
Summary
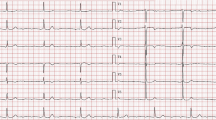
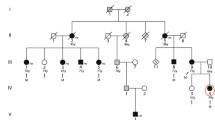
Even though mutations in LMNA have been reported in patients with typical dilated cardiomyopathy (DCM) and atrioventricular block (AVB) previously, the purpose of this study was to disclose this novel genetic abnormality in one Chinese family with the atypical phenotype of progressive AVB followed by DCM with normal QRS interval. Genome-wide linkage analysis mapped the AVB gene in this family to a marker at chromosome 1q21.2, where the LMNA gene was located. Direct DNA sequence analysis revealed a heterozygous G to A transition at nucleotide 244 in exon 1 of LMNA, which resulted in an E82K mutation. The E82K mutation co-segregated with all affected individuals in the family, and was not present in 200 normal controls. Further clinical evaluation of mutation carriers showed that 5 of 6 AVB patients exhibited mild DCM with a late onset of age in the fourth and fifth decades. Ejection fractions were documented in 5 patients with DCM, but 4 showed a normal value of ⩾50%. Echocardiography showed that atrial dilatation occurred earlier than ventricular dilatation in the patients. This study suggests that progressive AVB with normal QRS interval and accompanying DCM at later stages may represent a distinct type of DCM. The molecular mechanism by which the E82K mutation causes AVB as the prominent phenotype in DCM may be a focus of future studies.
Similar content being viewed by others

References
Lin F, Worman H. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem, 1993,268(22):16 321–16 326
Mounkes LC, Burke B, Stewart CL. The A-type lamins: nuclear structural proteins as a focus for muscular dystrophy and cardiovascular diseases. Trends Cardiovasc Med, 2001,11(7):280–285
Franz WM, Muller OJ, Katus HA. Cardiomyopathies: From genetics to the prospect of treatment. Lancet, 2001,358(9293):1627–1637
Bonne G, Di Barletta MR, Varnous S, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet, 1999,21(3):285–288
di Barletta R, Ricci E, Galluzzi G, et al. Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am J Hum Genet, 2000,66(4):1407–1412
Muchir A, Bonne G, van der Kooi AJ, et al. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet, 2000,9(9):1453–1459
De Sandre-Giovannoli A, Chaouch M, Kozlov S, et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet, 2002,70(3):726–736
Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet, 2000,9(1):109–112
Shackleton S, Lloyd DJ, Jackson SN, et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet, 2000,24(2):153–156
Novelli G, Muchir A, Sangiuolo F, et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet, 2002,71(2):426–431
Cao H, Hegele RA. LMNA is mutated in Hutchinson-Gilford progeria (MIM 176670) but not in Wiedemann-Rautenstrauch progeroid syndrome (MIM 264090). J Hum Genet, 2003,48(5):271–274
De Sandre-Giovannoli A, Bernard R, Cau P, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science, 2003,300(5628):2055
Garg A, Speckman RA, Bowcock AM. Multisystem dystrophy syndrome due to novel missense mutations in the amino-terminal head and alpha-helical rod domains of the lamin A/C gene. Am J Med, 2002,112(7):549–555
Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med, 1999,341(23):1715–1724
Arbustini E, Pilotto A, Repetto A, et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect related disease. J Am Coll Cardiol, 2002,39(6):981–990
Kirschner J, Brune T, Wehnert M, et al. p. S143F mutation in Lamin A/C: A new phenotype combining myopathy and progeria. Ann Neurol, 2005,57(1):148–151
Brodsky GL, Muntoni F, Miocic S, et al. Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation, 2000,101(5):473–476
Jakobs PM, Hanson EL, Crispell KA, et al. Novel lamin A/C mutations in two families with dilated cardiomyopathy and conduction system disease. J Card Fail, 2001,7(3):249–256
Hershberger RE, Hanson EL, Jakobs PM, et al. A novel lamin A/C mutation in a family with dilated cardiomyopathy, prominent conduction system disease, and need for permanent pacemaker implantation. Am Heart J, 2002,144(6):1081–1086
Taylor MR, Fain PR, Sinagra G, et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol, 2003,41(5):771–780
MacLeod HM, Culley MR, Huber JM, et al. Lamin A/C truncation in dilated cardiomyopathy with conduction disease. BMC Med Genet, 2003,4(1):4
Verga L, Concardi M, Pilotto A, et al. Loss of lamin A/C expression revealed by immuno-electron microscopy in dilated cardiomyopathy with atrioventricular block caused by LMNA gene defects. Virchows Arch, 2003,443(5):664–671
Kärkäinen S, Heliö T, Miettinen R, et al. A novel mutation, Ser143Pro, in the lamin A/C gene is common in Finnish patients with familial dilated cardiomyopathy. Eur Heart J, 2004,25(10):885–893
Wang L, Fan C, Topol SE, et al. Mutation of MEF2A in an inherited disorder with features of coronary artery disease. Science, 2003,302(5650):1578–1581
Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell, 1995,80(5):805–811
Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet, 1996,12(1):17–23
Schott JJ, Alshinawi C, Kyndt F, et al. Cardiac conduction defects associate with mutations in SCN5A. Nature Genet, 1999,23(1):20–21
Benson DW, Silberbach GM, Kavanaugh-McHugh A, et al. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest, 1999,104(11):1567–1573
Jay PY, Harris BS, Maguire CT, et al. Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J Clin Invest, 2004,113(8):1130–1137
Stuurman N, Heins S, Aebi U. Nuclear lamins: their structure, assembly, and interactions. J Struct Biol, 1998,122(1–2):42–66
Vytopil M, Benedetti S, Ricci E, et al. Mutation analysis of the lamin A/C gene (LMNA) among patients with different cardiomuscular phenotypes. J Med Genet, 2003,40 (12):e132
Genschel J, Bochow B, Kuepferling S, et al. A R644C mutation within lamin A extends the mutations causing dilated cardiomyopathy. Hum Mutat, 2001,17(2):154
Meune C, Van Berlo JH, Anselme F, et al. Primary prevention of sudden death in patients with lamin A/C gene mutations. New Eng J Med, 2006,354(2):209–210
Sebillon P, Bouchier C, Bidot LD, et al. Expanding the phenotype of LMNA mutations in dilated cardiomyopathy and functional consequences of these mutations. J Med Genet, 2003,40(8):560–567
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Wu, X., Wang, Q.K., Gui, L. et al. Identification of a new lamin A/C mutation in a chinese family affected with atrioventricular block as the prominent phenotype. J. Huazhong Univ. Sci. Technol. [Med. Sci.] 30, 103–107 (2010). https://doi.org/10.1007/s11596-010-0119-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11596-010-0119-z