
Abstract
HIV/AIDS is associated with significant morbidity, mortality, and financial burden. For these reasons, robust clinical evidence is critical. We aim to investigate the fragility index, fragility quotient, and risk of bias of clinical trial endpoints in HIV medicine. The fragility index represents the minimum amount of trial endpoint “nonevents” changed to “events” in one trial arm required to nullify statistical significance. The fragility quotient contextualized the fragility index by dividing the index by the total trial sample size. We selected eligible trials from the Department of Health and Human Services guideline for the use of antiretroviral agents in HIV-1-infected adults and adolescents. We calculated the fragility index and fragility quotient for all included trials. The Cochrane “risk of bias” Tool 2.0 was used to evaluate the likelihood and sources of bias in the included trials. Thirty-nine RCTs were included for our analysis of fragility. Thirty-six were included for our analysis of the risk of bias. The median fragility index was 5. Three RCTs were at high risk of bias, all due to the selection of the endpoint or statistical test. Twenty had some concerns for risk of bias. The analyzed HIV medicine RCT endpoints were fragile, overall. This indicates that a median of 5 patients across all included studies would nullify the statistical significance of the endpoints. Furthermore, we found evidence that concerns for bias are present at a high rate.
Similar content being viewed by others


INTRODUCTION
According to the US Centers for Disease Control and Prevention, the incidence of HIV in the USA in 2015 was 39,513, with an estimated prevalence of 1.1 million. Currently, HIV is the eighth leading cause of death in people aged 25–34 years and ninth for those aged 35–44 years1. In addition to substantial morbidity and mortality, individuals with HIV/AIDS experience significant financial burdens. The average annual cost for HIV care is estimated to be approximately $23,000, for an average lifetime cost of $367,134 (in 2010 dollars).2,3
The Department of Health and Human Services (DHHS) developed a clinical practice guideline (CPG) for the use of antiretroviral agents in HIV-1-infected adults and adolescents.4 Recommendations within the guidelines are graded on the underlying evidence, with grades ranging from I (high) to III (low). Randomized controlled trials (RCTs) often underpin recommendations because they can demonstrate cause–effect relationships and they are recognized by the DHHS to provide sufficient evidence for making high-quality recommendations.4,5 Therefore, the RCTs underpinning the DHHS guideline are likely to have the most robust results and are most likely to be important to practicing physicians.
In this study, we investigate the robustness of RCT endpoints in patients affected with HIV by applying the fragility index (FI)6 and the fragility quotient (FQ).7 The FI identifies the number of events required to change the statistical significance of a result to nonsignificance. An FQ is calculated by dividing the FI by the sample size of a trial, and it offers a relative measure of fragility. Some authors have suggested including the FQ in statistical analyses to ensure adequate interpretation.7 The knowledge that only a few additional events could nullify a statistically significant result may reduce health care providers’ confidence in the recommendation that rests on the result.6
The FI represents the robustness of the trial endpoint, while the FQ represents the robustness of the FI. The relationship is best explained by using two trials as examples. In the first trial, the FI was calculated to be 46, which indicates that 46 patient events would need to have been nonevents in the trial for statistical significance to be lost.8 The FQ for this trial was 0.008, indicating that that less than one patient (0.8) per 100 with an event changed to a nonevent would result in a nonsignificant difference for the endpoint. In contrast, the second trial had an FI of 10 and an FQ of 0.05, indicating that 5 patients per 100 with an event changed to a nonevent would nullify statistical significance.9 While the FI may make the second trial seem far more fragile, when sample size is accounted for, the larger FQ indicates that the second trial is actually much more robust. This example underpins the importance of reporting the FI alongside the FQ value.
Use of the FI and FQ will provide health care providers with a means to independently assess the recommendations arising from clinical trial results, thus ensuring applicability in decisions about the management and treatment of HIV/AIDS patients. The primary outcome of this study is the FI and FQ of eligible trial endpoints. Because this DHHS CPG is underpinned by a wide range of clinical evidence beyond the RCTs eligible for FI calculation, we do not attempt to judge the overall quality of evidence underpinning the DHHS recommendations. We chose trials from the DHHS guideline because of their importance to practicing physicians. The secondary outcome was to assess the RCT endpoints for risk of bias.
METHODS
Identification of Studies and Data Collection
We surveyed the Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents set forth by the DHHS.4 Investigators (CM, CW) screened the reference list for eligible studies using a combination of title/abstract and full text screen. Studies were eligible if they included the random assignment of patients to a condition using a 1:1 allocation ratio, used a parallel two-group design, and reported at least one significant, dichotomous endpoint.
Data extraction was then performed on each eligible RCT via a piloted electronic form. The data extracted were sample size, numbers lost to follow-up, endpoint reported, event rates of the endpoint, statistical significance for the endpoint, the statistical test used, and the trial’s Science Citation Index. When possible, the primary endpoint was prioritized for analysis. If that endpoint could not be used, the secondary or an unspecified dichotomous endpoint (e.g., an endpoint not designated as primary, secondary, or tertiary) was chosen. If a trial had multiple dichotomous endpoints, we used the GRADE Network’s10 approach to identify the most important endpoint. In this approach, board-certified internal medicine physicians (MS, DB) who work with HIV/AIDS patients were consulted. Endpoints were rated from 1 (low importance) to 9 (high importance), and the highest ranked endpoint was included for analysis.
Calculating the FI and the FQ
The FI for each endpoint was calculated using two-by-two contingency tables according to the method described by Walsh et al.6 Prior to this calculation, the P values of the included endpoints were recalculated using the two-sided Fisher exact test. We then iteratively added events to the group with the smaller number of events, while subtracting nonevents to keep the total number of participants constant. The smallest number of additional events needed to obtain a P value ≥ 0.05 represented the FI. The FQ for each endpoint was calculated by dividing the FI by the sample size of the trial.7 The FQ provided a method to evaluate fragility relative to the sample size, with a smaller FQ indicating a more robust trial endpoint.
Risk of Bias Assessment
We used the Cochrane “risk of bias” Tool 2.0 (RoB 2.0) to evaluate the likelihood and sources of bias in the included trials. RoB 2.0 is the newest version of the Cochrane RoB Tool, and it was updated to address concerns about interrater agreement, subjectivity in assigning risk of bias judgments, and bias judgments assigned at the trial level. RoB 2.0 redefined the bias domains from the original tool, and it now includes (1) bias arising from the randomization process, (2) bias due to deviations from intended interventions, (3) bias due to missing endpoint data, (4) bias in measurement of the endpoint, and (5) bias in selection of the reported result. Bias is evaluated on the endpoint level (with the exception of bias due to randomization), rather than on the trial level. Furthermore, RoB 2.0 contains decision algorithms to limit subjectivity in assigning bias judgments. The scaling was also modified from the earlier tool: the previously “unclear” risk of bias option has been replaced with “some concerns.” When three or more domains were classified as some concerns, we considered this trial endpoint to be at high risk. Because RoB 2.0 is new, no validity or reliability evidence is known to be available for review.
All investigators attended a risk of bias training, which included reviewing RoB 2.0 and performing evaluations on two trials from our sample. Following training, CM and CW independently evaluated all trials for risk of bias. We planned a priori for two additional investigators (MS and DB) to be consulted on difficult risk of bias judgments, and they were consulted three times with questions regarding bias domain 1 (randomization). After completing their separate risk of bias evaluations, CM and CW held a consensus meeting to resolve any disagreements.
Statistical Analysis
To determine the FI for each trial, we used an online calculator. We conducted a sensitivity analysis for trials whose included endpoint was less than .00125. We conducted power analyses on all included trials based on only primary endpoints identified. We used the observed effect size and sample size of each trial for our power analyses. We assumed that a power of less than 0.8 was underpowered. The median and interquartile ranges (IQRs) were calculated to characterize the dispersion and central tendency of the FI. A Pearson product-moment correlation was used to examine the relationship between the FI total sample size, event rate, and absolute risk difference. We used STATA 13.1, GPower 3.1, and Microsoft Excel to perform all calculations.
RESULTS
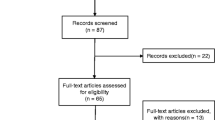
Our investigation of trials from the DHHS Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents yielded 533 citations, overall. Of these citations, 39 met all inclusion criteria: a 1:1 randomization, a parallel two-group design, and a statistically significant, dichotomous endpoint (Fig. 1). Three studies were published as continuation analyses of an already-published RCT in our sample, and therefore had identical methods (e.g., one RCT published two papers, one at 48 weeks and another at 96 weeks). We excluded these three studies for risk of bias evaluations to avoid duplicate risk of bias scores.

Flow diagram of included and excluded studies.
The 39 RCTs included in this study (Tables 1 and 2) had a median sample size of 610 (IQR 511) and a median loss to follow-up of 19 (IQR 30.75). We analyzed 22 primary endpoints and 17 secondary endpoints. The median FI for all endpoints was 5 (IQR 9) and the median FQ was 0.009 (IQR 0.015), or .9 (IQR 1.5) patients per 100. For primary endpoints, the median FI and FQ were 8 (IQR 10.25) and 0.01 (IQR 0.010), or 1 (IQR 1) patients per 100, respectively. For secondary endpoints, the median FI and FQ were 2 (IQR 9) and 0.009 (IQR 0.02), or .9 (IQR 2) patients per 100, respectively. Our sensitivity analysis, including only trials whose endpoints were less than .00125 (n = 12), resulted in a median FI of 10 (IQR 8) and median FQ of 0.0139 (IQR 0.0097), or 1.4 patients (IQR .97) per 100. The Pearson correlation between sample size and FI was r = 0.14; however, it may have been affected by three outliers. Removing these outliers revealed a correlation coefficient of r = 0.32. The correlation of FI and effect size (absolute risk reduction) was r = .71, whereas the correlation between FI and total number of events in each trial was r = .21. When we applied Fisher’s exact test, six endpoints resulted in an FI of 0, indicating that the statistical significance of the six endpoints was explained by choice of statistical test alone. The median Science Citation Index was 159 (IQR 133). The Science Citation Index values for the three most cited studies were 2961 (FI = 6), 2043 (FI = 10), and 1281 (FI = 46). The number lost to follow-up was greater than the FI in 30 of 39 trials (76.9%).
We further categorized the 39 endpoints into relevant domains: disease progression/death, viral load suppression, and adverse events (Table 2). Endpoints assessing disease progression or death (n = 7) had a median fragility of 3 (FQ = 0.006) and three (42.9%) were underpowered. Endpoints assessing viral load (n = 16) had a median fragility of 5.5 (FQ = 0.019) and eight (50.0%) were underpowered. Endpoints assessing adverse events (n = 16) had a median fragility of 7 (FQ = 0.005) and six (37.5%) were underpowered. Our power analyses revealed that 8 of 22 (36.4%) primary endpoints were underpowered. Compared to the 14 sufficiently powered trials with a median FI = 11 (IQR 6.5), the median FI of underpowered trials was 1.5 (IQR 1.75). The individual trial risk of bias assessments is detailed in Supplemental Table 1, publicly available via Open Science Framework (osf.io/d9n6z). Among the 36 trials included in the risk of bias assessments, high risk of bias was found in 3 (8.3%), some concerns for risk of bias was found in 20 (55.6%), and low risk of bias was found in the remaining 13 (36.1%). Bias in domain 5 (risk of bias from selection of the reported trial result) accounted for all three occasions of high risk of bias.
DISCUSSION
Our investigation demonstrates the fragility of trial endpoints from 39 RCTs from a prominent HIV clinical practice guideline. Our results show that a median of only five event alterations would be needed to nullify the statistical significance of trial endpoints. On 15 occasions, the FI was 2 or less. Moreover, there was concordance between the FI and FQ, reinforcing the fact that only a few events relative to sample size are necessary for loss of statistical significance. A low FI indicates that a subsequent trial may overturn results of a current trial.
Prior to calculating the FI, we used Fisher’s exact test to determine an endpoint’s P value based on the reported events and sample size in each arm. In six cases, our calculated P value was nonsignificant, despite the author’s reported value being less than 0.05. The discordance between our calculations and the author’s reported values is likely due to choice of statistical test, since both Fisher’s exact and chi-squared tests are used for categorical endpoints and may result in different levels of significance. This situation further calls into question the fragility of clinical trial endpoints in our sample since choice of statistical test alone may be the difference between a “positive” and “negative” endpoint.
We further investigated whether studies were adequately powered. Power analyses are used to ensure that a study will have a large enough sample size to detect statistical significance.11 If a study is inadequately powered, the magnitude of its results may be exaggerated.12 Our results show that 8 of the 22 trials whose primary endpoint was included in our analysis were underpowered. The median FI of the sufficiently powered trials was 11, while the median FI for the underpowered trials was 1.5. In other words, in the 8 underpowered trials, a median of only 1.5 events would have nullified the results of the trial. Although, given that our analyses of power were done after the fact (e.g., post hoc), caution is warranted in interpreting the results, since post hoc power analyses have been described as “noisy” and may give inaccurate estimates of study power.13 Thus, it may be the case that some studies we identified as underpowered are in reality sufficiently powered, though we are unable to confirm a priori study power. When conducted a priori, a power analysis informs the authors, readers, editors, and other interested parties whether a study has sufficient sample size to detect a true effect. In other words, a study that lacks sufficient power whose results show a statistically significant difference between groups cannot say whether the finding is a true or false positive. Thus, in our analysis of post hoc power, we avoid making accusations of true or false positive findings, since post hoc power analyses are inaccurate when compared to a priori power analyses. However, compounding the inadequate post hoc power of many trials we analyzed was the number of subjects lost to follow-up. Across all 39 trials, the median loss to follow-up across both arms was greater than the FI in 30 trials (76.9%). An open question is whether mitigating the loss to follow-up or powering a study to sufficiently account for estimated loss to follow-up would decrease the fragility of trial endpoints.
Our findings are consistent with previous FI studies that reported a range of FIs from 2 to 7.14,15,16,17,18,19,20 First, in general medicine, Walsh et al. reported an FI of 8 in trials published in general medicine journals15 and most recently, Shochet et al. reported an FI of 2 in nephrology trials.16 All of the previous studies compared the percentage of trials with loss to follow-up greater than the endpoint FI. None reported a percentage greater than ours (76.9%). Furthermore, we are the second study, after Bowers et al.21, to use the FQ as a measure of robustness of the FI.
While a low FI indicates fragile trial results, solutions that increase the robustness of trial results may prove difficult to identify. A recent editorial22 explored the FI using a simulation study with three different effect sizes and fixed sample sizes, two-sided alpha (p = .05), and beta (.2). They conclude that because RCTs must balance sample size with expected efficacy, the FI of most RCTs is necessarily fragile. They also rightly conclude, like we do, that the FI is not a measure of effect size. So, while we agree that fragile trial results may be a byproduct of RCT equipoise, the issue of fragile trial results—those that hinge on a relatively small number of patient events—will persist in biomedicine. If more robust RCT results (i.e., where significance does not rely on a few events) are desired, increased sample size, increased prespecified study power, or lower prespecified study alpha values are required. A recent call to lower the alpha threshold from p = .05 to p = .005 has gained momentum as a temporizing measure until better statistical frameworks that do not rely on p values are implemented.23 An analysis of the effect of a lower threshold found that 70% of phase 3 RCTs published in three major medical journals would maintain statistical significance, thus implying that major RCT results are more robust.24 The full benefits and harms of the new proposed p value threshold are a point of controversy25 and have not been completely explored, as of yet.
To complement our FI calculations, we evaluated the risk of bias of the included trial endpoints. We found three trial endpoints to be at high risk of bias, with all three at high risk in domain 5 (bias in the selection of the reported result). More specifically, these trials were labelled as high risk because of poorly defined statistical analyses. The authors reported the use of either chi-squared or Fisher’s exact test for categorical endpoints, but they did not specify which test they used when reporting their results. The variability in results from the two statistical tests has already been demonstrated by the number of FIs calculated to be zero. These three trials, aside from many of the trials in our sample, were scored as some concerns, indicating questionable aspects of the study design. For example, many trials were scored as some concerns in domain 1 (bias arising from the randomization process). We were unable to determine whether the concerns in this domain arise from poor reporting or poor methods, but nonetheless many trials did not have adequate description of their randomization and blinding procedures. The resulting baseline imbalances between the trials were cause for concern and drew into question the validity and generalizability of the results.
To conclude, we recommend the reporting of the FI and FQ alongside each trial endpoint. This practice would increase transparency and allow readers to determine the rigor of a statistically significant endpoint. Additionally, we recommend including an explanation of which statistical tests were used and avoiding ad hoc calculations with either Fisher’s exact or chi-squared test since this practice constitutes “P-hacking,” or the selective reporting of the results of one statistical test based on statistical significance.26 We believe that these recommendations will increase the quality of patient care by underscoring the time for caution or confidence in interpreting trial results.
Our study was not without limitations. The FI and FQ are only used to evaluate RCTs. Furthermore, only RCTs sorted in a 1:1 ratio with two parallel groups and a statistically significant dichotomous endpoint were eligible for this study. RCTs with an allocation ratio different from 1:1 are not eligible for analysis using the FI, which limited the number of RCTs from the DHHS guideline that we were able to include. Primary endpoints were used when available, but if they were not available, secondary endpoints were used.
References
Basic Statistics | HIV Basics | HIV/AIDS | CDC. https://www.cdc.gov/hiv/basics/statistics.html. Accessed 2 August 2017.
Gebo KA, Fleishman JA, Conviser R, et al. Contemporary costs of HIV healthcare in the HAART era. AIDS. 2010;24(17):2705–2715.
Schackman BR, Gebo KA, Walensky RP, et al. The lifetime cost of current human immunodeficiency virus care in the United States. Med Care. 2006;44(11):990–997.
Panel on Clinical Practices for Treatment of HIV Infection. Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. PsycEXTRA Dataset. https://doi.org/10.1037/e373632004-001
Sibbald B, Roland M. Understanding controlled trials. Why are randomised controlled trials important? BMJ. 1998;316(7126):201.
Walsh M, Srinathan SK, McAuley DF, et al. The statistical significance of randomized controlled trial results is frequently fragile: a case for a Fragility Index. J Clin Epidemiol. 2014;67(6):622–628.
Ahmed W, Fowler RA, McCredie VA. Does Sample Size Matter When Interpreting the Fragility Index? Crit Care Med. 2016;44(11):e1142-e1143.
Strategies for Management of Antiretroviral Therapy (SMART) Study Group, El-Sadr WM, Lundgren JD, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med. 2006;355(22):2283–2296.
Bunupuradah T, Chetchotisakd P, Ananworanich J, et al. A randomized comparison of second-line lopinavir/ritonavir monotherapy versus tenofovir/lamivudine/lopinavir/ritonavir in patients failing NNRTI regimens: the HIV STAR study. Antivir Ther. 2012;17(7):1351–1361.
Guyatt GH, Oxman AD, Kunz R, et al. GRADE guidelines: 2. Framing the question and deciding on important outcomes. J Clin Epidemiol. 2011;64(4):395–400.
Suresh K, Chandrashekara S. Sample size estimation and power analysis for clinical research studies. J Hum Reprod Sci. 2012;5(1):7–13.
Button KS, Ioannidis JPA, Mokrysz C, et al. Power failure: why small sample size undermines the reliability of neuroscience. Nat Rev Neurosci. 2013;14(5):365–376.
Gelman A. Post-hoc Power Using Observed Estimate of Effect Size is too Noisy to be Useful. Ann Surg. 2018. https://doi.org/10.1097/SLA.0000000000003089.
Ridgeon EE, Young PJ, Bellomo R, Mucchetti M, Lembo R, Landoni G. The Fragility Index in Multicenter Randomized Controlled Critical Care Trials. Crit Care Med. 2016;44(7):1278–1284.
Walsh M, Srinathan SK, McAuley DF, et al. The statistical significance of randomized controlled trial results is frequently fragile: a case for a Fragility Index. J Clin Epidemiol. 2014;67(6):622–628.
Shochet LR, Kerr PG, Polkinghorne KR. The fragility of significant results underscores the need of larger randomized controlled trials in nephrology. Kidney Int. 2017. https://doi.org/10.1016/j.kint.2017.05.011.
Evaniew N, Files C, Smith C, et al. The fragility of statistically significant findings from randomized trials in spine surgery: a systematic survey. Spine J. 2015;15(10):2188–2197.
Khan M, Evaniew N, Gichuru M, et al. The Fragility of Statistically Significant Findings From Randomized Trials in Sports Surgery: A Systematic Survey. Am J Sports Med. 2017;45(9):2164–2170.
Matics TJ, Khan N, Jani P, Kane JM. The Fragility Index in a Cohort of Pediatric Randomized Controlled Trials. J Clin Med Res. 2017;6(8). https://doi.org/10.3390/jcm6080079.
Shen Y, Cheng X, Zhang W. The fragility of randomized controlled trials in intracranial hemorrhage. Neurosurg Rev. 2017. https://doi.org/10.1007/s10143-017-0870-8
Bowers A, Meyer C, Tritz D, et al. Assessing quality of randomized trials supporting guidelines for laparoscopic and endoscopic surgery. J Surg Res. 2018;224:233–239.
Carter RE, McKie PM, Storlie CB. The Fragility Index: a P-value in sheep’s clothing? Eur Heart J. 2017;38(5):346–348.
Benjamin DJ, Berger JO, Johannesson M, et al. Redefine statistical significance. Nat Hum Behav. 2017;2(1):6–10.
Wayant C, Scott J, Vassar M. Evaluation of Lowering the P Value Threshold for Statistical Significance From .05 to .005 in Previously Published Randomized Clinical Trials in Major Medical Journals. JAMA. 2018;320(17):1813–1815.
Lakens D, Adolfi FG, Albers CJ, et al. Justify your alpha. Nat Hum Behav. 2018;2(3):168–171.
Head ML, Holman L, Lanfear R, Kahn AT, Jennions MD. The extent and consequences of p-hacking in science. PLoS Biol. 2015;13(3):e1002106.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they do not have a conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Table 1
(DOCX 20 kb)
Rights and permissions
About this article

Cite this article
Wayant, C., Meyer, C., Gupton, R. et al. The Fragility Index in a Cohort of HIV/AIDS Randomized Controlled Trials. J GEN INTERN MED 34, 1236–1243 (2019). https://doi.org/10.1007/s11606-019-04928-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11606-019-04928-5