
Abstract
Background
Mutations in the gene encoding the nuclear membrane protein lamin A/C have been associated with at least 7 distinct diseases including autosomal dominant dilated cardiomyopathy with conduction system disease, autosomal dominant and recessive Emery Dreifuss Muscular Dystrophy, limb girdle muscular dystrophy type 1B, autosomal recessive type 2 Charcot Marie Tooth, mandibuloacral dysplasia, familial partial lipodystrophy and Hutchinson-Gilford progeria.
Methods
We used mutation detection to evaluate the lamin A/C gene in a 45 year-old woman with familial dilated cardiomyopathy and conduction system disease whose family has been well characterized for this phenotype [1].
Results
DNA from the proband was analyzed, and a novel 2 base-pair deletion c.908_909delCT in LMNA was identified.
Conclusions
Mutations in the gene encoding lamin A/C can lead to significant cardiac conduction system disease that can be successfully treated with pacemakers and/or defibrillators. Genetic screening can help assess risk for arrhythmia and need for device implantation.
Similar content being viewed by others

Background
Dilated cardiomyopathy (DCM) is a disorder characterized by four-chamber dilation of the heart. DCM is often progressive and results in reduced heart function and congestive heart failure. Although etiologically diverse, inheritance is a significant contributor to the development of DCM. Supporting this, mutations in a number of different genes have recently been associated with familial forms of DCM [2], and mutations in the gene encoding lamin A/C appear to be a significant component. Lamins A and C are two different proteins produced by alternative splicing from the same gene. They are identical along their first 566 amino acids and differ only in their carboxyl terminus. As specialized proteins of the intermediate filament protein family, lamin A/C is found at the inner nuclear membrane where it forms a scaffold that interacts with a network of membrane bound and nucleoplasmic proteins in addition to chromatin [3–5]. Mutations in the gene encoding the nuclear membrane protein emerin produce autosomal recessive Emery Dreifuss Muscular Dystrophy, a disorder that phenotypically overlaps the neuromuscular and cardiovascular features seen with lamin A/C mutations [6]. Consistent with this, lamin A/C directly binds emerin [7, 8]. Lamins A and C are broadly expressed in many different cell types and are primarily expressed in terminally differentiated cells. It is not known how mutations in LMNA lead to tissue-specific phenotypes.
The first autosomal dominant form of cardiomyopathy was mapped in 1994 to the centromeric region of chromosome 1 [9]. Linkage was achieved using genetic and clinical material from a large, well-characterized kindred [1]. One of the striking characteristics from this family was the degree of cardiac conduction system disease that primarily targeted the atrioventricular node [1, 10]. Many members of the kindred exhibited sick sinus syndrome, atrial fibrillation and sudden death.
In addition to DCM with conduction system disease and variable skeletal muscle involvement, mutations in the Lamin A/C gene [OMIM 150330] have been shown to cause autosomal dominant [11] and autosomal recessive [12] Emery-Dreifuss Muscular Dystrophy (EDMD), autosomal dominant limb-girdle muscular dystrophy type 1B (LGMD) [13], autosomal recessive type 2 Charcot-Marie Tooth type 2 [14], familial partial lipodystrophy [15, 16], Mandibuloacral dysplasia [17] and Hutchinson-Gilford progeria [18]. The phenotypic spectrum from DCM to EDMD and limb girdle muscular dystrophy appears continuous. However, the mechanism by which LMNA mutations lead to cardiac versus skeletal muscle disease is unknown.
We now evaluated a woman with DCM and sinus bradycardia. Her immediate family history was significant for symptomatic bradycardia requiring pacemaker implantation. She is a member of the kindred initially described by Graber et al [1]. We screened this individual for mutations and found a novel mutation in exon 5 of LMNA. This two bp deletion is predicted to disrupt the reading frame and produce a truncated lamin A and C.
Methods
Patient recruitment
Written and informed consent was obtained in accordance with the University of Chicago's Institutional Review Board. Clinical data were obtained through evaluations performed at the University of Chicago.
Genetic Analysis
DNA extraction from blood was performed using PureGene according to the manufacturer's recommendations. PCR primers were designed to amplify all twelve protein coding exons of LMNA (Table I). DHPLC analysis of each exon was carried out on a WAVE™ (Transgenomic); exon 5 was analyzed at 65.2°C with 51%-56%-65% acetonitrile. SSCP was performed using MDE™ (FMC) as described [19]. Direct sequencing of PCR products was completed using cycle sequencing. To confirm the presence of the mutation, PCR products were purified and digested with AlwN I.
Results
Phenotype Analysis
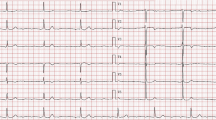
A 45 year-old Caucasian woman with a history of paroxysmal atrial fibrillation, sick sinus syndrome, dilated cardiomyopathy, and fatigue was seen in the University of Chicago's Cardiovascular Genetics Clinic for evaluation and treatment. The patient is a member of a large kindred previously described [1, 9, 10]. The patient was evaluated by EKG (Figure 1) that demonstrated marked sinus bradycardia and first degree atrioventricular block. An echocardiogram showed moderate dilation of the left ventricle (5.3 cm end diastolic internal diameter) and mild dilation of the right ventricle. The left ventricular performance was estimated as the lower end of normal with a normal fractional shortening (42%) (Figure 2). The patient reported no neuromuscular complaints, and on physical exam showed no overt muscle weakness.

EKG Shown is the 12 lead electrocardiogram performed prior to pacemaker-defibrillator implantation. Each box represents 200 msec. Sinus bradycardia is present; the proband's baseline heart rate was 45 beats per minute (normal is 80 beats per minute). First degree atrioventricular heart block is present (circled) as the conduction time between the atrial deflection and the ventricular deflection is 240 msec (normal is up to 200 msec).

Echo Shown is an M-mode echocardiogram through the diameter of the left ventricle revealing mild dilation. The left vertical line marks the dimension of the ventricle in diastole at its greatest, and this dimension is 5.3 cm (above the 95th percentile for body mass index). The fractional shortening was normal and the ejection fraction was read as low normal consistent with the mildly dilated left ventricle.
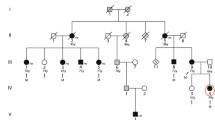
The pedigree for this proband is shown in Figure 3. All affected individuals had undergone pacemaker implantation for symptomatic bradycardia. Her mother reported fatigue and was also noted to have an abnormal skeletal muscle biopsy. Because of her marked fatigue and family history of sudden death, a dual chamber pacemaker and automatic defibrillator were implanted. Of note, the proband reported an improvement in her fatigue symptoms after pacer implantation. With pacemaker implantation, her baseline heart rate improved from 40 bpm to 80 bpm.

Pedigree Shown is the pedigree ascertained from the proband (indicated with an asterisk). Affected individuals are shown in black. Each affected individual had undergone pacemaker implantation for bradycardia. Shown in gray are individuals with a history of heart disease or sudden death.
Genotype Analysis
All twelve exons of LMNA were screened by denaturing high performance liquid chromatography (DHPLC) and single strand conformation polymorphism (SSCP) (Figure 4). In exon 5, a two bp (CT) deletion was detected in the LMNA gene (Figure 5). The two bp deletion falls into positions 908 and 909 of the NCBI Accession # NM_170708 where 1 is the first coding nucleotide. This frameshift mutation is expected to disrupt the lamin A and C sequences after the first 302 amino acids. In addition, 26 novel amino acids (CPAQPAPEAAGSQGGEASRPGGLTGP) are added after the truncation. The relatively high proline content of this additional sequence and the positioning of this truncation would be expected to disrupt the second half of the coiled-coil region of the protein. An additional 98 unrelated subjects with dilated cardiomyopathy were screened by SSCP and DHPLC and found not to carry the 908_909delCT mutation. Finally, 100 chromosomes from control DNA samples were checked and found not to carry the 908_909delCT mutation.

DHPLC A. DHPLC was used to detect an aberrant migration pattern (green) in the proband's DNA. The arrow indicates the polymorphism that correlates with the 2 bp deletion. B. Sequence analysis of LMNA exon 5 amplified directly from the proband's DNA revealed double-banding indicative of a frameshift mutation. To isolate the mutant allele, SSCP was used. Sequence of the mutant allele revealed a two bp deletion (top sequence). This deletion is predicted to truncate lamin A/C at amino acid 302 and add a novel 26 amino acids. The predicted protein product, if made, would be approximately 37 KDa and would disrupt the helical nature of lamin A/C and lack the carboxy-terminal globular region.

Digest with AlwN I to confirm the 908_909delCT mutation. The 908_909delCT mutation creates a novel AlwN I restriction site. Exon 5 was amplified from the proband and control genomic DNA and subsequently digested with AlwN I. Where control DNA lacks an AlwN I site (294 bp), the proband's exon 5 was digested with AlwN I generating fragments of 194 bp and 119 bp. DNA from 100 ethnically-matched control chromosomes was similarly checked for this mutation and did not have the 908_909delCT mutation. Lane 1, control; Lane 2, proband; Lane 3, size markers.
Discussion
Our findings demonstrate a novel mutation in lamin A/C in a proband whose family was previously linked to DCM with conduction disease and chromosome 1q21.2 locus [9]. Mutations in the LMNA gene associated with DCM have a more severe prognosis and higher-event rates compared to individuals with DCM that do not have LMNA mutations [20]. A recent report describing another family with a LMNA mutation causing DCM and conduction system disease documents the need for permanent pacemaker implantation [21]. Previous reports on the proband's kindred support a high-event rate in family members often resulting in sudden cardiac death even in family members with permanent pacemakers [10]. For example, ten out of 38 family members with pacemakers for high-grade atrioventricular block died suddenly. Subjects with LMNA mutations should be carefully evaluated for both pacemakers and defibrillators.
It is presently unknown whether autosomal dominant lamin A/C mutations impart their phenotype through dominant negative or haploinsufficient mechanisms. The majority of reported LMNA mutations are missense. In this case, the mutant protein is thought to be expressed and to act through a dominant interfering mechanism. A smaller number of LMNA mutations are frameshifting and are expected to create truncated lamin A and C. In at least one case, a nonsense mutation at amino acid position 6 was described [11]. This mutation is effectively a null allele and therefore, dominant mutations, at least in this case, would be expected to arise from haploinsufficiency of lamin A/C. Of note, mice with a null allele in lmna display a phenotype only in the homozygous state [22]. The mutation we now describe is predicted to generate a substantial protein fragment consisting of the first 37 KDa of lamin A/C. The novel, proline-rich sequence that is predicted at the carboxyl terminus of the truncated lamin A/C may further interfere with lamin function. Lamins assemble into a higher order structure and truncations may interfere with this assembly. The presence of aberrantly assembled lamin filaments may also interfere with other lamin-interacting proteins such as emerin or nesprin-1α [23].
The exact mechanism by which LMNA mutations cause disease is not clearly elucidated at this time. However, it is clear that mutations in LMNA cause tissue-specific disease affecting various differentiated cells including cardiac and skeletal muscle, adipocytes and neurons. This suggests that lamin's role at the nuclear membrane affects both cell-type and tissue-specific processes. Currently, there are at least two theories regarding the effects of LMNA [24]. The first postulates that disruptions of the inner nuclear membrane affect heterochromatin scaffolding and thereby gene expression. In this model, lamin A/C is necessary for the appropriate expression of tissue-specific genes. The second theory, the mechanical stress hypothesis, suggests weakness in the nuclear lamina leaves certain tissues more prone to damage by force. This model was first raised when LMNA mutations were primarily associated with disorders affecting striated muscle [24]. Interestingly, the preferential involvement of the specialized cells of the cardiac conduction system may support less the stress hypothesis as the cells of the atrioventricular node are less expected to undergo considerable deformation. The additional phenotypes now associated with LMNA mutations also favor alternative mechanisms of pathogenicity.
Conduction disease preceded the development of dilated cardiomyopathy in the proband's family [1, 10]. This indicates the necessity for careful documentation of a three-generation pedigree in the evaluation of patients with conduction system disease paying particular attention to sudden death, arrhythmia and bradycardia. Genotyping of patients with conduction disease and DCM may provide important information regarding device implantation. Genetic counseling for patients and family members should always accompany genetic testing as variable expressivity and age dependent penetrance may influence clinical decision making [25].
Conclusions
Individuals with DCM and conduction system disease should be screened by family history and genetic testing. Those found to have mutations in lamin A/C should be evaluated for placement of pacemakers and automatic defibrillators in order to prevent the sudden cardiac death associated with this disease.
Authors Contributions
HMM was responsible for gathering patient data. MRC and JMH carried out mutation detection. EMM conceived the study and evaluated the data. The manuscript was written and approved by all authors.
Abbreviations
- DCM:
-
dilated cardiomyopathy
- EDMD:
-
Emery Dreifuss Muscular Dystrophy
- LGMD:
-
limb Girdle muscular dystrophy
- SSCP:
-
single strand conformation polymorphism
- DHPLC:
-
denaturing high pressure liquid chromatography
- LMNA :
-
lamin A/C gene
References
Graber HL, Unverferth DV, Baker PB, Ryan JM, Baba N, Wooley CF: Evolution of a hereditary cardiac conduction and muscle disorder: a study involving a family with six generations affected. Circulation. 1986, 74: 21-35.
Watkins H: Genetic clues to disease pathways in hypertrophic and dilated cardiomyopathies. Circulation. 2003, 107: 1344-1346. 10.1161/01.CIR.0000057860.52586.9C.
Burke B, Stewart CL: Life at the edge: the nuclear envelope and human disease. Nat Rev Mol Cell Biol. 2002, 3: 575-585. 10.1038/nrm879.
Holmer L, Worman HJ: Inner nuclear membrane proteins: functions and targeting. Cell Mol Life Sci. 2001, 58: 1741-1747.
Mounkes LC, Burke B, Stewart CL: The A-type lamins: nuclear structural proteins as a focus for muscular dystrophy and cardiovascular diseases. Trends Cardiovasc Med. 2001, 11: 280-285. 10.1016/S1050-1738(01)00126-8.
Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D: Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994, 8: 323-327.
Clements L, Manilal S, Love DR, Morris GE: Direct interaction between emerin and lamin A. Biochem Biophys Res Commun. 2000, 267: 709-714. 10.1006/bbrc.1999.2023.
Vaughan A, Alvarez-Reyes M, Bridger JM, Broers JL, Ramaekers FC, Wehnert M, Morris GE, Whitfield WGF, Hutchison CJ: Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J Cell Sci. 2001, 114: 2577-2590.
Kass S, MacRae C, Graber HL, Sparks EA, McNamara D, Boudoulas H, Basson CT, Baker P. B., 3rd, Cody RJ, Fishman MC, et al.: A gene defect that causes conduction system disease and dilated cardiomyopathy maps to chromosome 1p1-1q1. Nat Genet. 1994, 7: 546-551.
Nelson SD, Sparks EA, Graber HL, Boudoulas H, Mehdirad AA, Baker P, Wooley C: Clinical characteristics of sudden death victims in heritable (chromosome 1p1-1q1) conduction and myocardial disease. J Am Coll Cardiol. 1998, 32: 1717-1723. 10.1016/S0735-1097(98)00424-0.
Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, Duboc D, Fardeau M, Toniolo D, Schwartz K: Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999, 21: 285-288. 10.1038/6799.
Raffaele Di Barletta M, Ricci E, Galluzzi G, Tonali P, Mora M, Morandi L, Romorini A, Voit T, Orstavik KH, Merlini L, Trevisan C, Biancalana V, Housmanowa-Petrusewicz I, Bione S, Ricotti R, Schwartz K, Bonne G, Toniolo D: Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am J Hum Genet. 2000, 66: 1407-1412. 10.1086/302869.
Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, de Visser M, Schwartz K: Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet. 2000, 9: 1453-1459. 10.1093/hmg/9.9.1453.
De Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, Kassouri N, Szepetowski P, Hammadouche T, Vandenberghe A, Stewart CL, Grid D, Levy N: Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet. 2002, 70: 726-736. 10.1086/339274.
Shackleton S, Lloyd DJ, Jackson SN, Evans R, Niermeijer MF, Singh BM, Schmidt H, Brabant G, Kumar S, Durrington PN, Gregory S, O'Rahilly S, Trembath RC: LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000, 24: 153-156. 10.1038/72807.
Cao H, Hegele RA: Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000, 9: 109-112. 10.1093/hmg/9.1.109.
Novelli G, Muchir A, Sangiuolo F, Helbling-Leclerc A, D'Apice MR, Massart C, Capon F, Sbraccia P, Federici M, Lauro R, Tudisco C, Pallotta R, Scarano G, Dallapiccola B, Merlini L, Bonne G: Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. 2002, 71: 426-431. 10.1086/341908.
De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Levy N: Lamin A Truncation in Hutchinson-Gilford Progeria. Science. 2003, 17: 17-
McNally EM, Duggan D, Gorospe JR, Bonnemann CG, Fanin M, Pegoraro E, Lidov HG, Noguchi S, Ozawa E, Finkel RS, Cruse RP, Angelini C, Kunkel LM, Hoffman EP: Mutations that disrupt the carboxyl-terminus of gamma-sarcoglycan cause muscular dystrophy. Hum Mol Genet. 1996, 5: 1841-1847. 10.1093/hmg/5.11.1841.
Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL, Boucek MM, Lascor J, Moss AC, Li WL, Stetler GL, Muntoni F, Bristow MR, Mestroni L: Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol. 2003, 41: 771-780. 10.1016/S0735-1097(02)02954-6.
Hershberger RE, Hanson EL, Jakobs PM, Keegan H, Coates K, Bousman S, Litt M: A novel lamin A/C mutation in a family with dilated cardiomyopathy, prominent conduction system disease, and need for permanent pacemaker implantation. Am Heart J. 2002, 144: 1081-1086. 10.1067/mhj.2002.126737.
Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B: Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999, 147: 913-920. 10.1083/jcb.147.5.913.
Mislow JM, Holaska JM, Kim MS, Lee KK, Segura-Totten M, Wilson KL, McNally EM: Nesprin-1alpha self-associates and binds directly to emerin and lamin A in vitro. FEBS Lett. 2002, 525: 135-140. 10.1016/S0014-5793(02)03105-8.
Wilson KL: The nuclear envelope, muscular dystrophy and gene expression. Trends Cell Biol. 2000, 10: 125-129. 10.1016/S0962-8924(99)01708-0.
Hanson E, Hershberger R: Genetic Counseling and Screening Issues in Familial Dilated Cardiomyopathy. J of Genetic Counseling. 2001, 10: 397-415. 10.1023/A:1016641504606.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/4/4/prepub
Acknowledgements
We would like to thank our patients for participating in these studies. This work is supported by NIH HL63783, American Heart Association, Burroughs Wellcome Fund and the Muscular Dystrophy Association (EMM).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing Interests
None declared.
Heather M MacLeod, Mary R Culley, Jill M Huber contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
MacLeod, H.M., Culley, M.R., Huber, J.M. et al. Lamin A/C truncation in dilated cardiomyopathy with conduction disease. BMC Med Genet 4, 4 (2003). https://doi.org/10.1186/1471-2350-4-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-4-4