
Abstract
The present work describes preparation of the reduced graphite oxide/nickel/palladium (rGO/Ni/Pd) composite and the examination of its basic electrochemical properties. The reduced graphite oxide was prepared by thermal reduction of graphite oxide beforehand obtained by electrochemical overoxidation of graphite in 8 M HClO4. In the second stage, Ni particles were electrodeposited onto rGO by the galvanostatic method. Finally, chemical deposition of palladium catalyst was performed to yield rGO/Ni/Pd composite. Transmission electron microscopy analysis (TEM) along with energy dispersive spectroscopy (EDS) technique has been used to determine the size as well as the distribution of Ni and Pd particles. The acquired electrochemical results revealed that rGO/Ni/Pd composite exhibits a good activity towards the processes of electrosorption of hydrogen. The highest calculated H/Pd atomic ratio for rGO/Ni/Pd electrode was equal 0.62. Electrocatalytic activity of the investigated material is also depicted by the reaction of methanol electrooxidation. Recorded current density of methanol electrooxidation was 15.04 A g−1 Pd.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nowadays, a lot of effort is devoted to design and production of highly efficient chemical power sources. One of the routes to reach the above mentioned goal is to use various carbon materials. They can serve as electrode templates for other electroactive substances like metals and metal oxides [1,2,3] or as an additional agent increasing the conductivity and stability of the applied electrode materials [4, 5]. Among them, one can distinguish mesoporous carbons [6, 7], carbon nanotubes and nanofibers [8], graphite and expanded graphite [9, 10], graphene [11], and carbon black [12]. In this term, graphite oxide (GO) and reduced graphite oxide (rGO) recently attract more attention [13,14,15,16].
GO is considered as a two-dimensional layered compound composed of oxidized graphene sheets decorated with oxygen-containing functional groups such as hydroxyl, carbonyl, carboxyl, and epoxyl [17, 18]. In consequence, an increase in the interlayer spacing between graphene sheets is noted [19]. GO can be prepared by chemical or electrochemical method of graphite overoxidation [20, 21]. Chemical routes require the use of strong oxidants, i.e., KMnO4 or KClO3 (Hummers, Brodie, and Staudenmaier methods) [17, 21]. An alternative method of GO synthesis is electrochemical overoxidation of graphite performed in sulfuric, nitric, or perchloric acid [22,23,24]. In these case, galvanostatic or voltammetric techniques are commonly used. Unfortunately, graphite oxide itself has a poor conductivity (~ 10−3 S cm−1) [25]. To solve this problem, graphite oxide is often subjected to reduction processes yielding reduced graphite oxide (rGO) with an enhanced electrical conductivity. GO reduction can be realized by chemical, electrochemical, or thermal methods [17, 26, 27]. To perform chemical processes, the use of various reducing agents such as sodium boron hydride, hydroquinone, hydrazine, or hydrogen is required [27,28,29]. Electrochemical route is usually based on the amperometric methods, in which GO is directly used as a working electrode [30] or may be added to the electrolyte solution to form suspension [31]. The process of thermal reduction of GO can be carried out in air or in an inert atmosphere (argon) at elevated temperature around 1000°C [32, 33]. The reduction of GO can be regarded as a partial removal of oxygen-containing functional groups accompanied by changes in the structure of the modified material. However, it is expected that the thickness of the rGO flakes will decrease due to removal of oxygen-containing functional groups; in many cases, contrary effect may be observed as a result of the exfoliation of the rGO flakes occurring during the reduction process of GO [18, 34]. The structure and conductivity of the final product strictly depend on the method and conditions used for reduction.
It is known that carbon based electrodes exhibit an interesting properties from the electrochemical point of view, but high level of electrocatalytical activity requires the presence of metallic catalysts [35,36,37,38,39,40]. The mentioned problem can be solved by the use of metallic catalyst alone, alloys or composite materials based on metallic components. The noble metals such as Pt, Pd, and Au are commonly used as catalysts for designing of electrode for electrochemical application including those associated with fuel cells [35,36,37,38,39,40]. In order to improve the electrochemical characteristics with accompaniment of catalyst costs reduction, the less expensive metals such as Ni, Fe, and Co are widely used to prepare electrode materials for fuel cells [38,39,40,41,42,43,44]. Among them, the systems based on Ni/Pd seem to be especially worth to mention because of their unique activity towards the processes of hydrogen production and storage as well as for alcohols electrooxidation [10, 41, 42]. On the other hand, the carbon/metal composites appear to be a very perspective for electrode production owing to limiting the use of expensive and/or often inaccessible metals. In such solutions, carbon commonly acts as a matrix, although there are reports in the literature that carbon can also be the active component of the composite, in which hydrogen can be accumulated [45]. Carbon/Ni/Pd systems exhibit also high stability as well as activity towards the processes of alcohol oxidation thus demonstrating possible application as anode materials for direct alcohol fuel cells [43, 44]. The main advantages of the considered systems upon the addition of carbon material include high specific surface area and electrical conductivity.
The aim of this work was to obtain an electrode material on the basis of reduced graphite oxide/nickel/palladium (rGO/Ni/Pd) composite which would exhibit an electrochemical activity in the processes of electrochemical hydrogen storage and methanol electro-oxidation. During the conducted measurements, the attention was paid on the electrochemical behavior of examined material in individual processes as well as on dual effect of both processes. Moreover, the reference point for tested rGO/Ni/Pd composite includes materials based containing palladium catalyst deposited onto graphite materials such as flaky graphite, expanded graphite or graphite and graphene oxides. This work is a continuation of our previous researches [10, 46,47,48] devoted to the use of such composites as EG/Ni/Pd and GIC-NiCl2-PdCl2-FeCl3 as potential materials for hydrogen storage and methanol oxidation. The reduced graphite oxide was prepared by the thermal reduction of graphite oxide previously obtained by the electrochemical overoxidation of graphite in 8 M HClO4. After electrochemical deposition of Ni onto rGO, the chemical deposition of palladium catalyst has been performed to yield final rGO/Ni/Pd composite. Electrochemical activity of the as prepared composite material was investigated in model processes of hydrogen electrosorption and methanol electrooxidation. The innovative element, and simultaneously, the most important aspect of this work is related to the synthesis of the examined composite material and combining hydrogen sorption processes with methanol electro-oxidation processes. In this work we propose a simple way of rGO/Ni/Pd preparation based on electrochemical overoxidation of pristine graphite followed by electrochemical and chemical deposition of nickel and palladium, respectively.
Materials and methods
Graphite oxide has been prepared by the electrochemical overoxidation of graphite (purity 99.5%, flake size 170–283 μm) in 8M HClO4 electrolyte realized by linear sweep voltammetry method (LSV). For this purpose, three-electrode cell was used, in which graphite powder (flakes) acts as a working electrode, whereas platinum wire and mercury sulfate electrode (Hg/Hg2SO4/1 M H2SO4) served as counter and reference electrode, respectively. Electrochemical investigations were started from the rest potential of electrode (ER) to 1.4 V with the scan rate equal to 0.01 mV s−1. The rest potential of graphite electrode from which its electrochemical overoxidation was started, was equal to 0.136 V. When the electrode reached 1.4 V, the as prepared GO was washed with distilled water until filtrate became neutral and then dried in air. In order to obtain reduced graphite oxide, the synthesized GO was heat treated in a muffle furnace for 4 min at 500 °C. The new-created rGO was rapidly removed from the furnace and cooled in air. More details about the process of GO synthesis may be found in our previous work [49]. To ensure good separation of the rGO flakes, the obtained product underwent a freeze-drying (lyophilization) process.
Process of electrochemical deposition of Ni onto the rGO was performed in bath composed of NiSO4∙ 7H2O (140 g dm−3), NiCl2∙6H2O (5 g dm−3), and H3BO3 (20 g dm−3). A galvanostatic deposition of Ni onto the rGO was carried out for 12 h with a constant current density equal to 90 mA g−1. In this process, mercury sulfate electrode (Hg/Hg2SO4/1 M H2SO4) was playing a role of reference electrode, whereas Ni plate was used as the counter electrode. In the final stage, the previously obtained rGO/Ni composite was subjected to chemical treatment in PdCl2/HCl water solution, in which the deposition of Pd catalyst occurred thus yielding rGO/Ni/Pd composite. The amount of Pd deposited was determined by the atomic absorption spectroscopy (AAS) and was 15.96 mg of Pd per 100 mg of rGO/Ni/Pd composite. The size as well as distribution of Ni and Pd particles was defined using transmission electron microscopy analysis (TEM) (Hitachi HT7700, Tokyo, Japan) along with energy dispersive spectroscopy (EDS) technique. The full process of rGO/Ni/Pd composite preparation has been depicted as a flow diagram in Scheme 1.

The flow diagram of the entire process of rGO/Ni/Pd composite preparation
In order to characterize the electrochemical properties of the obtained materials, the cyclic voltammetry (CV) and potentiostatic methods were used. All electrochemical measurements were performed at ambient temperature with potentiostat-galvanostat PGSTAT30 AutoLab (EcoChemie B.V.). For electrochemical tests, the rGO, rGO/Ni, or rGO/Ni/Pd composite were placed in porous polymer pocket along with graphite rod playing a role of current collector. Afterwards, the powder material was gently suppressed by a piece of polymer cloth placed at the top of the electrode to avoid washing out the previously inserted composite. The diameters of polymer pocket and graphite rod were 8 mm and 5 mm, respectively. Such geometry of the examined system allows forming uniform bed of tested material with a good electric conductivity without necessity of use of any additional binders. A mercury oxide electrode (Hg/HgO/6 M KOH, 0.098 V vs. NHE) and graphite rod serve as the reference and counter electrode, respectively. All potential values given in the manuscript have been calculated to reversible hydrogen electrode (RHE). As electrolytes, the water solution of 6 M KOH with or without addition of 1 M CH3OH have been used.
CV measurements were performed with scan rate 10 mV s−1 in the potential range of −0.13 ↔ 1.57 V, starting from the rest potential of electrode. In experiments devoted to the process of hydrogen electrosorption/desorption, when the electrode reached the potential of −0.13 V, the scanning was stopped for 15, 30, or 60 min to saturate the examined electrode with hydrogen. After that, the potential scanning was continued in the positive direction. In separate experiments, the lower limit potential was successively lowered to −0.43 V in order to examine the influence of the cathodic potential applied on the efficiency of hydrogen sorption.
To verify the electroactivity of examined electrodes in the reaction of methanol electrooxidation, the potentiostatic experiments were conducted under constant potential value gained from the voltammetric measurements.
Results and discussion
Characterization of the GO and rGO
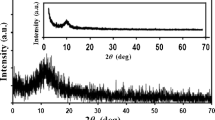
The structure of GO and the product of its thermal reduction was analyzed by X-ray diffraction (XRD) and Raman spectroscopy. Whereas, XPS and SEM techniques were used to determine their surface chemical composition and morphology. Results of the above mentioned investigations along with the relevant discussion have been presented in our previous work [49]. Briefly, XRD results (Fig. 1) showed that overoxidation of graphite in 8 M HClO4 led to increase in d spacing between graphene layers due to the formation of oxygen containing groups on their surface. This result was used to prove the GO formation.

XRD patterns of GO, rGO and rGO/Ni/Pd
Thermal treatment of GO contributed to the destruction of its crystal structure caused by transformation of graphene layers into non-crystalline graphene stacks with random interlayer spaces. Raman spectra (Fig. 2) have confirmed that along with the graphite transformation into GO, its structural order become worse and the formation of oxygen functionalities contributes to decrease in the concentration of sp2-hybridized bonds. The decrease in intensity of peak D together with the decrease of ID/IG ratio confirmed the above mentioned effect. It has been also shown that thermal reduction of GO causes the partial decomposition of oxygen groups being chemically bounded with graphene layers, thus allowing to partial recovery of the sp2-hybridized C–C bonds.

Raman spectra of GO and rGO
On the basis of the obtained SEM images, it can be concluded that thermal treatment of GO performed at 500 °C in air atmosphere also led to a significant increase in flakes volume. The violent nature of thermal exfoliation of GO flakes, which causes wrinkling and folding of the graphene layers is responsible for the abovementioned phenomenon. This behavior has been confirmed by the results of XPS analysis. The total content of carbon as well as oxygen are given in Table 1. It was observed that thermal reduction of GO results in a significant decrease in total oxygen content due to the decomposition of some oxygen functional groups.
On further investigations, thermally reduced GO was used for the preparation of rGO/Ni/Pd as a potential electrode material for energy conversion and storage devices.
TEM images showing the size and distribution of Ni and Pd particles over the matrix made of rGO are shown in Fig. 3. As one can see, metal particles are evenly distributed on the surface of reduced graphite oxide flakes. The size of Pd particles ranges from the several to 250 nm. The particle size of Pd catalysts deposited onto Ni surface has been determined using the Scherrer equation [50]:

TEM images of the rGO/Ni/Pd composite (a, b) along with the results of the EDS analysis (c, d)
where FWHM, K, θ, and λ represent full width at half maximum of peaks in radian, Scherrer constant (0.94), the Bragg angle in radians and X-ray wavelength (λ: 1.54056 Å for Cu Kα), respectively. The calculated average Pd particle size on the basis of XRD spectrum noted for rGO/Ni/Pd composite (insert in Fig. 1) equals 190 nm. It is worth to note that this value is comparable with that given from the observation of TEM images.
According to the mechanism of electroless deposition realized by a metal exchange reaction, the Pd particles can be accumulated only on the previously electrodeposited Ni substrate. Taking into the consideration the above mentioned rule, it is seen from EDS results (Fig. 3c and d) that the Ni particles are not completely covered with the Pd during the electroless process. In Fig. 3c and d, one can observe Ni components present on rGO surface.
Electrochemical measurements
Electrochemical characterization of the obtained materials
Figure 4 shows cyclic voltammograms (CV) for rGO, rGO/Ni, and rGO/Ni/Pd electrodes recorded in 6M KOH solution with a scan rate of 10 mV s−1. The CV measurements were started from the rest potential of electrode (ER) in the negative direction. For the CV curve recorded for rGO electrode, at the potential 0.67 V during cathodic polarization the current densities start to decrease. This effect is most likely attributed to the reduction process of oxygen functional groups originally present on the rGO surface. After reversing the polarization, no anodic peak is recorded up to 1.47 V. After reaching this potential, the oxygen evolution reaction starts causing a significant increase in anodic current density. In case of rGO/Ni electrode, during cathodic polarization, a small cathodic peak localized at 0.65 V followed by a further increase of the cathodic current density is noted. Both effects can be attributed to the reduction of oxygen functional groups present on rGO matrix. It is worth to note that in the potential range of 0.27 ↔ −0.13 V, the reduction reaction of Ni2+ to metallic form may also occur. After the reversal of polarization, a small anodic peak (0.22 V) related with Ni0 → Ni2+oxidation emerges on CV. When the potential of 1.47 V is reached, an increase of the anodic current density can be observed which depicts the reaction of oxygen evolution accompanied by the oxidation of nickel hydroxide to nickel oxyhydroxide (β-Ni(OH)2→β,γ-NiOOH) [51]. The reversal of polarization from anodic to cathodic reveals a small cathodic peak at 1.39 V illustrating the reduction of β-NiOOH to β-Ni(OH)2 [51]. A cyclic voltammogram recorded for rGO/Ni/Pd composite electrode has a typical character for palladium based electrodes. During potential sweep in the negative direction, a sharp cathodic peak at 0.71 V resulting from the reduction of PdO to Pd is noted. On further cathodic polarization, a significant increase in the cathodic current density can be observed arising from hydrogen sorption accompanied by PdH formation. On the anodic potential sweep, in the potential range of 0.09 ↔ 0.57 V, anodic peak depicting the reaction of hydrogen desorption (PdH decomposition) is recorded [48, 52]. When the potential of 1.47 V is exceeded, anodic current density rise due to reactions associated with the oxygen evolution as well as oxidation of nickel hydroxide to nickel oxyhydroxide. The reversal of polarization from anodic to cathodic brings about as mall cathodic peak related with the reduction of β-NiOOH to β-Ni(OH)2. The presence of signals originating from Ni component indicates that some part of Ni is not covered by Pd, that’s why the electrolyte can easily reach a Ni sublayer. It is worth to note that the above mentioned thesis well correlates with the observations of TEM images (Fig. 3).

Cyclic voltammograms for rGO, rGO/Ni and rGO/Ni/Pd electrodes recorded in 6 M KOH solution in the potential range of −0.13 ↔ 1.57 V with scan rate 10 mV s−1
Electrochemical hydrogen sorption/desorption
Based on the CV results described above, it can be concluded that only the electrode containing the palladium catalyst exhibits activity in the process of reversible electrosorption of hydrogen. Therefore, only rGO/Ni/Pd electrode was subjected to the further electrochemical investigations.
CV curves recorded for rGO/Ni/Pd electrode including the process of its potentiostatic saturation with hydrogen are presented in Fig. 5. Process of potentiostastic sorption was carried out by stopping the potential scanning for chosen time when the electrode reached the potential of −0.13 V. The saturation of the examined material with hydrogen was realized for 15, 30, or 60 min. After that, during the anodic scanning, current peak of hydrogen desorption has been recorded. As can be seen from the obtained curves, 15-min potentionstatic reduction treatment at −0.13 V results on appearance of a huge anodic peak associated with the process of hydrogen desorption. Simultaneously, the abovementioned peak becomes much wider and its current maximum shifts by about 0.4 V towards the more positive potentials with increasing the sorption time. This indicates that the amount of stored hydrogen enhances considerably along the extending the time of potentiostatic saturation.

Cyclic voltammograms for rGO/Ni/Pd electrode recorded in 6 M KOH solution in the potential range of −0.13 ↔ 1.57 V (involving the potentiostatic saturation of electrode with hydrogen at −0.13 V for 15, 30, and 60 min). Scan rate 10 mV s−1
The H/Pd atomic ratio calculated from the CV measurements for electrode potentiostatically saturated with hydrogen for 15 min is equal to 0.39. This value is remarkably higher than corresponding value for the electrode not subjected to the potentiostatic treatment (0.03) (see Table 2). From the above, it can be concluded that the examined composite material exhibits good activity in the process of electrochemical sorption/desorption of hydrogen. The extension of sorption time to 30 and further to 60 min causes a consecutive increase in the amount of sorbed/desorbed hydrogen. The H/Pd atomic ratio calculated for a longer time of potentiostatic hydrogen sorption (30 and 60 min) is 0.53 and 0.62, respectively. The latter value fits into the range of 0.6–0.7, mostly reported in the literature [52]. It should also be emphasized that, extending the time of the potentiostatic treatment from 15 to 30 and up to 60 min results in gradual decrease in intensity of anodic charge grow up associated with the hydrogen electro-oxidation (desorption) reaction. Therefore, the conclusion can be drawn, that process of 60-min potentiostatic sorption is very close to reach the stage of full saturation of the examined rGO/Ni/Pd electrode with hydrogen.
CV curves depicting the influence of the limit potential applied on the process of hydrogen sorption on rGO/Ni/Pd electrode are given in Fig. 6. As one can see, the gradual lowering of potential of hydrogen sorption from −0.13 to −0.43 V results a successive increase in cathodic current density related with hydrogen sorption. In consequence, during the anodic polarization, a significant enhancement of the hydrogen desorption peak is observed. Such a behavior is accompanied by the gradual shift of these peaks towards more positive potentials. In parallel, the H/Pd atomic ratios increase from 0.03 for −0.13 V up to 0.26 for −0.43 V (see Table 3). The latter value is close to the ratio noted for rGO/Ni/Pd electrode underwent potentiostatic saturation with hydrogen for 15 min. However, it should be noted, that on lowering the potential of hydrogen sorption down to −0.43 V, a competitive reaction of hydrogen evolution is likely to occur. It is also important to emphasize that the intensive and rapid evolution of gaseous hydrogen may be destructive both for the construction of the powder-type electrode as well as for the examined rGO/Ni/Pd composite itself.

Cyclic voltammograms for rGO/Ni/Pd electrode recorded in 6 M KOH solution depicting the influence of the applied cathodic potential on the process of hydrogen sorption/desorption. Scan rate 10 mV s−1
On the basis of the acquired results, it can be pointed out that high activity of rGO/Ni/Pd composite mainly arises from the Ni/Pd system, but it is plausible that in some part, during the electrochemical sorption, hydrogen diffuses through the all phases. Therefore, it cannot be excluded that a small amount of hydrogen can be also stored within the rGO phase.
Electrochemical oxidation of methanol
In the second part of this work, the activity of rGO/Ni/Pd composite in the process of methanol electrooxidation has been examined. Cyclic voltammagrams for the rGO/Ni/Pd electrode recorded in alkaline electrolyte containing 1 M of CH3OH are shown in Fig. 7.

Cyclic voltammograms for rGO/Ni/Pd electrode recorded in 1 M CH3OH/6 M KOH solution in the potential range of −0.13 ↔ 1.57 V. Scan rate 10 mV s−1
On the anodic polarization, at the potential of 0.85 V, a high and sharp anodic peak originating from the reaction of methanol oxidation is recorded. The appearance of this peak undoubtedly evidences the catalytic activity of Pd catalyst [53]. In the subsequent cycles, the intensity of above mentioned signal significantly increase. Most likely this effect is related with the improvement of electrode wettability. It cannot be excluded that for the increased electrochemical activity towards the methanol oxidation some changes in the structure of the examined compound are responsible. It can be explained in term of electrochemical exfoliation occurring on potential scanning, which makes easier access of the methanol molecules to the palladium particles. Contrary to many other works devoted to Pd catalyst, the recorded methanol oxidation peak is narrow and sharp with a symmetrical shape. These observations suggest a direct character of the methanol electrooxidation on the palladium catalyst surface, which means that methanol molecules are mostly oxidized to the final product—CO2, without the formation of stable intermediate products like aldehyde and formic acid.
As one can see, during the cathodic polarization a small anodic peak at 0.71 V corresponding to the reactivation of electrode surface is recorded [53].
In the next order, the durability and stability of rGO/Ni/Pd electrode in the process of methanol electrooxidation were examined by long-lasting CV measurements. The obtained results showing the dependence of current density of methanol oxidation on the number of performed cycles are present in Fig. 8. As can be seen, current density of methanol oxidation gradually increases and reaches nearly stable current density for 50th cycle. As previously mentioned, some structural changes in the examined material may be responsible for this behavior. It means that no decrease in electrocatalytic activity of rGO/Ni/Pd electrode caused by palladium catalyst poisoning by intermediate products of methanol oxidation (mostly CO) is observed.

Durability and stability of rGO/Ni/Pd electrode recorded during following cycles of voltammetric oxidation of methanol
To confirm the electrocatalytic activity of the rGO/Ni/Pd electrode the potentiostatic measurements under constant potential of 0.85 V were performed in 6 M KOH solution with and without addition of methanol. The value of the applied potential was chosen on the basis CV measurements observations. As can be seen in Fig. 9, for measurement in pure 6 M KOH solution, a very low and constant current density is recorded. Contrary, in case of electrolyte containing 1 M of CH3OH, rapid polarization gives initial increase in anodic current density up to 19.9 A gPd−1 associated with methanol oxidation. On further polarization, a gradual decrease in current density is observed until the current plateau is reached. After an hour and a half of potentiostatic oxidation of methanol on rGO/Ni/Pd electrode, the anodic current density equals 0.94 A gPd−1, whereas the calculated total charge of methanol oxidation is 3.64 Ah gPd−1.

Potentiostatic curves for rGO/Ni/Pd electrode recorded at 0.85 V in 6 M KOH with or without addition of 1 M CH3OH
Summarizing, the results of potentiostatic measurements are consistent with the observations acquired from CV investigations proving that the rGO/Ni/Pd composite exhibits good catalytic activity towards methanol electrooxidation.
Table 4 shows a comparison of electroactivity of different catalyst in processes of hydrogen electrosorption and methanol electrooxidation. As can be seen, rGO/Ni/Pd composite shows better performance in the process of hydrogen electrosorption than in electrochemical oxidation of methanol. This is because more emphasis in this work has been placed on finding a suitable material for electrochemical storage of hydrogen. In the next stage of the research, it was decided to prove the bifunctionality of the obtained composite electrode material by examining its catalytic activity in the process of methanol electro oxidation. In case of the process of hydrogen electrosorption, rGO/Ni/Pd composite shows hydrogen storage capacity (H/Pd = 062) close to the maximum value reported for similar materials, e.g., Pd/-rGO-EG (H/Pd = 0.14↔0.77) [58]. However, at this stage of the research, it should be emphasized that our composite material has not been optimized in terms of the amount of palladium catalyst introduced; therefore, achieving a slightly higher hydrogen capacity cannot be ruled out. In terms of methanol electrooxidation rGO/Ni/Pd composite exhibit catalytic activity of 15,04 A g−1 Pd falling in between other materials based on rGO and palladium catalysts, such as rGO/PDPA/Pd (2.7 mA cm−2) and Pd/rGO and Pd-Cu/rGO (200 and 550 mA mg−1 Pd).
Taking into account abovementioned information, it can be concluded that the palladium catalyst structure in the obtained rGO/Ni/Pd composite is favorable for hydrogen electrosorption process wchich has volumetric character. In order to increase the activity of the tested material in the methanol electrooxidation process, which takes place only on the surface of the Pd catalyst, it would be necessary to obtain a composite with a better distribution of the catalyst particles on the rGO matrix.
Conclusions
According to the obtained results, the proposed three step method was successfully used for preparation of rGO/Ni/Pd composite.
The study of electrochemical properties of rGO/Ni/Pd electrode conducted by cyclic voltammetry method showed that the examined composite material exhibits electrochemical activity typical for both Ni and Pd catalyst. We found that our material exhibits electrochemical activity at an acceptable level in two processes commonly used in various fuel cells, therefore, emphasizes its universal nature. It worth to note that, the examined material has a combined activity toward the hydrogen sorption processes and methanol electro-oxidation processes.
It has also been proved that the potentiostatic saturation of rGO/Ni/Pd electrode significantly increases the amount of stored hydrogen. Moreover, the extension of potentiostatic sorption duration leads to the further increase in its storage capacity. CV measurements revealed that 60-min potentiostatic treatment at −0.13 V allows for almost full saturation of the examined rGO/Ni/Pd electrode with hydrogen. The H/Pd atomic ratio calculated for rGO/Ni/Pd electrode potentiostatically saturated with hydrogen for 60 min equals to 0.62.
Additionally, it has been shown that hydrogen storage capacity of rGO/Ni/Pd increases with the lowering of the potential in which the hydrogen sorption is performed.
The CV experiments performed in the 1 M CH3OH in 6 M KOH solution indicated that rGO/Ni/Pd composite also exhibits catalytic activity in the reaction of methanol electrooxidation yielding the current density of 8.32 A g−1 Pd. The above mentioned activity increases on cycling up to 16.88 A g−1 Pd probably due to some structural changes caused by cyclic treatment of the examined material.
The results of potentiostatic measurements have confirmed electrocatalytic activity of the rGO/Ni/Pd electrode towards the methanol oxidation.
On the basis of the gained results, it can be concluded that rGO/Ni/Pd composite can be regarded as potential electrode material in direct methanol fuel cells.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
References
Rikkinen E, Santasalo-Aarnio A, Airaksinen S, Borghei M, Viitanen V, Sainio J, Kauppinen EI, Kallio T, Krause AOI (2011) Atomic layer deposition preparation of Pd nanoparticles on a porous carbon support for alcohol oxidation. J Phys Chem C 115:23067–23073
Shi W, Rui X, Zhu J, Yan Q (2012) Design of nanostructured hybrid materials based on carbon and metal oxides for Li ion batteries. J Phys Chem C 116:26685–26693
Khamsanga S, Nguyen MT, Yonezawa T, Thamyongkit P, Pornprasertsuk R, Pattananuwat P, Tuantranont A, Siwamogsatham S, Kheawhom S (2020) MnO2 heterostructure on carbon nanotubes as cathode material for aqueous zinc-ion batteries. Int J Mol Sci 21:4689
Sierczynska A, Lota K, Lota G (2010) Effects of addition of different carbon materials on the electrochemical performance of nickel hydroxide electrode. J Power Sources 195:7511–7516
Blecua M, Fatas E, Ocon P, Valenciano J, De la Fuente F, Trinidad F (2017) Influences of carbon materials and lignosulfonates in the negative active material of lead-acid batteries for microhybrid vehicles. J Energy Storage 11:55–63
Takamura T (2002) Trends in advanced batteries and key materials in the new century. Solid State Ionics 152–153:19–34
Nie Y, Yang H, Pan J, Li W, Sun Y, Niu H (2017) Synthesis of nano-Ni(OH)2/porous carbon composite as superior cathode materials for alkaline power batteries. Electrochim Acta 252:558–567
Benavides LA, Cuscueta DJ, Ghilarducci AA (2015) MWCNT as mechanical support during ball milling of an AB5 alloy used as negative electrode of a Ni-MH battery. Int J Hydrogen Energy 40:4925–4930
Luo P, Zheng C, He J, Tu X, Sun W, Pan H, Zhou Y, Rui X, Zhang B, Huang K (2022) Structural engineering in graphite-based metal-ion batteries. Adv Funct Mater 32:2107277
Krawczyk P, Rozmanowski T, Frankowski M (2019) Methanol electrooxidation at electrodes made of exfoliated graphite/nickel/palladium composite. Catal Lett 149:2307–2316
Ji L, Meduri P, Agubra V, Xiao X, Alcoutlabi M (2016) Graphene-based nanocomposites for energy storage. Adv Energy Mater 6:1502159
Ch LP, Hu CC, Ch LT, Chang WS, Wang TH (2014) Synthesis and characterization of carbon black/manganese oxide air cathodes for zinc-air batteries. J Power Sources 269:88–97
Maruyama J, Shinagawa T, Hayashida A, Matsuo Y, Nishihara H, Kyotani T (2016) Vanadium-ion redox reactions in a three-dimensional network of reduced graphite oxide. Chem Electro Chem 3:650–657
Dashairya L, Das D, Saha P (2020) Electrophoretic deposition of antimony/reduced graphite oxide hybrid nanostructure: a stable anode for lithium-ion batteries. Mater Today Commun 24:1011892
Subaşi Y, Somer M, Yağci MB, Slabon A, Afyon S (2020) Surface modified TiO2/reduced graphite oxide nanocomposite anodes for lithium ion batteries. J Solid State Electrochem 24:1085–1093
Zhang Y, Qin J, Lowe SE, Li W, Zhu Y, Al-Mamun M, Batmunkh M, Qi D, Zhang S, Zhong YL (2021) Enhanced electrochemical production and facile modification of graphite oxide for cost-effective sodium ion battery anodes. Carbon 177:71–78
Mauro M, Cipolletti V, Galimberti M, Longo P, Guerra G (2012) Chemically reduced graphite oxide with improved shape anisotropy. J Phys Chem C 116:24809–24813
Iurchenkova AA, Lobiak EV, Kobets AA, Kolodin AN, Stott A, Silva SRP, Fedorovskaya EO (2021) A complex study of the dependence of the reduced graphite oxide electrochemical behavior on the annealing temperature and the type of electrolyte. Electrochim Acta 370:137832
Bianco A, Cheng HM, Enoki T, Gogotsi Y, Hurt RH, Koratkar N, Kyotani T, Monthioux M, Park CR, Tascon JMD et al (2013) All in the graphene family - a recommended nomenclature for two-dimensional carbon materials. Carbon 65:1–6
Beck F, Jiang J, Krohn H (1995) Potential oscillations during galvanostatic overoxidation of graphite in aqueous sulphuric acids. J Electroanal Chem 389:161–165
Gurzęda B, Subrati A, Florczak P, Kabacińska Z, Buchwald T, Smardz L, Peplińska B, Jurga S, Krawczyk P (2020) Two-step synthesis of well-ordered layered graphite oxide with high oxidation degree. Appl Surf Sci 507:145049
Inagaki M, Iwashita N, Kouno E (1990) Potential change with intercalation of sulfuric acid into graphite by chemical oxidation. Carbon 28:49–55
Avdeev VV, Tverezovskaya OA, Sorokina NE (2000) Spontaneous and electrochemical intercalation of HNO3 into graphite. Mol Cryst Liq Cryst Sci Technol Mol Cryst Liq Cryst 340:137–142
Nakajima T, Matsuo Y (1994) Formation process and structure of graphite oxide. Carbon 32:469–475
Zhu Y, Murali S, Cai W, Li X, Won Suk J, Potts JR, Ruoff RS (2010) Graphene and graphene oxide: synthesis, properties, and applications. Adv Mater 22:3906–3924
Kobets AA, Iurchenkova AA, Asanov IP, Okotrub AV, Fedorovskaya EO (2019) Redox processes in reduced graphite oxide decorated by carboxyl functional groups. Phys Status Solidi B 256:1800700
Hussain IR, Radiah ABD, Kamil FH, Yasinb FM, Kamarudinb S, Yusoff HM (2018) Physical properties of reduced graphite oxide prepared via chemical reduction by using ammonia solution as a reducing agent. IOP Conf Ser Mater Sci Eng 454:012136
Kim J, Nam DG, Yeum JH, Suh S, Oh W (2015) Characterization of graphite oxide reduced by thermal and/or chemical treatments. Trans Electr Electron Mater 16(5):274–279
Gurzęda B, Krawczyk P (2019) Electrochemical formation of graphite oxide from the mixture composed of sulfuric and nitric acids. Electrochim Acta 310:96–103
Tateishi H, Koinuma M, Miyamoto S, Kamei Y, Hatakeyama K, Ogata C, Taniguchi T, Funatsu A, Matsumoto Y (2014) Effect of the electrochemical oxidation/reduction cycle on the electrochemical capacitance of graphite oxide. Carbon 76:40–45
Tong H, Zhu J, Chen J, Han Y, Yang S, Ding B, Zhang X (2013) Electrochemical reduction of graphene oxide and its electrochemical capacitive performance. J Solid State Electrochem 17:2857–2863
Gridnev AA, Gudkov MV, Bekhli LS, Melnikov VP (2015) Possible mechanism of thermal reduction of graphite oxide. Russ J Phys Chem B 12:1008–1016
Niyitanga T, Jeong HK (2018) Thermally reduced graphite oxide and molybdenum disulfide composite for enhanced hydrogen evolution reaction. Chem Phys Lett 706:266–272
Botas C, Alvarez P, Blanco C, Santamaría R, Granda M, Gutiérrez MD, Rodríguez-Reinoso F, Menéndez R (2013) Critical temperatures in the synthesis of graphene-like materials by thermal exfoliation–reduction of graphite oxide. Carbon 52:476–485
Shen PK, Xu C, Zeng R, Liu Y (2006) Electro-oxidation of methanol on NiO-promoted Pt/C and Pd/C catalysts. Electrochem Solid-State Lett 9(2):A39–A42
Zhu F, Ma G, Bai Z, Hang R, Tang B (2013) High activity of carbon nanotubes supported binary and ternary Pd-based catalyst for methanol, ethanol and formic acid electro-oxidation. J Power Sources 242:610–620
Lee J, Theerthagiri J, Nithyadharseni P, Arunachalam P, Balaji D, Kumar AM, Madhavan J, Mittal V, Choi MY (2021) Heteroatom-doped graphene-based materials for sustainable energy applications: a review. J Chem Eng 435:134790
Lee J, Theerthagiri J, Fonseca S, Pinto LMC, Maia G, Choi MY (2022) Reconciling of experimental and theoretical insights on the electroactive behavior of C/Ni nanoparticles with AuPt alloys for hydrogen evolution efficiency and Non-enzymatic sensor. J Chem Eng 143:110849
Jeong Y, Naik SS, Yu Y, Theerthagiri J, Lee SJ, Show PL, Choi HC, Choi MY (2023) Ligand-free monophasic CuPd alloys endow boosted reaction kinetics toward energy-efficient hydrogen fuel production paired with hydrazine oxidation. J Mater SciTechnol 143:20–29
Lee Y, Yu Y, Das HT, Theerthagiri J, Lee SJ, Min A, Kim G-A, Choi HC, Choi MY (2023) Pulsed laser-driven green synthesis of trimetallic AuPtCu nanoalloys for formic acid electro-oxidation in acidic environment. Fuel 332:126164
Miaoa F, Taoa B, Suna L, Liua T, Youa J, Wanga L, Chu PK (2010) Preparation and characterization of novel nickel–palladium electrodes supported by silicon microchannel plates for direct methanol fuel cells. J Power Sources 195:146–150
Song Y, Zhang X, Yang S, Wei X, Sun Z (2016) Electrocatalytic performance for methanol oxidation on porous Pd/NiO composites prepared by one-step dealloying. Fuel 181:269–276
Tan JL, De Jesus AM, Chua SL, Sanetuntikul J, Shanmugam S, Tongol BJV, Kim H (2017) Preparation and characterization of palladium-nickel on graphene oxide support as anode catalyst for alkaline direct ethanol fuel cell. Appl Catal A-Gen 531:29–35
Łukowiec D, Wasiak T, Janas D, Drzymała E, Depciuch J, Tarnawski T, Kubacki J, Wacławek S, Radoń A (2022) Pd decorated Co–Ni nanowires as a highly efficient catalyst for direct ethanol fuel cells. Int J Hydrogen Energy 47:41279–41293
Skowroński JM, Czerwiński A, Rozmanowski T, Rogulski Z, Krawczyk P (2007) The study of hydrogen electrosorption in layered nickel foam/palladium/carbon nanofibers composite electrodes. Electrochim Acta 52:5677–5684
Skowroński JM, Rozmanowski T, Krawczyk P (2013) Enhancement of electrochemical hydrogen storage in NiCl2-FeCl3-PdCl2-graphite intercalation compound effected by chemical exfoliation. Appl Surf Sci 275:282–288
Rozmanowski T, Krawczyk P (2018) Influence of chemical exfoliation process on the activity of NiCl2-FeCl3-PdCl2 graphite intercalation compound towards methanol electrooxidation. Appl Catal B 224:53–59
Krawczyk P, Rozmanowski T, Osińska M (2016) Electrochemical sorption of hydrogen in exfoliated graphite/nickel/palladium composite. Int J Hydrogen Energy 41:20433–20438
Gurzęda B, Florczak P, Wiesner M, Kempiński M, Jurga S, Krawczyk P (2016) Graphene material prepared by thermal reduction of the electrochemically synthesized graphite oxide. RSC Adv 6:63058–63063
Hüner B, Demir N, Kaya MF (2023) Ni-Pt coating on graphene based 3D printed electrodes for hydrogen evolution reactions in alkaline media. Fuel 331:125971
Urbanczyk E, Maciej A, Stolarczyk A, Basiaga M, Simka W (2019) The electrocatalytic oxidation of urea on nickel-graphene and nickel-graphene oxide composite electrodes. Electrochim Acta 305:256–263
Grdeń M, Piaścik A, Koczorowski Z, Czerwiński A (2002) Hydrogen electrosorption in Pd/Pt alloys. J Electroanal Chem 532:35–42
Khuntia H, Bhavani KS, Anusha T, Trinadh T, Stuparu MC, Brahman PK (2021) Synthesis and characterization of corannulene-metal-organic framework support material for palladium catalyst: an excellent anode material for accelerated methanol oxidation. Colloids Surf A Physicochem Eng Asp 615:126237
Subbaraman R, Danilovic N, Lopes PP, Tripkovic D, Strmcnik D, Stamenkovic VR, Markovic NM (2012) Origin of anomalous activities for electrocatalysts in alkaline electrolytes. J Phys Chem C 116:22231–22237
Bakos I, Paszternák A, Zitoun D (2015) Pd/Ni synergestic activity for hydrogen oxidation reaction in alkaline conditions. Electrochim Acta 176:1074–1082
Smiljanić M, Bele M, Moriau L, Ruiz-Zepeda F, Šala M, Hodnik N (2021) Electrochemical stability and degradation of commercial Pd/C catalyst in acidic media. J. Phys Chem C 125(50):27534–27542
Hubkowska K, Pająk M, Czerwiński A (2022) Hydrogen electrosorption properties of electrodeposited Pd-Ir alloys. J Solid State Electrochem 26:103–109
Boateng E, van der Zalm J, Chen A (2021) Design and electrochemical study of three-dimensional expanded graphite and reduced graphene oxide nanocomposites decorated with Pd nanoparticles for hydrogen storage. J Phys Chem 125:22970–22981
Hu C, Wang X (2015) Highly dispersed palladium nanoparticles on commercial carbon black with significantly high electro-catalytic activity for methanol and ethanol oxidation. Int J Hydrogen Energy 40:12382–12391
Tao Y, Dandapat A, Chen L, Huang Y, Sasson Y, Lin Z, Zhang J, Guo L, Chen T (2016) Pd-on-Au supra-nanostructures decorated graphene oxide: an advanced electrocatalyst for fuel cell application. Langmuir 32:8557–8564
Liu Q, Lin Y, Fan J, Lv D, Min Y, Wu T, Xu Q (2016) Well-dispersed palladium nanoparticles on three-dimensional hollow N-doped graphene frameworks for enhancement of methanol electro-oxidation. Electrochem Commun 73:75–79
Kiyani R, Parnian MJ, Rowshanzamir S (2017) Investigation of the effect of carbonaceous supports on the activity and stability of supported palladium catalysts for methanol electro-oxidation reaction. Int J Hydrogen Energy 42:23070–23084
Li H, Zhang Y, Wan Q, Li Y, Yang N (2018) Expanded graphite and carbon nanotube supported palladium nanoparticles for electrocatalytic oxidation of liquid fuels. Carbon 131:111–119
Yang M, An J, Zhang S, Gao L, Yan S (2022) High electrochemical activity of Pd/C catalyst with trace amounts of PdxHy. Int J Electrochem Sci 17:1–17
Madaswamy SL, Keertheeswari NV, Alothman AA, Al-Anazy MM, Alqahtani KN, Wabaidur SM, Dhanusuraman R (2022) Fabrication of nanocomposite networks using Pd nanoparticles/polydiphenylamine anchored on the surface of reduced graphene oxide: an efficient anode electrocatalyst for oxidation of methanol. Adv Ind Eng Polym Res 5:18–25
Chandra Sekhar Y, Raghavendra P, Sri Chandana P, Maiyalagan T, Subramanyam Sarma L (2023) Graphene supported Pd–Cu bimetallic nanoparticles as efficient catalyst for electrooxidation of methanol in alkaline media. J Phys Chem Solids 174:111133
Funding
Financial support from the National Science Centre of Poland (grant No. 2017/25/B/ST8/01634) is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
T. R. investigation, conceptualization, methodology, writing-original draft preparation, writing- reviewing and editing. P. K. conceptualization, supervision, writing-reviewing and editing, project administration, funding acquisition. B. G. investigation, methodology. K. R. investigation. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rozmanowski, T., Krawczyk, P., Gurzęda, B. et al. New composite materials based on reduced graphite oxide for electrochemical sorption of hydrogen and methanol electrooxidation. Ionics 30, 1531–1542 (2024). https://doi.org/10.1007/s11581-023-05354-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-023-05354-8