
Abstract
Zinc ferrite nanoparticles were obtained by chemical methods (co-precipitation and thermal decomposition of metalorganic compounds) and systematically probed with volume (XRD, VSM), microscopic (TEM) and element sensitive probes (ICP-OES, Mössbauer Spectroscopy, XPS, XAFS). Magnetic studies proved the paramagnetic response of stoichiometric ZnFe2O4 (ZF) nanoparticles, while superparamagnetic behavior was observed in as-synthesized, non-stoichiometric ZnxFe3−xO (NZF) nanoparticles. Upon annealing up to 1400 °C in an inert atmosphere, a significant change in the saturation magnetization of NZF nanoparticles was observed, which rose from approximately 50 up to 140 emu/g. We attribute this effect to the redistribution of cations in the spinel lattice and reduction of Fe3+ to Fe2+ during high-temperature treatment. Iron reduction is observed in both ZF and NZF nanoparticles, and it is related to the decomposition of zinc ferrite and associated sublimation of zinc oxide.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Nanocrystalline magnetic nanoparticles are characterized by unique properties, such as superparamagnetism, stable core/shell structures, high catalytic activity and chemical reactivity. These reveal enhanced physical properties, e.g., magnetization and permeability, compared to their bulk counterparts.[1,2,3,4,5,6,7,8] Selected systems, for example, zinc ferrite, have low toxicity, which is advantageous in terms of applications in biotechnology/biomedicine (drug delivery, magnetic hyperthermia, imaging),[9,10,11,12] as magnetic fluids[13,14,15] and in environmental remediation.[16,17,18,19] In addition, zinc ferrites are also promising candidates for application in magnetic storage[20] or as a photo-absorber material for light-driven water splitting.[21]
Stoichiometric zinc ferrite ZnFe2O4 (ZF) belongs to a group of ferrites, i.e., iron oxides of the general formula (Me 2+1−x Fe 3+x )Td [Me 2+x Fe 3+2−x ]Oh O4, where ()Td denotes tetrahedral sites and []Oh denotes octahedral sites. Bulk ZFs have a paramagnetic nature around room temperature, when it is the normal spinel with Zn incorporated mostly in the tetrahedral lattice sites, so the super exchange interaction between Fe magnetic ions located in Td and Oh sublattices is excluded. When the particle size decreases, the composition of ZF is altered, leading to the occurrence of superparamagnetism or ferrimagnetism at room temperature.[5,22,23,24,25,26,27,28,29] This is mainly related to the redistribution of cations in the lattice, which occurs when grain sizes are decreased to nanoscale.[24,30,31,32] In contrast to bulk ZF (of normal spinel structure), nanoparticles of stoichiometric zinc ferrite (ZF NPs) reveal a mixed spinel structure, where Zn2+ and Fe3+ cations are distributed between ()Td and []Oh sites. Non-stoichiometry of nanoparticles is usually compensated for by point defects in the crystal structure.[22,33] This is why in case of ZN NPs even a small deviation of Zn content from the stoichiometric composition causes significant changes of physical-chemical properties, especially magnetic[33,34,35,36,37] and structural.[22,23,31,36,38]
Several doping models of Fe3O4 nanoparticles by zinc ions Zn2+ leading to the formation of non-stoichiometric zinc ferrite (NZF), ZnxFe3−xO4 are proposed in the literature.[23,24,34,35,39,40,41,42]
Ma et al.[34] discuss two models of doping that are dependent on the concentration of Zn2+ ions in NZF NPs. The first model, proposed for x ≤ 0.2, assumes that non-magnetic Zn2+ ions are taking Td sites, substituting partially Fe3+ ions, while Fe2+ ions in Oh sites are substituted by Fe3+ ions. The second model is proposed for higher Zn2+ content, i.e., x > 0.2. It assumes that a part of the Zn2+ ions takes the place of Fe3+ in Td sites, while the other part takes the place of Fe2+ in Oh sites.[34] Both mechanisms lead to an increased magnetic moment of nanoparticles. On the other hand, Srivastava et al.[24] proposed the mechanism in which all Zn2+ ions are in Td sites, reducing the amount of Fe3+, while Fe2+ ions are distributed in Td as well as in Oh sites. Control of the doping provides an efficient tool for designing nanoparticles of required magnetic properties.[24,34,35]
It is well known that thermal treatment of as-prepared magnetic nanoparticles at elevated temperatures can reduce the surface disorder and improve the magnetic properties, but this leads to significant grain growth.[43,44] Several papers were devoted to studying the changes of the physical-chemical and especially magnetic properties of ZF NPs, which are associated with phase and size changes during thermal treatment.[22,32,43,45,46,47,48,49,50] In high-temperature applications of zinc ferrite the most interesting is the temperature range of 1100 °C to 1200 °C, at which the thermal decomposition of zinc ferrite occurs; the final products of this reaction are both solid and gaseous. Various decomposition temperatures are reported in the literature, which depend on several factors, such as the stoichiometry of zinc ferrite, grain size, atmosphere, heating rate, constant temperature annealing time and initial material purity, etc.[21,51,52,53,54,55]
Thermal decomposition of ZF is crucial in high-temperature processes such as metal casting,[51,56] clean energy production (H2 and/or O2) using water splitting by solar/chemical energy conversion systems (solar fuels),[21,52,53] use as an absorbent material for hot-gas desulfurization and to remove sulfur-containing impurities from coal gas because of its enhanced chemical reactivity[45,57] and as a catalyst.[58,59,60] The investigation of the influence of high temperature (up to a temperature of 1200 °C) on the magnetic and structural properties of ZF NPs was discussed in several papers.[22,30,32,43,45,47,48,49,50,51,54,55,61,62] However, the influence of higher temperature on the properties mentioned is less known.[52,53]
The result of the annealing process at high temperature depends on the atmosphere in which the process takes place. Philip et al.[45] studied ZF NP annealing in vacuum. The authors found dynamic changes of the lattice parameter and an increase of magnetic properties of particles related to the cation redistribution in the lattice due to increasing temperature. In addition, the authors assume that these changes result from Fe3+ reduction to Fe2+.[45] Singh et al.[48] investigated the influence of temperature (from 400 °C to 700 °C, in air) and sintering time of ZF NPs on the structural and magnetic properties. They found that the higher the heating temperature the lower the saturation magnetization of particles. Ayyappan et al.[61] observed that ZF NPs heated up to 1000 °C in air change their magnetic properties from superparamagnetic to paramagnetic, whereas particles heated in vacuum become ferromagnetic. This indicates that the oxygen vacancies during the annealing process play an important role in the migration of cations between interstitials leading to robust magnetic ordering. Finally, Makovec et al.[22] pointed out that heating of ZF and NZF NPs to a temperature of approximately 600 °C causes significant changes in their lattice parameter and magnetization. These changes are related to the change in the nanoparticle composition and structure.
The effect of the thermochemical decomposition of ZF is especially important for H2 production in water splitting using solar energy. Kaneko et al.[53] showed that this process starts at a temperature > 1227 °C, and at 1477 °C about 40 pct molar of the ZF decomposes.
Stopić et al.[55] determined the decomposition temperature of the technical zinc ferrite (1011 °C to 1120 °C in an inert atmosphere) and proved the presence of Fe3O4 in the final products. The results were used to create a stochastic geometric model of the decomposition process of zinc ferrite from neutral leach residues.[54]
While ZF is a common subject of scientific work, systematic research on NZF NPs is still rare. In view of potential high-temperature applications, it is crucial to establish the thermal resistance of such nanoparticles. Here, the influence of Zn stoichiometry on the structural and magnetic changes of the final solid products of the thermal decomposition of NZF NPs is discussed. Nanoparticles heated up to 1400 °C in inert atmosphere were studied. The decomposition degree and corresponding concentrations of Fe2+ ions were determined. Also, the distribution of cations and magnetic properties was determined. Our results show that high temperature thermal treatment of NZF NPs in inert atmosphere leads to significant Fe3+ reduction and similar structural transformations as in the case of ZF NPs, but magnetic properties are greatly enhanced, contrary to ZF NPs.
2 Materials
Iron(III)chloride hexahydrate (FeCl3·6H2O), iron(II)chloride tetrahydrate (FeCl2·4H2O), iron(III)acetylacetonate (Fe(acac)3), zinc(II)acetylacetonate hydrate (Zn(acac)2·xH2O) and benzyl alcohol (BA) (C7H8O) were from Sigma-Aldrich. Zinc chloride (ZnCl2) and sodium hydroxide (NaOH) were from POCH. All chemicals were used without further purification
2.1 Synthesis of ZnxFe3−xO4 NPs via Co-precipitation Method
ZF and NZF NPs (ZnxFe3−xO4, 0 < x ≤ 1) were synthesized via the co-precipitation method, which was redesignated based on the synthesis in works.[5,63] In this process, adequate amounts of FeCl3·6H2O, FeCl2·4H2O and ZnCl2 were dissolved in deionized water. The ratio of the metal ions was Fe2+:Fe3+:Zn2+ = (1 − x):2:x. A water solution of NaOH was used as the precipitating agent. The precipitate that formed was separated and washed with deionized water. Based on ICP-OES the composition of these particles was ZnFe2O4 and Zn0.35Fe2.65O4 named respectively as S1 (x = 1NaOH) and S4 (x = 0.35NaOH). The dried samples were utilized for further characterizations.
2.2 Synthesis of ZnxFe3−xO4 NPs via the Thermal Method in Non-aqueous Solvent
NZF NPs were synthesized via thermal method, which was redesignated based on the synthesis in References 23, 64,65,66,67. In a typical reaction, Fe(acac)3 and Zn(acac)2 (molar ratio 2:1) were dissolved in 3.5 mL of benzyl alcohol (BA), which was used as the solvent, capping agent and reducing agent for the combined reduction and thermal decomposition reaction. The mixture was heated to 120 °C and kept for 1 hour and finally heated to 200 °C to reflux the mixture and kept for 2 hours. The mixture with precipitates was cooled down to ambient temperature and then washed by organic solvents (ethanol, diethyl ether) several times. Based on ICP-OES, the composition of these particles was Zn0.86Fe2.14O4 and Zn0.53Fe2.47O4 named respectively as S2 (x = 0.86BA3.5) and S3 (x = 0.53BA7). The dried samples in powder form were utilized for further investigations.
Nanoparticles were heated from 30 °C to 1400 °C under inert atmospheres at a constant heating rate ß = 10 °C/min using a TA Instruments Q600 thermobalance.
3 Characterization Methods
The determination of the zinc and iron concentration in the as-prepared samples S1–S4 were analyzed by inductively coupled plasma optical emission spectroscopy (ICP-OES) using a Thermo Scientific CAP 7000 Plus spectrometer.
The X-ray diffraction (XRD) was used to detect the phase of all samples before (S1–S4) and after their thermal treatment (S1′–S4′). The data were collected using a Panalytical Empyrean diffractometer (Royston, UK) with Cu Kα radiation (1.541874 Å) at 40 kV and 40 mA in the Bragg-Brentano (θ/2θ) horizontal geometry (0.026 step at 3000 s/step). The crystallite size of the samples was estimated from the X-ray line broadening of the most intense peak using Scherrer′s formula, while the lattice parameters of the samples were estimated by the Rietveld method.
The morphology, size distribution and average particle size of all samples were measured using micrographs from a TEM Tecnai TF 20 X-TWIN (FEI) transmission emission microscope equipped with a field emission gun, having a point resolution > 0.25 nm.
Determination of defects of the output material and the degree of decomposition of zinc ferrite nanoparticles was performed using:
-
X-ray photoelectron spectroscopy (XPS) PHI 5000 VersaProbe II spectrometer with an Al Ka monochromatic x-ray beam. The spectra were recorded using a PHI 5000 VersaProbe II (ULVAC-PHI, Chigasaki, Japan) spectrometer with a Al Kα monochromatic X-ray beam (E = 1486.6 eV). The operating pressure in the analytical chamber was < 3 × 10–7 Pa. The X-ray source was operated at 25 W and 15 kV beam voltage. Spectra were collected from three points each of 100 μm diameter for each sample. A dual-beam charge neutralizer was used to compensate the charge-up effect. The pass energy of the hemispherical analyzer for iron (Fe 2p) spectra was fixed at 23.5 eV and for other elements at 46.95 eV. The calibration energy used was C1s = 284.8 eV of the atmospheric carbon and hydrocarbon adsorbed on the sample surface. The spectrum background subtraction was performed using the Shirley method. Data analysis software from PHI MultiPak was used to calculate elemental compositions from the peak areas.
-
X-ray absorption spectra (XAS) were collected at room temperature using the high-energy resolution fluorescence detection (HERFD) method at ID26 beamline of the European Synchrotron Radiation Facility (Grenoble, France). A Si(311) double-crystal monochromator was employed to select incident photon energy with the resolution of approximately 0.2 eV, while a set of four Ge(440) spherically bent crystal analyzers and avalanche photodiodes was used to detect Fe Kα emission intensity;
-
57Fe Mössbauer spectroscopy in the transmission geometry at room (300 K) and cryogenic (80 K) temperatures. Measurements were performed using a constant acceleration type spectrometer Renon MS-4 with 57Co source in Rh matrix kept at room temperature.
Volume magnetic properties were probed using a Lakeshore 7407 Vibrating Sample Magnetometer (VSM) equipped with cryostat cooling with liquid nitrogen. Measurements were collected at room (300 K) and cryogenic (80 K) temperatures.
4 Results and Discussion
4.1 Chemical Characterization of the As-prepared Samples
The as-prepared NPs were subjected to chemical analysis by the ICP-OES method. Ferrites of the following chemical composition were obtained: ZnFe2O4 (S1), Zn0.86Fe2.14O4 (S2), Zn0.53Fe2.47O4 (S3) and Zn0.35Fe2.65O4 (S4).
4.2 X-ray Diffraction analysis
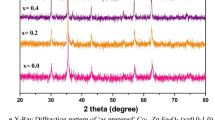
X-ray diffraction (XRD) studies confirmed that S1–S4 samples are zinc ferrite with varying degrees of non-stoichiometry with the FCC spinel structure (Figure 1). The following diffraction peaks were identified: (022), (113), (004), (224), (115) and (044).[24,68]

Rietveld refinement of XRD patterns for: (a) as-prepared samples (S1–S4) and (b) for solid products (S1–S4′) after thermal treatment up to 1400 °C
The average mean size of the crystallites for all samples was calculated using Scherrer’s formula (Table I). In light of the presented results (Figure 1 and Table I), it can be concluded, that for all samples the as-synthesized crystallite size is decreasing from 6.5 nm (x = 0.35) to 3.15 nm (x = 1) with an increase of the Zn doping concentration in the zinc ferrite structure. This is a known phenomenon in this group of ferrites, attributed to thermodynamic factors rather than to nucleation features.[69,70] In the solid product, formed after heating up to 1400 °C (samples S1’–S4’), phases with a spinel structure such as zinc ferrite and/or magnetite were identified. Additionally, a trace amount of ZnO (at the limit of detection of 0.5 wt pct) was identified in sample S1’ (Figure 1(b)). The stronger broadening of S1–S4 XRD peaks suggests that S1′–S4′ samples consist of significantly larger nanocrystallites (Figures 1(a) and (b)). During annealing, the primary grains of solid products expand by coalescence[43,45,50] (visible sharp peaks in XRD spectra, Figure 1(b)).
The crystal cell parameters as a function of Zn concentration for the as-prepared samples (S1–S4) and for solid products after thermal treatment (S1’–S4’) were determined by Rietveld refinement and are given in Table I.
The XRD analysis indicates that the lattice constant for samples S1–S4 and for S1–S4′ decreases with Zn content in the structure (Table II, Figure 2). For the as-prepared samples, the observed changes are the result of a variable Zn concentration and distribution,[69] which was controlled by a mutual Zn/Fe ratio during synthesis. However, in the case of annealed samples, the lattice parameter a was decreasing and approaching values expected for the lattice constant of magnetite Fe3O4 substituted with Zn in the tetrahedral site, depicted as normal (spinel) in Figure 2. This is in line with the hypothesis of a partial thermochemical decomposition of the zinc ferrite including sublimation and partial reduction to produce “new” ZnxFe3−xO4(s), which is a solid solution between ZnFe2O4 and Fe3O4. Similar results were obtained in the previous study.[53]

Expected evolution of the lattice parameters of NZF vs. Znx doping concentration (lines) compared to the results of XRD analysis (points). The solid line approximates the lattice constant dependence on x between franklinite and magnetite (as for Zn2+ locating in Td sites), while the dashed line estimates the dependence between the S1 sample and magnetite (as for Zn2+ locating in Oh sites)
4.3 Morphology and Composition of Zinc Ferrite
The TEM pictures for all as-prepared nanoparticles and the corresponding selected area electron diffraction patterns (SAED) are shown in Figure 3. SAED patterns confirm the FCC phase (Fd–3m space group). The morphology of the S1–S4 samples is spherical in shape.

TEM micrographs of as-prepared nanoparticles with size histograms shown in the insets and corresponding SAED patterns
The crystal size values determined by XRD (Table I) are in good agreement with the particle size determined by TEM using ImageJ software. With a decreasing Zn content in the structure the average grain size increases (Table I). Generally, all samples after thermal treatment present a common tendency to increase in size by the coalescence process,[43,44] which is in agreement with the TEM results (Figure 4) and XRD investigation (Figures 1(a) and (b)) and Table I). The EDX qualitative analysis of the chemical composition of the final solid products indicates significant changes of peak intensity, especially originating from oxygen, zinc or iron, which suggests that the annealing process leads to essential structural evolution in nanoparticles.

TEM images for solid products after thermal treatment of sample S3′ up to 1400 °C (average crystal size calculated using Scherer’s formula was about 290 nm)
4.4 XPS Analysis
XPS spectra of as-prepared ferrite nanoparticles S1–S4 and for materials after thermal treatment S1’–S4’ that allowed quantifying their composition (C, Si, O, Fe and Zn elements) collected for all samples are presented in Figure 5. Chemical composition of the as-prepared ferrites was determined based on the XPS data and compared with the ICP-OES results (Table II). The XPS formulas match well with the ICP-OES results only for the S4 sample, with a small amount of zinc. For samples S1–S3, the amount of zinc in the structure is larger than that obtained via ICP-OES. Thus, such a large amount of Zn may suggest a higher accumulation of zinc in the surface layer of the nanoparticles,[6] because the XPS information depth is about 4.5 nm for Fe3O4 and 10 nm for the organic layers.[71,72]

Fe2p3/2 XPS high-resolution spectra of (a through d) obtained for ferrite samples S1–S4 and (e through h) samples S1’–S4’ after thermal treatment
In addition, the chemical composition of the surface of particles was examined before and after thermal treatment. Table III summarizes the results for the ferrite samples before and after heating. The ratio of zinc to iron in the samples increases from S1–S4. A trend in the zinc content is similar in the samples after heat treatment. However, the zinc content decreases noticeably (Table III). This decrease in zinc concentration in samples after heating may be due to the release of zinc oxide from the nanocrystalline structure. It is possible because ZnO sublimes congruently and then dissociates to Zn(g) and O2(g) under the influence of high temperature.[73,74,75,76] Such results are in agreement with the XRD analysis (Figure 1, Table I).
Furthermore, high-resolution spectra of Zn 2p and Fe 2p regions were analyzed to determine the chemical states of the elements in all the samples before and after heating. Analyses of Fe 2p spectra (Figure 5) for all samples, except S1, showed the presence of Fe2 + (709.2 eV) and Fe3 + (710.5 eV) in octahedral [ ]Oh sites and Fe3+ (712.0 eV) in tetrahedral ()Td interstices.[77,78] The Fe 2p3/2 peak observed at 709.3 eV is assigned to Fe2+ species present in the ferrite, and 2p3/2 peaks at 710.5 and 712.0 eV are attributed to Fe3+ species, respectively.[79,80]
After the heating process, an increase in the Fe2+ content in the all tested ferrites was observed (Table IV). These observations display agreement between the XPS measurements and Mössbauer’s data.
4.5 HERFD-XAS Analysis
Fe 1s2p HERFD-XAS shows a typical shape of spinel oxide for all the studied samples. In the near-edge range (Figure 6(a)), the only significant evolution due to thermal treatment is a considerable sharpening of the features (multiple scattering peaks). This is consistent with the increase of structural order due to the increase of crystallite size as derived from XRD. On the other hand, a significant change is visible in the pre-edge range (Figures 6(b) and (c)), which reflects a weighted average of the spectral shape of all the excited iron sites. It is predominantly sensitive to density of unoccupied 3d states of transition metal ions and their multiplet structure.[82] In the case of spinel ferrites, it consists of combined signals from Td and Oh coordinated iron ions in various oxidation states.

Fe 1s2p HERFD-XAS in the near-edge region of S1 and S1′ (a) and pre-edge region of S1–S3 (b) and S1–S3′ (c). Reference spectra of magnetite (Fe3O4, mixed Fe2+/3+, mixed Oh/Td) and hematite (α-Fe2O3, pure Fe3+, pure Oh) are shown for comparison in (b, c)
The K pre-edge spectrum of each of the as-synthesized samples studied (S1–S3) is different. The spectrum of S1 agrees well with that of the hematite reference (pure Fe3+ in octahedral sites), except that the minimum between the two characteristic peaks is weaker; this is attributed to the small admixture of Fe3+ in tetrahedral sites. A single pre-edge peak is present in the spectrum of sample S2. It is similar to that of magnetite; however, it is shifted to higher photon energy as in the case of maghemite (γ-Fe2O3).[83] This pre-edge shape suggests that sample S2 is formed predominantly from Fe3+ ions, which are distributed between Oh and Td sites. The spectrum of S3 shows a characteristic maximum at the same energy (7.1139 keV) as magnetite, which is an indication of the presence of tetrahedrally coordinated Fe3+ ions and octahedrally coordinated Fe2+. However, the lower peak intensity and a noticeable bump at the high energy shoulder agree with lower Fe occupation of Td with respect to Oh sites by Fe3+ ions compared to pristine Fe3O4. As such, the evolution observed in the Fe K pre-edge spectra (Figure 6(b)) of the as-synthesized samples agrees qualitatively with the hypothesis of formation of inverse (or mixed) spinel in NZF nanoparticles.
Noticeable evolution upon thermal treatment is present in the Fe K pre-edge range, which is revealed in a gradual shift of the spectral weight towards lower photon energy visible in Figure 6(c). This is a clear indication of the increasing number of Fe2+ ions in the total population of iron. The strongest evolution is observed for the samples with high Zn content (S1′ vs. S1 and S2′ vs. S2), while it is much weaker in case of the S3′ (vs. S3) sample. This observation agrees with the scenario of significant reduction of iron ions caused by annealing, which is suggested by XPS. However, HERFD-XAS proves that the effect is present also in the volume of particles. To quantify the relative amount of ferrous to ferric iron ions, we performed a detailed Mössbauer spectroscopy study, which is discussed in the next section.
4.6 Mössbauer Spectroscopy
57Fe Mössbauer spectroscopy was performed to determine the structural and chemical properties of iron (i.e., cation distribution in the spinel lattice and oxidation state) for both as-prepared ZnxFe3−xO4 nanoparticle samples (S1–S4) and their solid products after thermal treatment (S1′–S4′). The Mössbauer spectra (MS) were recorded at room (300 K) and cryogenic (80 K) temperatures (Figure 7).


57Fe Mössbauer spectra recorded at 300 K and 80 K for: (a) as-prepared nanoparticle samples (S1–S4) and (b) solid products after thermal treatment up to 1400 °C (S1′–S4′)
Mössbauer spectra samples S1–S4 were analyzed in terms of two components. For some components superparamagnetic relaxation effects were taken into account in the frame of the relaxation Blume-Tjon model,[84] assuming fluctuation frequency anisotropy.[85,86] Component relative contributions, isomer shift IS and mean 〈IS〉 values for as-prepared ZnxFe3−xO4 samples measured at 300 K and 80 K are presented in Table V.
The inversion parameters σ were calculated (Table V) assuming that spectrum component contributions correspond to the relative iron occupation at tetrahedral Td and octahedral Oh positions,[24] respectively: (Znx−σ Fe1−x+σ)Td [Zn σ Fe2−σ]Oh O4
where r = C2/C1 and x = Zn amount.
The results in Table VI show that the as-prepared nanoparticles (S1–S4) have a mixed spinel structure. Thermally treated samples S2′–S4′ exhibit large magnetic splitting indicating strong magnetic iron interactions. The spectra were analyzed in terms of a number of magnetic components and the products of ferrite nanoparticle decomposition. The results of the analysis for spectra measured at 300 K and 80 K are presented in Table VI.
Mean isomer shift 〈IS〉 as a function of Zn content for measurements at 300 K and 80 K is presented in Figure 8. Isomer shift IS represents mutual proportions of Fe3+ and Fe2+. Generally IS < 0.5 mm/s corresponds to Fe3+ while IS > 0.5 mm/s corresponds to Fe2+.[34] For zinc ferrite nanoparticles of non-stoichiometric composition, especially for small nanoparticles (d < 10 nm), the determined above boundaries are not always identified in practice.[5,24] 〈IS〉 for the as-prepared samples is characteristic for low-spin Fe3+ and does not change with Zn content (approximately IS300 K = 0.35 mm/s, IS80 K = 0.45 mm/s). They are close to 〈IS〉 values for maghemite, γ-Fe2O3, at these temperatures (approximately 〈IS〉300 K = 0.32 mm/s, 〈IS〉80 K = 0.42 mm/s). An increase of 〈IS〉 is observed for thermally treated (S1′–S4′) samples as a function of Zn ion concentration (Figure 8). This probably testifies to a partial reduction of Fe3+ ions to Fe2+ ions as a result of thermal decomposition of zinc ferrite nanoparticles [53] due to reaction (1):

Mean isomer shift 〈IS〉, determined for the as-prepared (S1–S4) samples and after thermal treatment (samples S1′–S4′), measured at 300 K (a) and 80 K (b)
This behavior of the nanoparticles of zinc ferrite at high temperature (partially thermochemical decomposition) means that zinc ferrite nanoparticles can be potentially used in the functionalization of binding materials in the foundry industry to improve the quality of the castings.[87]
Making use of isomer shift scaling between maghemite and magnetite nanoparticles,[88] and assuming that zinc ferrite and maghemite have nearly the same isomer shift, the degree of Fe3+ reduction in decomposition process (1) was estimated (Table VII). The detailed data concerning the decomposition process of the considered (S1′–S4′) samples are listed in Table VII.
As an example, when approximately 6 pct moles of Fe2+ ions were formed in solid products after decomposition of Zn0.35Fe2.65O4 at a temperature of 1400 °C, this means that approximately 24 pct moles of initial ferrite (Zn0.35Fe2.65O4)—because of its thermochemical decomposition—underwent conversion to Fe3O4 forming “new ferrite” Zn0.19Fe2.81O4, which is a solid solution between Zn0.35Fe2.65O4 and Fe3O4.[53]
4.7 Magnetic Properties
The evolution of magnetic properties accompanying the chemical changes of S1–S4 and S1′–S4′ samples was determined based on measurements of magnetization M vs. applied field H at room (300 K) as well as at cryogenic (80 K) temperatures (Figure 9). Saturation magnetization, Ms, was estimated at the maximum field applied, i.e., 15 kOe, and further refined based on the law of approach to saturation (LAS)[36,89] according to Eq. [1]:
where MS = saturation magnetization the at a particular temperature, \( {{\chi }}_{\text{dia}} \) = high field susceptibility, kB = the Boltzmann constant.

VSM results at 80 K and 300 K for the as-prepared (a, c) and after thermal treatment samples (b, d)
In this way, it is possible disentangle the relative signal that originates from the superparamagnetic phase characteristic for NZF from that of paramagnetic ZF. The results obtained indicate the superparamagnetic character of the as-prepared samples (S1–S4) (Figure 9). However, a significant fraction of the paramagnetic signal recorded on the S1 sample agrees well with the expected ZF composition. The magnetization saturation (Ms) falls along with increasing zinc content in a tested range of 0.35 ≤ x ≤ 1.0, coming to near zero value for x = 1 (S1). The highest Ms value equal to 52.8 emu/g (at 300 K) is exhibited by nanoparticle S4, for which x = 0.35 (Figure 9, Table VIII). Similar behavior is observed at 80 K (Figure 9, Table VIII).
After annealing up to 1400 °C, the S1–S4′ samples indicated Ms values increased from 2 to 3 times (at 80 K) and from 2 to 4 times (at 300 K) in relation to the initial samples (Table VIII, Figures 9 and 10).

Ms values at 80 K for nanoparticles of the as-prepared (S1–S4) and for after annealing (S1–S4′) samples as a function of Zn content
Generally, for superparamagnetic nanoparticles, the field-cooled (FC) magnetization monotonically increases with decreasing temperature.[36,90] Such temperature dependence of FC confirms that the as-prepared samples, e.g., S3 (Figure 11(a)), are formed by superparamagnetic and paramagnetic phases, which is characteristic for distribution of NZF NPs of < 10 nm mean diameter. After annealing (Figure 11(b)), the temperature dependence of magnetization changes to ferri-/ferromagnetic, which is expected after an increase of Fe2+ ion content, decrease in Zn content and significant growth of grains, in line with the observations of XRD, Mössbauer, XPS, HERFD-XAS and other investigations.[91]

Field cool (FC) magnetization curves of ZF NPs: (a) as-prepared (S3, x = 0.53) and (b) after thermal treatment (S3′) in the applied field of 100 Oe
Based on the systematic investigation of non-stoichiometric zinc ferrite nanoparticles by means of XRD, XPS, Mössbauer spectroscopy, VSM and HERFD-XAS, the probable cation distribution in the spinel structure can be proposed. It is discussed on the basis of the expected magnetic properties of nanoparticles, as-prepared (S1–S4) and solid products, after their decomposition at 1400 °C (S1–S4′) following the models proposed in References 24, 34,35.
-
for the as-prepared (S1–S4) samples:
the observed decrease of the Ms value accompanying the Zn concentration increase, 0.35 ≤ x ≤ 1 is probably the effect of the domination of non-magnetic Zn2+ ions, which are in ()Td as well as [ ]Oh sites, according to the model below:
where “x” denotes the concentration of Zn2+ ions; in the ()Td sites, the concentration of Zn2+ is smaller than in the [ ]Oh sites.
The mutual fraction of Zn2+ cations in the Td and Oh sites is changing along with Zn concentration changes in ferrite. These changes occur in such a way that at higher concentrations the fraction of Zn2+ ions in the Oh sites is larger than in the Td sites. This is in agreement with investigations presented in References 24, 34, 41, 42.
-
for the solid products after decomposition at 1400 °C (S1–S4′):
the clearly observed increase of the Ms value is probably caused by non-magnetic Zn2+ ions existing only in the Td sites, in which they are partially substituting Fe3+ ions, according to the model:
where “x” denotes the concentration of Zn2+ ions.
Thus, to maintain the charge equilibrium, the number of Fe2+ ions in the octahedral sites decreases, according to (1 − x). Since the magnetic moment of Fe3+ ions is higher than that of Fe2+ ions, the total magnetic moment increases.
Investigations in References 34, 35 confirm that this distribution of cations in the spinel lattice of zinc ferrite NPs, at relatively low concentrations of Zn 0.2 ≤ x ≤ 0.4, is responsible for the high Ms values (e.g., 154 emu/g for x = 0.2).[35]
5 Conclusion
As revealed by XAS and Mossbauer spectroscopy, the stoichiometric ZF NPs (S1) have mixed spinel structure, i.e., Zn2 + ions occupy both tetrahedral and octahedral sites. Mossbauer spectroscopy of the NZF NPs (S2–S4) also reveals a mixed spinel structure, i.e., the inversion degree σ was respectively 0.80, 0.77 and 0.57 (Table V).
Upon systematic investigation of the evolution of the structural and magnetic properties of ZF and NZF NPs upon annealing to 1400 °C in the inert atmosphere (at a constant heating rate ß = 10 °C/min), we can draw the following conclusions:
-
annealing of ZF and NZF NPs leads to re-growing of grains by means of coalescence and sintering (XRD, TEM, Mössbauer spectroscopy);
-
the tested NZF NPs samples (S2–S4) are undergoing thermochemical decomposition in approximately 20 pct under the applied conditions (annealing up to 1400 °C, inert atmosphere), as shown by Mössbauer spectroscopy, XPS, XANES and VSM;
-
for ZF NPs of stoichiometric composition (S1), the decomposition degree under the applied conditions is twice lower than for NZF NPs;
-
Mössbauer spectroscopy proves that after annealing NZF NPs are enriched in Fe2+ ions and depleted of Zn2+ ions (shown by XRD and XPS);
-
zinc ferrite of a different composition than that of the initial sample is a solid solution of “initial zinc ferrite” and magnetite Fe3O4 (cf. Table VI, Mössbauer spectroscopy, XRD, XANES, XPS);
-
XRD investigations indicate that annealing at a temperature up to 1400 °C causes a decrease of the lattice constant of samples;
-
zinc ferrites formed after annealing indicate a significant increase of the magnetization saturation (in relation to their initial values), probably being the effect of reduction and/or relocation of cations in the spinel lattice.
References
1 R.H. Kodama: J. Magn. Magn. Mater., 1999, vol. 200, pp. 359–72.
J.Z.J. Changwa Yao, Qiaoshi Zeng G. F. Goya, T. Torres, Jianfang Liu, Haiping Wu, Mingyuan Ge, Yuengu Zeng, Youwen Wang: J. Phys. Chem. C, 2007, vol. 111, pp. 12274–78.
3 S. Bullita, A. Casu, M.F. Casula, G. Concas, F. Congiu, A. Corrias, A. Falqui, D. Loche, and C. Marras: Phys. Chem. Chem. Phys., 2014, vol. 16, p. 4843.
A. Najafi Birgani, M. Niyaifar, and A. Hasanpour: J. Magn. Magn. Mater., 2015, vol. 374, p. 179.
5 W. Szczerba, J. Zukrowski, M. Przybylski, M. Sikora, O. Safonova, A. Shmeliov, V. Nicolosi, M. Schneider, T. Granath, M. Oppmann, M. Straßer, and K. Mandel: Phys. Chem. Chem. Phys., 2016, vol. 18, pp. 25221–9.
D. Lachowicz, R. Wirecka, W. Górka-Kumik, M.M. Marzec, M. Gajewska, A. Kmita, J. Żukrowski, M. Sikora, S. Zapotoczny, and A. Bernasik: Phys. Chem. Chem. Phys., 2019, vol. 21, p. 23473.
7 R.M. Borade, S.B. Somvanshi, S.B. Kale, R.P. Pawar, and K.M. Jadhav: Matarials Res. Express, 2020, vol. 7, p. 016116.
8 L. Kaykan, A.K. Sijo, A. Żywczak, J. Mazurenko, and K. Bandura: Appl. Nanosci., 2020, vol. 10, pp. 4577–4583.
Z. Nemati, J. Alonso, I. Rodrigo, R. Das, E. Garaio, J.Á. García, I. Orue, M.H. Phan, and H. Srikanth: J. Phys. Chem. C, 2018, vol. 122, p. 2367.
10 A.J. Giustini, A.A. Petryk, S.M. Cassim, J.A. Tate, I. Baker, and P.J. Hoopes: Nano Life, 2010, vol. 01, pp. 17–32.
11 Y. Xu, Y. Qin, S. Palchoudhury, and Y. Bao: Langmuir, 2011, vol. 27, pp. 8990–7.
12 R.P. Sharma, S.D. Raut, A.S. Kadam, R.M. Mulani, and R.S. Mane: Appl. Phys. A, 2020, vol. 126, p. 824.
13 K. Raj and R.J. Boulton: Mater. Des., 1987, vol. 8, pp. 233–6.
14 M.P. Pileni: Adv. Funct. Mater., 2001, vol. 11, pp. 323–36.
15 A.S. Drozdov, V. Ivanovski, D. Avnir, and V. V. Vinogradov: J. Colloid Interface Sci., 2016, vol. 468, pp. 307–12.
T.H. Shin, Y. Choi, S. Kim, and J. Cheon: Chem. Soc. Rev., 2015, 44, vol. 44.
17 L. Vékás, D. Bica, and M. V. Avdeev: China Particuology, 2007, vol. 5, pp. 43–9.
18 L. Mohammed, H.G. Gomaa, D. Ragab, and J. Zhu: Particuology, 2017, vol. 30, pp. 1–14.
19 A. Afzal, A. Mujahid, N. Iqbal, R. Javaid, and U.Y. Qazi: Nanomaterials, 2020, vol. 10, p. 2133.
M. Qin, Q. Shuai, G. Wu, B. Zheng, Z. Wang, and H. Wu: Mater. Sci. Eng. B Solid-State Mater. Adv. Technol., 2017, vol. 224, pp. 125–38.
21 A.G. Hufnagel, K. Peters, A. Müller, C. Scheu, D. Fattakhova‐Rohlfing, and T. Bein: Adv. Funct. Mater., 2016, vol. 26, pp. 4435–43.
22 D. Makovec and M. Drofenik: J. Nanoparticle Res., 2008, vol. 10, pp. 131–41.
23 Y. Yang, X. Liu, Y. Yang, W. Xiao, Z. Li, D. Xue, F. Li, and J. Ding: J. Mater. Chem. C, 2013, vol. 1, pp. 2875–85.
24 M. Srivastava, S.K. Alla, S.S. Meena, N.Gupta, R.K. Mandala, and N.K. Prasad: New J. Chem., 2018, vol. 42, pp. 7144–53.
25 G. Muscas, N. Yaacoub, G. Concas, F. Sayed, R.S. Hassan, J.M. Greneche, C. Cannas, A. Musinu, V. Foglietti, S. Casciardi, C. Sangregoriohe, and D. Peddis: Nanoscale, 2015, vol. 7, pp. 13576–85.
26 S.J. Stewart, I.A. Al-Omari, F.R.Sives, and H.M.Widatallah: J. Alloys Compd., 2010, vol. 495, pp. 506–8.
S.J. Stewart, S.J.A. Figueroa, J.M. Ramallo-Lopez, F.G. Reguejo, S.G. Marchetti, J.F. Benoga, and R.J. Prado: Cond. Mat. Mtrl. Sci.
28 G.F. Goya and H.R. Rechenberg: J. Magn. Magn. Mater., 1999, vol. 196–197, pp. 191–2.
J. Kuciakowski, A. Kmita, D. Lachowicz, M. Wytrwal-Sarna, K. Pitala, S. Lafuerza, D. Koziej, A. Juhin, and M. Sikora: Nanoscale, 2020, pp. 16420–26.
30 M. Vucinic-Vasic, E.S. Bozin, L. Bessais, G. Stojanovic, U. Kozmidis-Luburic, M. Abeykoon, B. Jancar, A. Meden, A. Kremenovic, and B. Antic: J. Phys. Chem. C, 2013, vol. 117, pp. 12358–65.
J. Hölscher, L.A. H., M. Saura-Múzquiz, P.G. Garbusa, and M. Christensen: CrystEngComm, 2020, vol. 22, pp. 515–24.
B. Antic, M. Perovic, A. Kremenovic, J. Blanusa, V. Spasojevic, P. Vulic, L. Bessais, and E.S. Bozin: J. Phys. Condens. Matter.
33 J. Yao, Y. Li, X. Li, and X. Zhu: Metall. Mater. Trans. A, 2016, vol. 47A, pp. 3753–60.
34 Y. Ma, J. Xia, C. Yao, F. Yang, S.G. Stanciu, P.Li, Y. Jin, T. Chen, J. Zheng, G. Chen, H. Yang, and L. Luo: Chem. Mater., 2019, vol. 31, pp. 7255–64.
35 J. Jang, H. Nah, J. Lee, S. Moon, M. Kim, and J. Cheon: Angew. Chem. Int. Ed., 2009, vol. 48, pp. 1234–8.
36 P.T. Phong, P.H. Nam, N.X. Phuc, B.T. Huy, L.T. Lu, D.H. Manh, and I.J. Lee: Metall. Mater. Trans. A, 2019, vol. 50, pp. 1571–1581.
37 Y. Hadadian, A.P. Ramos, and T.Z. Pavan: Sci. Rep., 2019, vol. 9, p. 18048.
38 N.J. Orsini, M.M. Milić, and T.E. Torres: Nanotechnology, 2020, vol. 31, pp. 1–33.
39 H. Kavas, A. Baykal, M.S. Toprak, Y. Koseoglu, M. Sertkol, and B. Aktas: J. Alloys Compd., 2009, vol. 479, pp. 49–55.
40 C. Upadhyay, H.C. Verma, V. Sathe, and A. V. Pimpale: J. Magn. Magn. Mater., 2007, vol. 312, pp. 271–9.
41 Z. Beji, A. Hanini, L.S. Smiri, J. Gavard, K. Kacem, F. Villain, J.-M. Grenèche, F. Chau, and S. Ammar: Chem. Mater., 2010, vol. 22, pp. 5420–9.
A.H. Morrish: The Physical Principles of Magnetism., Wiley-IEEE Press, 2001.
43 S. Mallesh and V. Srinivas: J. Magn. Magn. Mater., 2018, vol. 475, pp. 290–303.
44 Y. El Mendili, J.-F. Bardeau, N. Randrianantoandro, F. Grasset, and J.M. Greneche: J. Phys. Chem. C, 2012, vol. 116, pp. 23785–92.
45 J. Philip, G. Gnanaprakash, G. Panneerselvam, M.P. Antony, T. Jayakumar, and B. Raj: J. Appl. Phys., 2007, vol. 102, p. 054305.
46 P. Sivagurunathan and K. Sathiyamurthy: Can. Chem. J., 2016, vol. 4, pp. 244–54.
47 M. Amir, H. Gungunes, A. Baykal, M.A. Almessiere, H. Sozeri, I. Ercan, M. Sertkol, and S. Asiri: J. Supercond. Nov. Magn., 2018, vol. 31, pp. 3347–3356.
48 S. Singh, N. Kumar, A. Jha, M. Sahni, R. Bhargava, A. Chawla, R. Chandra, S. Kumar, and S. Chaubey: J. Supercond. Nov. Magn., 2014, vol. 27, pp. 821–826.
49 M.A. Cobos, P. Presa, I. Llorente, J.M. Alonso, A. García-Escorial, P. Marín, A. Hernando, and J. Jiménez: J. Phys. Chem. C, 2019, vol. 123, pp. 17472–82.
J.P. Singh, R.S. Payal, R.C. Srivastava, H.M. Agrawal, P. Chand, A. Tripathi, and R.P. Tripathi: in Journal of Physics: Conference Series. International Conference on the Applications of the Mössbauer Effect (ICAME 2009), Vienna, Austria, 2009.
51 A. Kmita, J. Żukrowski, K. Hodor, H. Smogór, and M. Sikora: Metalurgija, 2017, vol. 56, pp. 29–32.
52 Y. Tamaura and H. Kaneko: Sol. Energy, 2005, vol. 78, pp. 616–22.
53 H. Kaneko, T. Kodama, N. Gokon, Y. Tamaura, K. Lovegrove, and A. Luzzi: Sol. Energy, 2004, vol. 76, pp. 317–22.
54 B. Janković, S. Stopić, A. Güven, and B. Friedrich: J. Magn. Magn. Mater., 2014, vol. 358–359, pp. 105–18.
S. Stopić and B. Friedrich: in Proceedings of EMC, 2009, pp. 1–14.
56 A. Kmita, A. Pribulova, M. Holtzer, P. Futas, and A. Roczniak: Arch. Metall. Mater., 2016, vol. 61, pp. 2141–6.
57 V.Š. P. Baláz, M. Bálintová, Z. Bastl, J. Briančin: Solid State Ionics, 1997, vol. 101–103, pp. 45–51.
58 G. Fan, Z. Gu, L. Yang, and F. Li: Chem. Eng. J., 2009, vol. 155, pp. 534–41.
59 Z. Jia, D. Ren, Y. Liang, and R. Zhu: Mater. Lett., 2011, vol. 65, pp. 3116–9.
60 J. Zhao, L. Mi, H. Hou, X. Shi, and Y. Fan: Mater. Lett., 2007, vol. 61, pp. 4196–8.
61 S. Ayyappan, S.P. Raja, C. Venkateswaran, J. Philip, and B. Raj: Appl. Phys. Lett., 2010, vol. 96, p. 143106.
62 S. Ayyappan, G. Paneerselvam, M.P. Antony, and J. Philip: Mater. Chem. Phys., 2011, vol. 128, pp. 400–4.
R. Raeisi Shahraki, M. Ebrahimi, S.A. Seyyed Ebrahimi, and S.M. Masoudpanah: J. Magn. Magn. Mater., 2012, vol. 324, pp. 3762–65.
N. Pinna, S. Grancharov, P. Beato, P. Bonville, M. Antonietti, and M. Niederberger: Chem. Mater.
S.E. Gilliland, E.E. Carpenter, and M.D. Shultz: Nanobiomedicine, 2014, vol. 1, pp. 1–15.
N. Pinna and M. Niederberger: Angew. Chemie, 2008, vol. 47, pp. 5292 – 5304.
I. Bilecka, I. Djerdja, and M. Niederberger: Chem. Commun., 2008, vol. 7, pp. 886–8.
A. Kmita, D. Lachowicz, J. Żukrowski, M. Gajewska, W. Szczerba, J. Kuciakowski, S. Zapotoczny, and M. Sikora: Materials (Basel)., 2019, vol. 12, p. 1048.
M. Rivero, A. Campo, Á. Mayoral, E. Mazario, J. Sánchez-Marcos, and A. Muñoz-Bonilla: RSC Adv., 2016, vol. 6, pp. 40067–76.
B.T. Naughton and D.R. Clarke: J. Am. Ceram. Soc., 2008, vol. 91, pp. 1253–7.
H. Shinotsuka, S. Tanuma, C.J. Powell, and D.R. Penn: Surf. Interface Anal., 2015, vol. 47, pp. 1132–1132.
Z. Beji, M. Sun, L.S. Smiri, F. Herbst, C. Mangeneya, and S. Ammar: RSC Adv., 2015, vol. 80, pp. 65010–22.
L. Brewer and D.F. Mastick: J. Chem. Phys., 1951, vol. 19, pp. 835–43.
D.F. Anthrop and A.W. Searcy: J. Phys. Chem., 1964, vol. 68, pp. 2335–42.
A. Weidenkaff: Ph.D. Thesis, ETH Zurich, PSI, Zurich., 1998.
A. Weidenkaff, A. Steinfeld, A. Wokaun, P.O. Auer, B. Eichler, and A. Reller: Sol. Energy Vol. 65, 1999, vol. 65, pp. 59–69.
D. Wilson and M.A. Langell: Appl. Surf. Sci., 2014, vol. 303, pp. 6–13.
T. Radu, C. Iacovita, D. Benea, and R. Turcu: Appl. Surf. Sci., 2017, vol. 405, pp. 337–43.
Y. Joly, J.E. Lorenzo, E. Nazarenko, J.-L. Hodeau, D. Mannix, and C. Marin: Phys. Rev. B, 2008, vol. 78, p. 134110.
T. Baidya, T. Murayama, S. Nellaiappan, N.K. Katiyar, P. Bera, O. Safonova, M. Lin, K.R. Priolkar, S. Kundu, B.S. Rao, P. Steiger, S. Sharma, K. Biswas, S.K. Pradhan, N. Lingaiah, K.D. Malviya, and M. Haruta: J. Mater. Chem. C, 2019, vol. 123, pp. 19557–19571.
S. Poulin, R. França, L. Moreau-Bélanger, and E. Sacher: J. Phys. Chem. C, 2010, vol. 114, pp. 10711–10718.
F. de Groot, G. Vankó, and P. Glatzel: J. Phys. Condens. Matter., 2009, vol. 21, p. 104207.
M. Wilke, F. Farges, P.-E. Petit, G.E.J. Brown, and F. Martin: Am. Mineral., 2001, vol. 86, pp. 714–612.
M. Blume and A. Tjon: Phys. Rev.
D.G. Rancourt and M.J. Daniels: Phys. Rev. B, 1984, vol. 29, pp. 2410–4.
D.G. Rancourt: Hyperfine Interact., 1998, vol. 117, pp. 3–38.
M. Holtzer and A. Kmita: Mold and Core Sands in Metalcasting: Chemistry and Ecology. Sustainable Development., Springer Nature Switzerland AG, 2020.
J. Fock, L.K. Bogart, D. González-Alonso, J.I. Espeso, M.F. Hansen, M. Varón, C. Frandsen, and Q.A. Pankhurst: J. Phys. D. Appl. Phys., 2017, vol. 50, p. 265005.
J.P. Wang, D.-H. Han, H.-L. Luo, Q.-X. Lu, and Y.-W. Sun: Appl. Phys. A, 1995, vol. 61, pp. 407–15.
M. Suzuki, S.I. Fullem, I.S. Suzuki, L. Wang, and C.-J. Zhong: Phys. Rev. B, 2009, p. 024418.
T. Kamiyama, K. Haneda, T. Sato, S. Ikeda, and H. Asano: Solid State Commun., 1992, vol. 81, pp. 563–6.
Acknowledgments
This work was supported by the National Science Centre, Poland, Grant number 2016/23/D/ST8/00013.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Manuscript submitted September 10, 2020; accepted January 2, 2021.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kmita, A., Żukrowski, J., Kuciakowski, J. et al. Effect of Thermal Treatment at Inert Atmosphere on Structural and Magnetic Properties of Non-stoichiometric Zinc Ferrite Nanoparticles. Metall Mater Trans A 52, 1632–1648 (2021). https://doi.org/10.1007/s11661-021-06154-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-021-06154-3