
Abstract
Cancer stem cells (CSCs), which are critical targets for cancer therapy as they are involved in drug resistance to anticancer drugs, and metastasis, are maintained by angiocrine factors produced by particular niches that form within tumor tissue. Secreted frizzled-related protein 1 (Sfrp1) is an extracellular protein that modulates Wnt signaling. However, the cells that produce Sfrp1 in the tumor environment and its function remain unclear. We aimed to elucidate angiocrine factors related to CSC maintenance, focusing on Sfrp1. Although Sfrp1 is a Wnt pathway-related factor, its impact on tumor tissues remains unknown. We investigated the localization of Sfrp1 in tumors and found that it is expressed in some tumor vessels. Analysis of mice lacking Sfrp1 showed that tumor growth was suppressed in Sfrp1-deficient tumor tissues. Flow cytometry analysis indicated that CSCs were maintained in the early tumor growth phase in the Sfrp1 knockout (KO) mouse model of tumor-bearing cancer. However, tumor growth was inhibited in the late tumor growth phase because of the inability to maintain CSCs. Real-time PCR results from tumors of Sfrp1 KO mice showed that the expression of Wnt signaling target genes significantly decreased in the late stage of tumor growth. This suggests that Sfrp1, an angiocrine factor produced by the tumor vascular niche, is involved in Wnt signaling-mediated mechanisms in tumor tissues.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The vascular network is not only responsible for the transport of oxygen and nutrients but also plays a critical role in organogenesis and homeostasis through the production of angiocrine factors. Angiocrine factors are secreted molecules expressed by vascular endothelial cells (ECs) and other components of blood vessels that act on cell populations in the tissue microenvironment (Rafii et al. 2016). Blood vessels that produce angiocrine factors are called vascular niches and regulate the differentiation and proliferation of stem cells, including neural and hematopoietic stem cells, in various tissues (Ramasamy et al. 2016; Karakatsani et al. 2023). In cancer tissues, blood vessels form a niche that contributes to the maintenance of cancer stem cells and regulates their proliferation (Alsina-Sanchis et al. 2021). Therefore, the tumor vascular niche and angiocrine factors are potential targets for cancer therapies. CSCs in tumor tissues represent a highly malignant, specialized population of cancer cells (CCs). The frequencies of metastasis and recurrence, which determine the malignant potential of cancer, are largely dependent on the degree of CSC control (Batlle and Clevers 2017; Atkins et al. 2019). Tumor blood vessels in tumor tissue not only supply nutrients to CCs but also form a niche in which CSCs are maintained (Krishnamurthy et al. 2010; Hira et al. 2015). Direct targeting of CSCs is challenging because of their drug resistance (Ishimoto et al. 2011). Therefore, there is a need to develop indirect therapies targeting the CSC niche.
Secreted frizzled-related protein 1 is an extracellular protein that modulates Wnt signaling. The Wnt pathway is involved in cell proliferation, differentiation, anchoring, apoptosis, and cell cycle regulation (Clevers 2006; Niehrs 2012; Skronska-Wasek et al. 2017; Perugorria et al. 2019). The Sfrp1 protein harbors two structural domains: a carboxy-terminal netrin (NTR) domain and an amino-terminal cysteine-rich domain (CRD). It has been postulated that Sfrp1 serves as an antagonist of Wnt because the CRD of Sfrp1 is similar to that of the frizzled receptor (Finch et al. 1997; Bhat et al. 2007). However, Sfrp1 has been shown to directly activate intracellular signaling by binding to Frizzled receptors and may modulate the Wnt signaling cascade in positive and negative ways in a context-dependent manner (Rodriguez et al. 2005). The expression of Sfrp1 is downregulated in cancer tissues and is presumed to function in cancer suppression by dysregulating cell proliferation and invasion (Atschekzei et al. 2012). However, the cells that produce Sfrp1 in the tumor microenvironment and the function of Sfrp1 remain underexplored. In this study, we clarified the effects of Sfrp1 on tumor tissues and analyzed its interactions with tumor blood vessels.
Materials and methods
Generation of Sfrp1−/− mice using CRISPR-Cas9 technology
C57BL/6N mice were purchased from Japan SLC (Shizuoka, Japan) and Sankyo Labo Service Corporation (Toyama, Japan). Sfrp1 knockout mice were generated using the CRISPR-Cas9 genome editing system (Hashimoto et al. 2016). C57BL/6N female mice were superovulated by intraperitoneal injection of pregnant gonadotropin, and after 48 h, human chorionic gonadotropin. One-cell-stage zygotes were collected from the oviduct ampulla in M2 medium, followed by injection of a combination of single guide (sgRNA) and Cas9 mRNA through electroporation. These were then implanted into the oviducts of pseudopregnant females. The sgRNA target sequences for Sfrp1 are as follows: gRNA1, 5′-ACA TCG GCT CGT ATC AGA GC-3′; gRNA2, 5′-CTG AGG CTG TGC CAC AAC GT-3′; and gRNA3, 5′-CAA ATG TGA CAA GTT CCC CG-3′. The animals were housed in environmentally controlled rooms of animal experimentation facilities at Osaka and Fukui Universities. All experiments were carried out according to the Osaka and Fukui University Committee for Animals guidelines and approved by the Osaka and Fukui University Institutional Review Boards.
Genotype identification
Sfrp1−/− mutation was identified via Sanger sequencing. Polymerase chain reaction and Sanger sequencing were performed using the following primers: forward primer1, 5′-GCC AGC GAG TAC GAC TAC GTG AG-3′; reverse primer1, 5′-CCA AGG TAA GGG TAT GCC TTC CCA-3′ and forward primer2, 5′-AGG ACC CCA TCG ATC GGA GAC-3′; reverse primer2, 5′-CCA GTC TGG CGT TTT CCA TAC CTG-3′.
Cell lines, plasmid construction, and transfection
Cell lines, including Lewis lung carcinoma (LLC) and MC38, were purchased from the Riken BRC Cell Bank (Tsukuba, Japan). Both cell lines were cultured in Dulbecco’s modified Eagle’s medium (Sigma, St. Louis, MO) containing 10% fetal bovine serum (FBS; Gibco, Grand Island, NY) and 1% penicillin/streptomycin (100 U/mL, PS; Life Technologies, Tokyo, Japan). Full-length Sfrp1 cDNA was isolated from total mouse RNA using PCR-based cloning methods, as described previously (Hayashi et al. 2019). Primers used for cloning were as follows: sense, 5′-TCT ATC CGA ATT CAG CAA CAT GGG CGT CGG GCG-3′ and anti-sense, 5′-TCT ATC CGT CGA CTC ACT TAA AAA CAG ACT GGA-3′. The PCR product containing the EcoRI/SalI fragment was inserted into the pIRES-EGFP vector (Clontech, Mountain View, CA). LLC cells were transfected with pCMV-mSfrp1-IRES2-EGFP containing mouse Sfrp1 or a mock vector as a control. According to the manufacturer’s instructions, all cells were stably transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). GFP+ cells were selected based on antibiotic resistance to G418 (Geneticin; Gibco) by addition to the culture medium and sorted using flow cytometry (FACSAria, BD, San Jose, CA).
Tumor transplantation model
LLC or MC38 cells (1 × 106 cells per mouse in 100 µL phosphate-buffered saline [PBS]) were inoculated subcutaneously into wild-type (WT; C57BL/6) or Sfrp1−/− mice (8–9 wk of age). Tumor volumes were measured using calipers every 2–3 d and calculated as follows: length × width × width × 0.52.
Immunostaining analysis
Tumors were fixed using 4% paraformaldehyde in PBS, treated with 15% sucrose in PBS, followed by 30% sucrose in PBS, and embedded in an optimal cutting temperature compound (Sakura Finetek, Tokyo, Japan). Frozen blocks were sectioned into slices measuring 20 or 40 µm. The following primary antibodies (Abs) were used: rat anti-mouse CD31 Ab (BD Bioscience, Franklin Lakes, NY), hamster anti-mouse CD31 Ab (Merck Millipore, Darmstadt, Germany), rabbit anti-mouse Sfrp1 Ab (Sigma), and αSMA-Cy3 (Merck Millipore). Alexa Fluor 488-conjugated anti-rat IgG (Invitrogen), Alexa Fluor 488-conjugated anti-hamster IgG (Jackson ImmunoResearch Laboratories, West Grove, PA), and Alexa Fluor 546-conjugated anti-rabbit IgG (Invitrogen) were used as secondary Abs. Cell nuclei were visualized using TO-PRO-3 (Invitrogen). The sections were examined using a STELLARIS (Leica, Wetzlar, Germany) instrument. More than four images were captured from the vascular area of each sample and analyzed using ImageJ software for quantitative measurements.
Cell preparation and flow cytometric analysis
Flow cytometry and cell isolation were performed as described previously (Hu et al. 2021). Fluorescently labeled anti-CD44, -CD133 mAbs (BioLegend) were used. The stained cells were sorted using FACSAria (BD Biosciences) or Sonysh800 (Sony, Tokyo, Japan) and analyzed using FlowJo software (Tree Star Software, San Carlos, CA).
Quantitative reverse transcription PCR (qRT-PCR)
Total RNA was extracted from cells using RNeasy-plus mini kits (Qiagen, Hilden, Germany) and reverse-transcribed using the PrimeScript RT reagent kit (Takara, Tokyo, Japan). Real-time PCR was performed using TB Green Premix Ex Taq II (Takara) on an Mx3000p QPCR system (Agilent, Santa Clara, CA). The primers used for PCR are as follows: mouse Sfrp1, sense 5′-CAG CTT TTG AAC TGG CCA CC-3′, anti-sense 5′- CCT TGC CTG GCA TCC TTG TA-3′; mouse Oct4, sense 5′- TGT TCA GCC AGA CCA CCA TC-3′, anti-sense 5′-GCT TCC TCC ACC CAC TTC TC-3′; mouse Abcg2, sense 5′- CTC ACC TTA CTG GCT TCC GG -3′, anti-sense 5′-ATC CGC AGG GTT GTT GTA GG-3′; mouse Sox2, sense 5′-ACA ACT CCA TGA CCA GCT CG-3′, anti-sense 5′-ACT TGA CCA CAG AGC CCA TG-3′; mouse Bmi1, sense 5′-GAC TCT GGG AGT GAC AAG GC-3′, anti-sense 5′-GTG AGG GAA CTG TGG GTG AG-3′; mouse Ssea1, sense 5′-ACA TCA CCG AGA AGC TGT GG-3′, anti-sense 5′-GCA CGA AGC GCT CAT AGT TG-3′; mouse Ccnd1, sense 5′-CCC TGG AGC CCT TGA AGA AG-3′, anti-sense 5′-AGA TGC ACA ACT TCT CGG CA-3′; mouse Axin2, sense 5′-CCT GAC CAA ACA GAC GAC GA-3′, anti-sense 5′-GCT TCT GCC TCG ATC TCC TC-3′; mouse Lef1, sense 5′-GGC ATG AGG TGG TC AGA CAA-3′, anti-sense 5′-TTG TTG TAC AGG CCT CCG TC-3′; mouse GAPDH, sense 5′-TGG CAA AGT GGA GAT TGT TGC C-3′, anti-sense 5′-AAG ATG GTG ATG GGC TTC CCG-3′. The results were normalized to those of GAPDH using the comparative threshold cycle method.
Statistics analysis
The data are presented as the mean ± SEM. GraphPad Prism9 software was used for statistical analysis. Data were analyzed using a t-test. The level of statistical significance was set at p < 0.05 (*p < 0.05, **p < 0.01).
Results
Sfrp1 is expressed in a part of tumor ECs
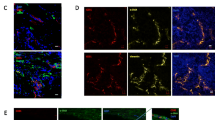
We previously reported that Sfrp1 is expressed in fetal apelin receptor (APJ)–positive veins and is involved in arteriovenous alignment (Kidoya et al. 2015). To determine the localization of Sfrp1, an angiocrine factor critical for angiogenesis, in tumor tissues, we evaluated its expression. We performed immunostaining to determine the distribution of Sfrp1 within the tumors. Immunostaining revealed that Sfrp1 was expressed in a subset of CD31+ vascular ECs (Fig. 1A). Positive Sfrp1 signals were not detected in the area of the mouse LLC cancer cells. Further analysis using ImageJ revealed that approximately 8% of CD31+ cells expressed Sfrp1. These results suggest that Sfrp1 is expressed in a fraction of the vascular ECs in tumor tissues.

Sfrp1 is expressed in a part of tumor ECs. Immunofluorescence staining of CD31 (green), Sfrp1 (red, arrowhead), and TO-PRO-3 (purple) in the frozen section of LLC tumors from WT mice (A). Scale bar = 100 µm. CRISPR/Cas9-mediated gene engineering strategy for Sfrp1 and schematic presentation of the Sfrp1 strategy (B). PCR analysis of DNA from the tail of Sfrp1 KO and WT mice (C). Immunofluorescence staining of CD31 (green), Sfrp1 (red, arrowhead), and TO-PRO-3 (purple) in frozen sections of LLC tumors from WT and Sfrp1 KO mice (D). Scale bar = 100 µm.
Generation of Sfrp1 KO mice
To investigate the effect of Sfrp1 on tumor tissues, the CRISPR/Cas9 gene editing system was used to introduce a deletion into exon 1 of Sfrp1 (Fig. 1B). Sanger sequencing (details shown in Supplementary Fig. 1) and PCR were performed to confirm the deletion of Sfrp1 (Fig. 1C). We confirmed the absence of Sfrp1 protein expression in LLC tumors of Sfrp1 KO mice via immunostaining (Fig. 1D).
Sfrp1 is required for tumor growth
To examine whether Sfrp1 in the tumor environment affected tumor growth, we inoculated LLC cells into WT and Sfrp1−/− (Sfrp1 KO) mice and evaluated their growth. Compared to the WT group, tumors in Sfrp1 KO mice showed reduced tumor growth (Fig. 2A, p < 0.01). In addition, the tumor weight was significantly lower in Sfrp1 KO mice than in WT mice (Fig. 2B, p < 0.05). A similar transplantation experiment was performed using MC38 cells, and tumor growth was suppressed in Sfrp1 KO mice compared with that in WT mice (Fig. 2C, p < 0.01). To further examine the effect of Sfrp1 on tumor growth in tumor tissues, LLC cells overexpressing Sfrp1 were generated using lipofectamine. Real-time PCR confirmed that the expression of Sfrp1 was significantly upregulated (Fig. 2D, p < 0.01). Using these Sfrp1-overexpressing cells, we performed tumor transplantation experiments and found that tumor growth was promoted in Sfrp1-overexpressing cells, and tumor weight was markedly increased (Fig. 2E–F, p < 0.05). These data suggest that Sfrp1 is required for the process of tumor growth in tumor tissues.

Sfrp1 is required for tumor progression. Tumor growth curves and tumor weight of LLC cells subcutaneously (s.c.) inoculated into WT or Sfrp1 KO mice (n = 7 for each group) (A, B). Tumor growth curves of MC38 cells s.c. inoculated into WT or Sfrp1 KO mice (n = 7 for each group) (C). Sfrp1 expression was evaluated via qRT-PCR and normalized against that of GAPDH (D). Tumor growth curves and tumor weight of Sfrp1-overexpressing LLC cells or LLC cells s.c. inoculated into WT mice (n = 7 for each group) (E, F). Error bars indicate ± SEM. *p < 0.05. **p < 0.01.
Sfrp1 deficient tumors fail to maintain CSCs in the late stages of tumor growth
Some CSCs in tumor tissues exist in a state of arrested cell proliferation, and their presence contributes to tumor growth and resistance to anticancer drugs. Tumor blood vessels are a niche for CSCs. Therefore, we examined the effect of Sfrp1, which is expressed in some tumor ECs, on CSCs. To test the effect of Sfrp1 on tumor tissues, we determined the percentage of CD44+ CD133+ tumor cells in Sfrp1 KO and WT mice. Initially, we generated cells expressing GFP in LLC cells and transplanted them subcutaneously. On examining the percentage of GFP+ CD44+ CD133+ CSCs via FACS 14 d after transplantation, a higher percentage of CSCs was noted in Sfrp1 KO mice (Fig. 3A–B, p < 0.05). In addition, tumor retrieval 24 d after transplantation showed a drastic decrease in the percentage of CSCs in Sfrp1 KO mice. These results suggest that Sfrp1 KO mice exhibit an abundance of CSCs in the early stages of tumor growth; however, as tumor growth progresses, the proportion of CSCs decreases significantly, and tumor growth is suppressed.

Sfrp1 deficient tumors have a reduced proportion of CSCs in the late stages of tumor growth. Flow cytometry was used to isolate GFP+ CD44+ CD133+ LLC cells from WT and Sfrp1 KO mice bearing tumors at days 14 and 24 (A). Quantification of GFP+ CD44+ CD133+ CSCs (n = 3) (B). Error bars indicate ± SEM. *p < 0.05.
Sfrp1 does not affect the structure of tumor blood vessels
We previously reported that Sfrp1 affects the vascular structure of fetal skin (Kidoya et al. 2015). Therefore, immunostaining was performed to examine the effect of Sfrp1 on the structure of blood vessels in tumor tissues. LLC cells were transplanted into Sfrp1 KO and WT mice, and tumors were collected 14 and 23 d later. Collected tissues were stained with the vascular EC marker CD31 and the vascular pericyte marker αSMA. A comparison of the tumor vasculature between Sfrp1 KO and WT mice showed no significant differences. Tumor growth was suppressed 23 d after transplantation in Sfrp1 KO mice, but no changes were observed in the structure of tumor vasculature (Fig. 4A). The CD31+ vascular area was measured and compared between the WT and KO mice, but no changes were observed (Fig. 4B). The areas of αSMA+/CD31+ were also analyzed, showing no significant difference between WT and KO mice (Fig. 4C). These results suggest that the effect of Sfrp1 on tumor growth is unrelated to the structure tumor vasculature.

Sfrp1 does not affect the structure of tumor blood vessels in tumor tissues. Immunostaining of sections from LLC tumor-bearing WT or Sfrp1 KO mice 14 or 23 d after implantation. ECs were stained with anti-CD31 antibody (green), and perivascular cells were stained with anti-αSMA antibody (red) (A). Scale bar = 100 µm. Quantification of the CD31+ area per field in Sfrp1 KO and WT mice (n = 4) (B). Quantification of the area of αSMA+ per CD31+ area in Sfrp1 KO and WT mice (n = 4) (C). Error bars indicate ± SEM.
Sfrp1 contributes to CSC maintenance via Wnt signaling
The mechanism of action of Sfrp1 in tumor tissues is not well understood. It has been reported that Sfrp1 binds to Frizzled, a Wnt signaling receptor, and competitively inhibits Wnt signaling. However, the mechanism by which Sfrp1 acts directly on Frizzled remains unknown (Zhan et al. 2017). Therefore, we focused on Wnt signaling in CCs during the early and late stages of tumorigenesis. Flow cytometry was used to collect GFP+ CD44+ CCs 14 d after tumor cell transplantation, and qRT-PCR was used to examine the expression of CSC markers (Oct4, Abcg2, Sox2, Bmi1, and Ssea1). Sfrp1-deficient cancer tissues showed decreased expression of CSC markers in CCs (Fig. 5A). Moreover, GFP+ CD44+ CCs were obtained and investigated 23 d after tumor transplantation. After 14 and 23 d, the expression of most CSC markers was significantly reduced in Sfrp1 KO mice (Fig. 5B). Next, we examined the expression of Wnt signaling target genes (Ccnd1, Axin2, and Lef1) in GFP+ CD44+ CCs at 14 and 23 d after inoculation. In the early stages of tumor transplantation, the expression of Wnt signaling target genes was observed in the CCs of Sfrp1 KO mice. On day 14, there was no significant decrease in the expression of Wnt signaling target genes in Sfrp1 KO mice compared with that in WT mice. In contrast, the expression of Wnt signaling target genes was significantly decreased in the late stage after tumor inoculation in the CCs of Sfrp1 KO mice (Fig. 5C–D).

Examination of Wnt signaling by Sfrp1 in tumor tissues. qRT-PCR analysis of mRNA expression of CSC markers (Oct4, Abcg2, Sox2, Bmi1, and Ssea1) in GFP+ CD44+ LLC cells collected from WT or Sfrp1 KO mice 14 d after transplantation via FACS, relative to GAPDH expression (A). Twenty-three d after transplantation (B). qRT-PCR analysis of mRNA expression of Wnt signaling target genes (Ccnd1, Axin2, and Lef1) relative to that of GAPDH (n = 3). GFP+ CD44+ LLC cells collected from WT or Sfrp1 KO mice 14 d after transplantation (C). Twenty-three d after transplantation (D). Error bars indicate ± SEM. ** shows p < 0.01, * shows p < 0.05, statistical significance; n.s., not significant.
Discussion
In this study, we demonstrated that Sfrp1, an angiocrine factor produced by tumor blood vessels in the tumor environment, is involved in tumor growth. Analysis using WT and Sfrp1 KO tumor model mice suggested that Sfrp1 is involved in maintaining the stemness of CCs without affecting tumor vasculature. Although the detailed effects of Sfrp1 in tumor tissues remain unknown, we demonstrated that Sfrp1 contributes to tumor growth by regulating CSC proliferation.
Sfrp1 reportedly serves as an antagonist by binding to Wnt ligands. However, our findings suggest that it is not an antagonist but a regulator of Wnt signaling in the tumor environment. Another possible signaling mechanism is the direct binding of Sfrp1 to Frizzled receptors. Preliminary studies have examined Frizzled expression in CSCs, revealing significant expression of Fzd 1, 4, and 5. It is possible that these receptor-mediated signals support stem cell maintenance mechanisms. As for Fzd expression in CCs, Fzd1 expression showed a declining rate compared to that in CSCs (data not shown). However, the difference in sensitivity to the receptor might have influenced the results of this study, which should be clarified in future studies.
Our results indicate that Sfrp1 may regulate CSC self-renewal and transient malignant growth and act to maintain a dormant state (Fig. 3). In the absence of Sfrp1 in the tumor environment, CSC growth increases in the early stages of tumor growth but decreases in the late stages, which may attenuate tumor progression. The transcript levels of canonical Wnt signaling targets in tumor tissues of Sfrp1-deficient mice were similar to those in WT mice in the early phase of tumor development but were markedly reduced in the late phase (Fig. 5). However, the proportion of CSCs in tumor tissue increased in Sfrp1-deficient mice in the early phase (Fig. 3). This suggests that canonical Wnt signaling may not be involved in the maintenance of CSC dormancy during the early phase of tumor development. Nonetheless, further analysis is needed to elucidate the mechanism of regulatory signaling by Sfrp1 in CSCs.
The results of this study suggest that Sfrp1 is expressed in a portion of tumor vessels in the CSC niche, indicating that the blood vessels within the tumor are not homogeneous but rather heterogeneous. Such a vascular niche has been previously proposed in glioblastomas, indicating that CSCs reside near blood vessels and are maintained by Notch signaling (Calabrese et al. 2007). A stem cell maintenance mechanism in response to Wnt signaling has also been proposed in colorectal cancer. GLI1-positive mesenchymal cells in colorectal cancer tissues secrete Wnt, aiding in the maintenance and promotion of stem cell self-renewal (Degirmenci et al. 2018). This report describes the mechanism of stem cell maintenance by mesenchymal cells. However, the mechanism of CSC niche formation by Sfrp1-positive ECs observed in the present study was also regulated by Wnt signaling and is similarly relevant.
It has also been reported that Sfrp1 promotes vascular maturation by enhancing the number of mesenchymal stem cells around neovascular vessels (Dufourcq et al. 2008). In contrast, the present study showed that Sfrp1 did not affect tumor angiogenesis or vascular structure. Sfrp1 plays a vital role in the formation of the dense structure of arteries and veins during fetal development (Kidoya et al. 2015). While Sfrp1 was expressed in many APJ-positive veins during this period, only approximately 8% of tumor vessels showed Sfrp1 expression. Perhaps changes in vascular structure could be observed at the time of tumor implantation or in the early stages of angiogenesis; however, we did not analyze this because there was no significant difference in tumor size at that time.
Several cancer therapies have been developed; however, developing anticancer drugs that directly target proliferating cells has been challenging due to significant side effects and the ability of CSC to develop drug resistance. Further, the development of anticancer agents targeting blood vessels has been explored, but these compounds have exhibited lower-than-expected efficacy and are not discriminatory, affecting blood vessels throughout the body, resulting in significant side effects (Ebos and Kerbel 2011; Liu et al. 2023). It has recently been proposed that ECs that constitute blood vessels do not represent a uniform cell population, which applies to tumor vessels as well. Therefore, targeting specialized vascular ECs involved in niche formation is expected to destroy the CSC niche, establishing a method for inhibiting tumor growth with minimal side effects.
Conclusions
Sfrp1 in tumor tissues is expressed in a subset of vascular ECs. Furthermore, Sfrp1 may promote tumor growth by contributing to the maintenance of CSCs via Wnt signaling.
Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
References
Alsina-Sanchis E, Mülfarth R, Fischer A (2021) Control of tumor progression by angiocrine factors. Cancers (basal) 13:2610
Atkins RJ, Stylli SS, Kurganovs N, Mangiola S, Nowell CJ, Ware TM, Corcoran NM, Brown DV, Kaye AH, Morokoff A, Luwor RB, Hovens CM, Mantamadiotis T (2019) Cell quiescence correlates with enhanced glioblastoma cell invasion and cytotoxic resistance. Exp Cell Res 374:353–364
Atschekzei F, Hennenlotter J, Jänisch S, Großhennig A, Tränkenschuh W, Waalkes S, Peters I, Dörk T, Merseburger AS, Stenzl A, Kuczyk MA, Serth J (2012) SFRP1 CpG island methylation locus is associated with renal cell cancer susceptibility and disease recurrence. Epigenetics 7:447–457
Batlle E, Clevers H (2017) Cancer stem cells revisited. Nat Med 23:1124–1134
Bhat RA, Stauffer B, Komm BS, Bodine PV (2007) Structure-function analysis of secreted frizzled-related protein-1 for its Wnt antagonist function. J Cell Biochem 102:1519–1528
Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, Frank A, Bayazitov IT, Zakharenko SS, Gajjar A, Davidoff A, Gilbertson RJ (2007) A perivascular niche for brain tumor stem cells. Cancer Cell 11:69–82
Clevers H (2006) Wnt/beta-catenin signaling in development and disease. Cell 127:469–480
Degirmenci B, Valenta T, Dimitrieva S, Hausmann G, Basler K (2018) GLI1-expressing mesenchymal cells form the essential Wnt-secreting niche for colon stem cells. Nature 558:449–453
Dufourcq P, Descamps B, Tojais NF, Leroux L, Oses P, Daret D, Moreau C, Lamazière JM, Couffinhal T, Duplàa C (2008) Secretes frizzled-related protein-1 enhances mesenchymal stem cell function in angiogenesis and contributes to neovessel maturation. Stem Cells 26:2991–3001
Ebos JM, Kerbel RS (2011) Antiangiogenic therapy: impact oninvasion, disease progression, and metastasis. Nat Rev Cli Oncol 8:210–221
Finch PW, He X, Kelley MJ, Uren A, Schaudies RP, Popescu NC, Rudikoff S, Aaronson SA, Varmus HE, Rubin JS (1997) Purification and molecular cloning of a secreted, Frizzled-related antagonist of Wnt action. Proc Natl Acad Sci U S A 94:6770–6775
Hashimoto M, Yamashita Y, Takemoto T (2016) Electroporation of Cas9 protein/sgRNA into early pronuclear zygotes generates non-mosaic mutants in the mouse. Dev Biol 418:1–9
Hayashi Y, Jia W, Kidoya H, Muramatsu F, Tsukada Y, Takakura N (2019) Galectin-3 inhibits cancer metastasis by negatively regulating integrin β3 expression. Am J Pathol 189:900–910
Hira VV, Ploegmakers KJ, Grevers F, Verbovšek U, Silvestre-Roig C, Aronica E, Tigchelaar W, Turnšek TL, Molenaar RJ, Van Noorden CJ (2015) CD133+ and nestin+ glioma stem-like cells reside around CD31+ arterioles in niches that express SDF-1α, CXCR4, osteopontin and cathepsin K. J Histochem Cytochem 63:481–493
Hu L, Hayashi Y, Kidoya H, Takakura N (2021) Endothelial cell-derived apelin inhibits tumor growth by altering immune cell localization. Sci Rep 11:14047
Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, Yagi H, Masuko T, Shimizu T, Ishikawa T, Kai K, Takahashi E, Imamura Y, Baba Y, Ohmura M, Suematsu M, Baba H, Saya H (2011) CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc (-) and thereby promotes tumor growth. Cancer Cell 19:387–400
Karakatsani A, Álvarez-Vergara MI, Ruiz de Almodóvar C (2023) The vasculature of neurogenic niches: properties and function. Cells Dev 174:203841
Kidoya H, Naito H, Muramatsu F, Yamakawa D, Jia W, Ikawa M, Sonobe T, Tsuchimochi H, Shirai M, Adams RH, Fukamizu A, Takakura N (2015) APJ regulated parallel alignment of arteries and veins in the skin. Dev Cell 33:247–259
Krishnamurthy S, Dong Z, Vodopyanov D, Imai A, Helman JI, Prince ME, Wicha MS, Nör JE (2010) Endothelial cell-initiated signaling promotes the survival and self-renewal of cancer stem cells. Cancer Res 70:9969–9978
Liu ZL, Chen HH, Zheng LL, Sun LP, Shi L (2023) Angiogenic signaling pathway and anti- angiogenic therapy for cancer. Singal Transduct Target Ther 8:198
Niehrs C (2012) The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol 13:767–779
Perugorria MJ, Olaizola P, Labiano I, Esparza-Baquer A, Marzioni M, Marin JJG, Bujanda L, Banales JM (2019) Wnt-β-catenin signalling in liver development, health and disease. Nat Rev Gastroenterol Hepatol 16:121–136
Rafii S, Butler JM, Ding BS (2016) Angiocrine functions of ogan-specific endothelial cells. Nature 529:316–325
Ramasamy SK, Kusumbe AP, Itkin T, Gur-Cohen S, Lapidot T, Adams RH (2016) Regulation of hematopoiesis and osteogenesis by blood vessel-derived signals. Annu Rev Cell Dev Biol 32:649–675
Rodriguez J, Esteve P, Weinl C, Ruiz JM, Fermin Y, Trousse F, Dwivedy A, Holt C, Bovolenta P (2005) SFRP1 regulates the growth of retinal ganglion cell axons through the Fz2 receptor. Nat Neurosci 8:1301–1309
Skronska-Wasek W, Mutze K, Baarsma HA, Bracke KR, Alsafadi HN, Lehmann M, Costa R, Stornaiuolo M, Novellino E, Brusselle GG, Wagner DE, Yildirim AÖ, Königshoff M (2017) Reduced frizzled receptor 4 expression prevents WNT/β-catenin-driven alveolar lung repair in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 196:172–185
Zhan T, Rindtorff N, Boutros M (2017) Wnt signaling in cancer. Oncogene 36:1461–1473
Acknowledgements
We thank C. Yamazaki and N. Aikawa for their technical assistance.
Funding
Open Access funding provided by University of Fukui. This study was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant numbers: 23H03860, 23H03862, and 19H03503), PRESTO-JST (grant number: JPMJPR1944), JST FOREST Program (grant number: JPMJFR2160), University of Fukui Research Farm-Firm, a grant for the next generation of outstanding researchers project of the University of Fukui, and Joint Usage and Joint Research Programs, the Institute of Advanced Medical Sciences, Tokushima University.
Author information
Authors and Affiliations
Contributions
YH and HK contributed to the study conception and data analysis. YH conducted most experiments. MH, KT, and TT established Sfrp1 knockout mice. YH and HK wrote the manuscript, and NT supervised the study. All authors approved this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hayashi, Y., Hashimoto, M., Takaoka, K. et al. Tumor endothelial cell-derived Sfrp1 supports the maintenance of cancer stem cells via Wnt signaling. In Vitro Cell.Dev.Biol.-Animal (2024). https://doi.org/10.1007/s11626-024-00899-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11626-024-00899-y