation of a scatter band (dashed lines) around gnet of the mean composition. This means that segregation effects result in a significant scatter band of the austenite stability, which accounts for the fact that some microstructural sites can transform to α-martensite at lower plastic deformations, i.e., additional driving force, than others. It can further be observed that the scatter band constricts towards lower temperatures. This means that with decreasing temperature, the influence of segregation effects on α-martensite formation becomes less pronounced, which serves as an explanation for the more homogeneous α-martensite distribution in the specimen tested at − 196 °C compared to the specimen tested at 0 °C.

Calculated net driving forces for the regions of microstructure depicted in Fig. 7 with scatter bands representing the influence of element segregations on the austenite stability (Color figure online)
As stated in Section II–A, the investigated AISI 304L material was subjected to an electroslag-remelting during production. This process step is performed in order to improve the steels cleanliness and to improve the chemical homogeneity for applications with advanced requirements.[67] Even in steel grades with a rather high chemical homogeneity due to ESR processing, the assumption of a constant driving force throughout the whole microstructure is too much of a simplification. This applies both to the experimental investigations and to the theoretical considerations of austenite stability.
4 Conclusions
The deformation-induced α-martensite formation in an AISI 304L steel was investigated in the temperature range between 75 °C and − 196 °C. The experimental assessment of the α-martensite evolution during tensile testing at different temperatures revealed an initial increase in the volume content of α-martensite upon a reduction of the temperature to 0 °C. A further reduction of the deformation temperature until − 75 °C results merely in a shift of the α-martensite formation to lower strain levels, as an equal saturation content of α-martensite is reached in this temperature range. Both the increase in volume content of α-martensite and the shifting to lower plastic deformations are indicators for a reduction of the austenite stability. Further reductions of the deformation temperature do not result in significant changes of the α-martensite formation, thus proving that no further reduction of the austenite stability occurs between − 75 °C and − 196 °C. The theoretical considerations of the austenite stability according to the model proposed by Ghosh and Olson[35] suggest that this is a result of an increasingly high thermal friction work at the bcc/fcc interface, which counteracts the phase transformation and balances the increasing chemical driving force, which promotes the phase transformation.
The theoretical assessment of the austenite stability correctly indicates a slowing decrease and even stagnation of austenite stability with decreasing temperatures and thus provide a reasonable description of the austenite stability over a wide temperature range. The calculations therefore offer significant advantages over simple considerations solely based on chemical driving forces. In addition, it has been shown that taking segregation-induced fluctuations in alloy composition into account results in a scatter band of austenite stability. This aids to clarify the effect of element segregations on the phase transformation tendency and the distribution of the arising α-martensite. However, a re-stabilization of the austenite at temperatures below − 120 °C, which was indicated by the calculation results is not reflected in the experimental results. A revision of the temperature-dependence of the thermal friction work could probably bring improvements in the accuracy of the calculations.
Concerning the temperature-dependence of hydrogen embrittlement, the experimental results of this study suggest a connection between the hydrogen embrittlement maximum that is reported to exist at − 50 °C to − 70 °C and deformation-induced α-martensite formation.[18,19] The common theory stating that an increase in α-martensite content from room temperature on to low temperatures increases embrittlement is consistent with the observations of increasing α-martensite contents and the shift of α-martensite formation to lower strains until − 75 °C.[6] The absence of a further destabilization of the austenite at even lower temperatures, which was observed experimentally, certainly contributes to the fact that the tendency to hydrogen embrittlement does not further increase. Nevertheless, it can be assumed that the usual explanation, incorporating a decrease of the mobility of diffusible hydrogen atoms,[20] is mainly accountable for the reduced embrittlement tendency, rather than a re-stabilization of the austenite.
Ongoing investigations are devoted to testing of hydrogen-charged tensile specimens in the relevant temperature range. These target the experimental verification of the proposed, and previously observed, links between low-temperature austenite stability and hydrogen embrittlement of austenitic stainless steels.
References
T. Michler, J. Naumann, M. Hock, K. Berreth, M.P. Balogh, and E. Sattler: Mater. Sci. Eng. A, 2015, vol. 628, pp. 252–61.
T. Michler, J. Naumann, S. Weber, M. Martin, and R. Pargeter: Int. J. Hydrogen Energy, 2013, vol. 38, pp. 9935–41.
M. Martin, S. Weber, W. Theisen, T. Michler, and J. Naumann: Int. J. Hydrogen Energy, 2011, vol. 36, pp. 15888–98.
S. Weber, M. Martin, and W. Theisen: J. Mater. Sci., 2012, vol. 47, pp. 6095–6107.
T. Michler, M. Lindner, U. Eberle, and J. Meusinger: Gaseous Hydrogen Embrittlement of Materials in Energy Technologies: Volume 1: The Problem, Its Characterisation and Effects on Particular Alloy Classes, Woodhead Publishing Ltd, Cambridge, 2012, pp. 94–125.
L. Zhang, M. Wen, M. Imade, S. Fukuyama, and K. Yokogawa: Acta Mater., 2008, vol. 56, pp. 3414–21.
C. Izawa, S. Wagner, M. Deutges, M. Martín, S. Weber, R. Pargeter, T. Michler, H.-H. Uchida, R. Gemma, and A. Pundt: Int. J. Hydrogen Energy, 2019, vol. 44, pp. 25064–75.
B.S. Kumar, V. Kain, M. Singh, and B. Vishwanadh: Mater. Sci. Eng. A, 2017, vol. 700, pp. 140–51.
G. Han, J. He, S. Fukuyama, and K. Yokogawa: Acta Mater., 1998, vol. 46, pp. 4559–70.
L. Kaufman and M. Cohen: Prog. Met. Phys., 1958, vol. 7, pp. 165–246.
I. Tamura: Met. Sci., 1982, vol. 16, pp. 245–53.
C. Quitzke, C. Schröder, C. Ullrich, M. Mandel, L. Krüger, O. Volkova, and M. Wendler: Mater. Sci. Eng. A, 2021, vol. 808, p. 140930.
U. Krupp, C. West, and H.-J. Christ: Mater. Sci. Eng. A, 2008, vol. 481–482, pp. 713–17.
A. Zergani, H. Mirzadeh, and R. Mahmudi: Steel Res. Int., 2020, vol. 91, p. 2000114.
N. Tsuchida, Y. Morimoto, T. Tonan, Y. Shibata, K. Fukaura, and R. Ueji: ISIJ Int., 2011, vol. 51, pp. 124–29.
C. Zheng and W. Yu: Mater. Sci. Eng. A, 2018, vol. 710, pp. 359–65.
A.K. De, J.G. Speer, D.K. Matlock, D.C. Murdock, M.C. Mataya, and R.J. Comstock: Metall. Mater. Trans. A, 2006, vol. 37A, pp. 1875–86.
G.R. Caskey: Hydrogen Degradation of Ferrous Alloys, Noyes Publications, Park Ridge, NJ, 1985, pp. 822–62.
H.K. Birnbaum and P. Sofronis: Mater. Sci. Eng. A, 1994, vol. 176, pp. 191–202.
T. Michler, A.A. Yukhimchuk, and J. Naumann: Corros. Sci., 2008, vol. 50, pp. 3519–26.
V.Y. Ilichev, L.V. Skibina, and M.M. Chernik: Austenitic Steels at Low Temperatures, Springer, Boston, MA, 1983, pp. 69–82.
M. Hauser, M. Wendler, O. Fabrichnaya, O. Volkova, and J. Mola: Mater. Sci. Eng. A, 2016, vol. 675, pp. 415–20.
Z. Wang and M.X. Huang: Metall. Trans. A, 2019, vol. 50, pp. 5650–55.
I. Janeiro, O. Hubert, and J.-H. Schmitt: Int. J. Plast., 2022, vol. 154, p. 103248.
K. Nohara, Y. Ono, and N. Ohashi: ISIJ Int., 1977, vol. 63, pp. 772–82.
K.W. Andrews: J. Iron Steel Inst., 1965, vol. 203, pp. 721–27.
W. Steven and A.G. Haynes: J. Iron Steel Inst., 1956, vol. 183, pp. 349–59.
H.-S. Yang, J.H. Jang, H. Bhadeshia, and D.W. Suh: Calphad, 2012, vol. 36, pp. 16–22.
S. Martin, O. Fabrichnaya, and D. Rafaja: Mater. Lett., 2015, vol. 159, pp. 484–88.
M. Martin, S. Weber, and W. Theisen: Int. J. Hydrogen Energy, 2013, vol. 38, pp. 14887–95.
A. Borgenstam and M. Hillert: J. Phys. IV France, 1997, vol. 07, pp. C5-23–C5-28.
M. Hauser, M. Wendler, A. Weiß, O. Volkova, and J. Mola: Adv. Eng. Mater., 2019, vol. 21, p. 1800676.
S. Benito, G. Egels, A. Hartmaier, and S. Weber: Mater. Today Commun., 2023, vol. 34, p. 105016.
G. Ghosh and G.B. Olson: Acta Metall. Mater., 1994, vol. 42, pp. 3361–70.
G. Ghosh and G.B. Olson: JPE, 2001, vol. 22, pp. 199–207.
G. Egels, L. Mujica Roncery, R. Fussik, W. Theisen, and S. Weber: Int. J. Hydrogen Energy, 2018, vol. 43, pp. 5206–16.
T. Michler, Y. Lee, R.P. Gangloff, and J. Naumann: Int. J. Hydrogen Energy, 2009, vol. 34, pp. 3201–09.
M. Martin, S. Weber, C. Izawa, S. Wagner, A. Pundt, and W. Theisen: Int. J. Hydrogen Energy, 2011, vol. 36, pp. 11195–206.
M. Radajewski, R. Eckner, S. Decker, M. Wendler, and L. Krüger: Adv. Eng. Mater., 2019, vol. 21, p. 1800617.
M. Martin, S. Weber, W. Theisen, T. Michler, and J. Naumann: Int. J. Hydrogen Energy, 2013, vol. 38, pp. 5989–6001.
C. Müller-Bollenhagen. Verformungsinduzierte Martensitbildung bei mehrstufiger Umformung und deren Nutzung zur Optimierung der HCF- und VHCF-Eigenschaften von austenitischem Edelstahlblech. Dissertation, Siegen, 2011.
R. Fussik. Untersuchung der mikrostrukturellen Stabilität in Relation mit der Wasserstoffversprödung von austenitischen FeCrNi-Legierungen. Dissertation, Bergische Universität Wuppertal, 2020.
G.B. Olson and M. Cohen: Metall. Trans. A, 1976, vol. 7, pp. 1905–14.
F.C. Nascimento, C.E. Foerster, S.L.R. Da Silva, C.M. Lepienski, C.J. de Mesquita Siqueira, and C. Alves Junior: Mater. Res., 2009, vol. 12, pp. 173–80.
G. Ghosh and G.B. Olson: Acta Mater., 2002, vol. 50, pp. 2655–75.
G. Ghosh and G.B. Olson: Acta Metall. Mater., 1994, vol. 42, pp. 3371–79.
R.L. Tobler, D.H. Beekman, and R.P. Reed: Austenitic Steels at Low Temperatures, Springer, Boston, MA, 1983, pp. 135–57.
L. Krüger, T. Halle, L.W. Meyer, U. Brüx, and G. Frommeyer: Mat.-wiss. u. Werkstofftech., 2005, vol. 36, pp. 299–306.
J. Talonen. Effect of Strain-Induced α′-Martensite Transformation on Mechanical Properties of Metastable Austenitic Stainless Steels. Dissertation, Espoo, Finland, 2007.
G.B. Olson and M. Cohen: Metall. Trans. A, 1975, vol. 6, pp. 791–95.
T. Angel: J. Iron Steel Inst., 1954, vol. 177, pp. 165–74.
P.M. Ahmedabadi, V. Kain, and A. Agrawal: Mater. Des., 2016, vol. 109, pp. 466–75.
H.M. Ledbetter and E.W. Collings: Mater. Sci. Eng., 1985, vol. 68, pp. 233–37.
H. Ledbetter: Physica B, 1990, vol. 161, pp. 91–95.
S. Chen, C. Chung, C. Yan, and T. Hsu: Mater. Sci. Eng. A, 1999, vol. 264, pp. 262–68.
L. Warnes and H.W. King: Cryogenics, 1976, vol. 16, pp. 659–67.
J.R. Patel and M. Cohen: Acta Metall., 1953, vol. 1, pp. 531–38.
D. San Martín, K. Aarts, P. Rivera-Díaz-del-Castillo, N.H. van Dijk, E. Brück, and S. van der Zwaag: J. Magn. Magn. Mater., 2008, vol. 320, pp. 1722–28.
R. Fussik and S. Weber: J. Mater. Sci. Eng. A, 2016, vol. 6, pp. 243–53.
R. Fussik, G. Egels, W. Theisen, and S. Weber: MSF, 2018, vol. 941, pp. 263–68.
F. Sadeghi, T. Zargar, J.W. Kim, Y.-U. Heo, J.S. Lee, and C.H. Yim: Mater. Charact., 2021, vol. 175, p. 111063.
J. Man, M. Smaga, I. Kuběna, D. Eifler, and J. Polák: Eng. Fract. Mech., 2017, vol. 185, pp. 139–59.
J. Talonen and H. Hänninen: Acta Mater., 2007, vol. 55, pp. 6108–18.
J.L. Wang, M.H. Huang, X.H. Xi, C.C. Wang, and W. Xu: Mater. Charact., 2020, vol. 163, p. 110234.
J. Venables: J. Phys. Chem. Solids, 1964, vol. 25, pp. 693–700.
O. Grässel, L. Krüger, G. Frommeyer, and L. Meyer: Int. J. Plast., 2000, vol. 16, pp. 1391–409.
S. Ahmadi, H. Arabi, A. Shokuhfar, and A. Rezaei: J. Mater. Sci. Technol., 2009, vol. 25, pp. 592–96.
Conflict of interest
The authors declare that they have no conflict of interest.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was funded by the German Research Foundation (DFG) through the Grant Number 456380481 - WE 4436/7-1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Egels, G., Bussmann, M., Benito, S. et al. On the Temperature-Dependence of Deformation-Induced Martensite Formation in AISI 304L Type Steel. Metall Mater Trans A 54, 4222–4232 (2023). https://doi.org/10.1007/s11661-023-07175-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-023-07175-w