
Abstract
The work is devoted to a computational study of three types of cationic polymeric membranes in Li+-ionic form, in water and methanol environments, at various solvation levels. The studied membranes Nafion, IonClad, and M3 possess the perfluorinated backbone; however, various side chains were terminated with the functional groups of distinctly different ionic strength. The paper discusses the structural features of the membrane-solvent clusters as well as an influence of the side chain nature on the dissociation of the functional groups and the binding energy of the solvent molecules. Additionally, the paper compares the obtained results for Li+-Nafion membrane in water with the results published earlier for H+ and Na+ forms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ion-exchange membranes are widely used in fuel cell technology, electrodialysis, medical devices, and other various membrane separation processes [1–4]. Numerous papers presented also experimental investigations of ion-exchange membranes applied in the separation of liquid mixtures by pervaporation [5–9]. Cabasso et al. [5, 6] and Godino et al. [7] showed that separation of alcohol/water mixtures using Nafion membrane depends on the type of the counter-ions. The experiments indicated that water is preferentially transported across the Nafion membrane than alcohol. The water flux decreases in the following sequence: H+ > Li+ > Na+ > K+ > Cs+. Zhao et al. [8] studied an influence of water, methanol, and ethanol on the sorption and transport properties of Nafion membrane in different ionic forms. It was pointed out that the counter-ions affect the amount of the solvents absorbed by the membrane.
Kujawski et al. [9, 10] investigated the influence of the character of the sulfonic group and size of the counter-ion on aliphatic alcohols transport across the membranes Nafion, IonClad, and PESS (based on polyethylene/poly(styrene-co-divinylbenzene)). Both, the stronger acidic character of the sulfonic group and the smaller radius of the counter-ion, provide an easier dissociation of the ion-pairs and enhance the alcohol transport. The dissociation of the ion-pair is easier in the perfluorinated membrane Nafion than in aromatic membranes IonClad and PESS. Also, the diffusion coefficients of aliphatic alcohol are higher in perfluorinated membranes than in aromatic ones [9, 10].
Hamann et al. [11], Koter [12], and Villaluenga et al. [13] investigated experimentally water and methanol uptake by sulfonated cation-exchange membranes. It was reported that Nafion 117 membrane in sodium form can uptake up to 15 water and 21 methanol molecules per sulfonic group. Nandan et al. [14] indicated that Nafion shows unusually large sorption of methanol for the membrane in H+, Li+, and Na+ ionic forms. The solvent uptake decreases with increasing size of the counter-ion. Moreover, studies carried out in 50:50 mol% methanol-water mixture confirmed the selective enrichment of water in the membrane phase. Struis et al. [15] found out that permselectivities for methanol and water depend on the counter-ion in the polymer. It was shown that lithium is the best suited as a counter-ion with respect to long-term stability and membrane performance.
Numerous theoretical investigations of perfluorinated sulfonic cationic membranes in the water environment have been performed using quantum chemical and molecular dynamics (MD) methods [16–27].
The aggregation of the sulfonic acid groups of Nafion membrane at different hydration levels was studied by other authors using computer simulations [16–21]. Elliott et al. [16] and Khalatur [17] performed the NVT molecular dynamics simulations and the semi-empirical molecular orbital SCF calculations, respectively. Yamamoto et al. [18] used dissipative particle dynamics (DPD) simulations, whereas Urata et al. [19] and Devanahan et al. [20, 21] carried out the investigation of the hydrated Nafion membrane model using the molecular dynamic (MD) simulations. The hydrophilic sulfonic domains of the membrane bind the water, thus increasing aggregation size, whereas the hydrophobic pendant side chain does not bind the water even at the high hydration level. It was established that the diffusion coefficient of water and counter-ions increases with growing hydration level. These results are found in a good agreement with experiments [19–24]. Khalatur et al. [17] showed that polar sulfonic acid (–SO3H) groups and water molecules build three-layer structure aggregates. The aggregates stability rises with increased hydration. The results of the spectroscopic studies and time-correlated single-photon counting experiments of Moilanen et al. [22], as well as the dissipative particle dynamics (DPD) simulations of Wu et al. [23] and Dorenbos et al. [24], indicate that hydrophilic domains of Nafion membrane grow and the dispersion of water regions around sulfonic acid groups increase with increased hydration level.
Nechaeva et al. [25] performed a computational study based on molecular dynamics (MD) simulations with IR spectral analysis of the dissociation and vibrational frequencies of carboxyl and sulfonic acid groups in the aromatic membranes. The analysis of the obtained results demonstrated that ion transport takes place by breaking the bonds between the acid group and the counter-ion.
The papers [28–33] presented the computational studies using the molecular dynamics simulations (MD) of an interaction of methanol and water with perfluorosulfonated membranes. Vishnyakov et al. [28] and Abroshan et al. [29] demonstrated that both water and methanol formed stable hydrogen bonds with the oxygen atoms of the hydrophilic sulfonic group (–SO3 −). It was noticed that solvent molecules bonded to the sulfonic group form the first solvation shell. The other part of the side chain, including ester oxygen atoms, has hydrophobic character. Urata el al. [30, 31] and Abroshan et al. [29] pointed out, despite the fact that both water and methanol molecules have the similar attractive energy to the sulfonic groups, that more methanol molecules are located in the vicinity of the hydrophobic polymer matrix than water molecules. Water molecules are preferentially situated closer to the sulfonic group. The papers [32, 33] presented experimental study on the transport properties of hydrocarboned CM2 membrane and perfluorinated sulfonic Nafion membrane in water-methanol mixtures. It was shown that, at methanol mass fraction 30–40 % in the external solution, the water content in perfluorinated membrane Nafion 117 increases. The authors assumed that it happens due to methanol presence, which in opposite to water can interact with the hydrophobic parts of side chains thus expanding the space between them. It leads to an increase in membrane volume and in this way promotes water absorption.
The simulations presented in [34–36] were focused on solvation of the poly(ether ether ketone) (SPEEK) membranes. The simulations indicated that water and methanol clusters can be formed with increasing solvation level. However, in the SPEEK membrane, the solvent clusters are smaller and more isolated than in the perfluorinated the Nafion membrane [35]. In a proton ionic form of the SPEEK membrane, the hydrogen ion is located pronouncedly closer to the sulfonic group than in the Nafion one, what suggests weaker acidic character of sulfonic group in the SPEEK membrane.
In our previous work [26], quantum-mechanical calculations were carried out with the analysis of conductivity for the hydrated membranes: aromatic MK-40 and perfluorinated MF-4SK (analogue of Nafion). The transport properties of the membranes were investigated for Na+ and H+ ionic forms. It was found that the dissociation of the ion-pairs and strong water network around the functional group occur in MF-4SK membrane. This finding is consistent with high diffusion coefficient and molar conductivity of counter-ions in MF-4SK membrane compared to the MK-40 aromatic membrane.
The focus of the present paper is to discuss the differences between the interactions of water and methanol molecules with the ion-exchange membranes by quantum chemical calculations. Moreover, taking into account the influence of the nature of the functional groups and the polymeric matrix, we focused on understanding the solvation and dissociation phenomena of the ion-pairs in the equilibrium clusters of ion-exchange membrane models.
The numerical calculations were carried out as a function of the number of solvent (water or methanol) molecules (X, with X = 0–10), allowing to get information regarding (i) structural properties of the Nafion, IonClad, and M3 ion-exchange membranes and (ii) binding energy of the solvent molecules (water or methanol) to the functional groups in the investigated ion-exchange membranes. The calculations were performed for the lithium ionic form of the membranes. Moreover, in order to investigate the counter-ion effect, the simulation results obtained for the Nafion-Li+ membrane in water environment are compared with the earlier reported results for H+ and Na+ ionic forms at various hydration level [26].
Membranes studied
In the study, three cation-exchange membranes were investigated: Nafion, IonClad, and M3. The chemical structures of the membranes are shown in Fig. 1. These membranes possess identical perfluorinated backbone but different side chains and the functional groups with varying ionic strength [9, 10]. Nafion membrane contains ether-linked side chains terminated with sulfonic groups. The backbone chains of IonClad membrane is grafted with poly(styrene sulfonic acid) side chains also terminated with sulfonic group [37]. Membrane M3 is a virtual ionomer, modeled with the same polymer matrix as IonClad, but possessing the carboxyl functional groups. The lithium ionic form of all these membranes was chosen for investigations.

Chemical structure of the membranes: a Nafion, b IonClad, c M3. The membrane models used in the quantum calculations are marked by the squares
Methods
The theoretical investigation of the membranes was carried out on the reduced models including the lithium counter-ion. For the Nafion membrane, a structural model consists of the reduced backbone and the side chain [(CF3)2–CF–OF2–CF–(CF3)–O–CF2–CF2–SO3–Li+]. The model of IonClad membrane is represented by the reduced backbone and another reduced side chain [(CF3)2–CF–CH(CH3)–C6H5–SO3–Li+]. The M3 membrane is represented by the following model [(CF3)2–CF–CH(CH3)–C6H5–COO–Li+]. The membrane models used for the numerical calculations are indicated by the rectangular boxes in Fig. 1.
The theoretical simulations were performed for the membrane models with lithium counter-ion in the dry state and at various solvation levels with the solvents water or methanol. The number of solvent molecules (water or methanol) per sulfonic group has been varied from 1 to 10 for all membranes.
The geometries were optimized using the Restricted Hartree Fock (RHF) method with the 6–31++G(d,p) basis set [38]. The calculations of the binding energy of the membrane-solvent complexes were carried out using the local pair natural orbital—coupled-electron pair approximation (LPNO-CEPA/1) with the def2-TZVP basis set [39]. The LPNO-CEPA/1 method was referenced in order to accurately describe weak interactions [40]. The computational study was carried out using the program ORCA [41].
The binding energy E b was computed from the electronic energy values (E) of the dry and solvated membranes by the following expression:
where X is the number of water molecules in the solvated membrane; E cluster, E dry, E solvent are the electronic energies of the solvated membrane with X solvent molecules, of the dry membrane and of one solvent molecule, respectively. In order to compare the different clusters, the binding energy per solvent molecule E b /X will be also discussed.
Results and discussion
Nafion, IonClad, and M3 membranes in Li+ form in water and methanol
Using the quantum chemical calculations, the following data were analyzed in detail: the geometries of the membrane models and the binding energy of the solvent per solvent molecule as a function of the solvation level (water/or methanol). The distinct attention was focused on the state of the functional groups (also called ion-pairs) consisting of the fixed groups and counter-ions and the water molecules interactions with the functional groups.
Figure 2 displays the optimized model structures of investigated membranes in the dry state, with three water molecules, and with three methanol molecules. The geometry parameters of these membrane systems are gathered in Table 1. The comparison of the geometries of the dry sulfonic cationic membranes shows that, on the average, distances S…O in the –SO3 − groups are slightly larger in the aromatic membrane IonClad (1.46 Å) than in the perfluorinated Nafion membrane (1.45 Å). In the membrane M3, the average value of the bonds C…O in the carboxyl group was equal to 1.25 Å.

Calculated geometries of Li+ form membranes: a dry state, b with three water molecules, c and three methanol molecules. Nafion—on the left, IonClad—in the middle, M3 —on the right
Figure 3 presents the distances between the nearest oxygen atom of the functional group and Li+ ion as a function of the number of the absorbed water molecules (on the left) and methanol molecules (on the right). During advancing solvation by water, it was found that uniquely in the perfluorinated Nafion membrane, the lithium ion shifts away from the sulfonic group. The distance –SO3 −…Li+ dramatically increases from 1.90 to 3.70 Å after the membrane absorbed seventh water molecule (Fig. 3). This observed movement of the lithium ion from the sulfonic group in the hydrated Nafion membrane proves the dissociation of the functional group [9]. Further buildup of the water molecules in the Nafion membrane leads to additional slight increase of the–SO3 −…Li+ distance. In aromatic membranes IonClad and M3, the shortest distances between the lithium and the fixed groups practically do not change (1.85–2.06 Å) over the whole investigated range of the hydration level (Fig. 3). It was also found that in course of solvation by methanol, for all three membranes, the distance between the lithium ion and fixed groups was practically invariant over the whole investigated range of the solvation level.

Minimal distance between sulfonyl group and Li+ counter-ion in Nafion, IonClad, and M3 membranes vs. number of absorbed water (left) and methanol (right) molecules
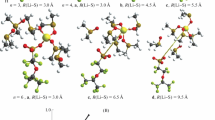
Figure 4 illustrates the calculated geometries of all the three membranes solvated by 10 molecules of water (Fig. 4a) or methanol (Fig. 4b). In the hydrated Nafion membrane, the ion-pair rests in the dissociated state, and some water molecules separate the sulfonic group and lithium counter-ion. In the hydrated aromatic membranes IonClad and M3, the lithium ion remains close to the fixed group in the whole investigated range of the solvation level. The same situation is observed in the case of all three membranes solvated by methanol: the ion-pairs do not dissociate over the whole studied solvation range.

Calculated geometries of solvated membranes in Li+ form: a with ten water molecules, b with ten methanol molecules. Nafion—on the left, IonClad—in the middle, M3—on the right
Figure 5 schematically displays a difference in the formation of the solvation shells around the dissociated and non-dissociated ion-pairs [42]. When the ion-pair dissociates, the solvent molecules are located between the fixed group and the counter-ion separating them. When the ion-pair is non-dissociated, the solvent molecules surround the non-separated ion-pair forming the outer solvation shell [9, 42]. From the calculated geometries of the membranes with ten solvent molecules presented in Fig. 4, the distances between the ion-pairs and the solvent molecules of the first solvation shells were measured. The obtained values are gathered in Table 2.

Scheme of the solvation for dissociated (left) and non-dissociated (right) ion-pairs in the ion-exchange membranes
The average distances between the ion-pairs and water molecules in the hydrated membranes increase in the following order Nafion < M3 < IonClad, while at the solvation by methanol molecules, these distances follow the sequence Nafion < IonClad < M3. It should be mentioned that in the case of the hydrated Nafion membrane, the first hydration shell of the sulfonic group consists of five water molecules, while the first hydration shell of the counter-ion Li+ is built by four molecules, revealing a tetrahedral coordination. A similar observation is reported in the papers [43, 44] devoted to the computational study of Li+ in aqueous solution, as well as in a study [45] which investigated the water sorption in Nafion.
The occurrence of the ion-pair dissociation in the hydrated Nafion membrane in comparison with aromatic membranes has already been discussed in earlier works [9, 35]. The sulfonic groups in Nafion membrane are bound to the fluorocarbon vinyl ether side chains. The presence of electron-withdrawing –CF2- groups in the side chains of Nafion membrane generates stronger acidic and polarizable character of the functional group facilitating its dissociation. Mahajan et al. [35] indicated that polymer matrix in SPEEK and Nafion membranes has some influence on the separation of the functional groups from another one. It was shown that the separation between the sulfonic groups in aromatic membrane induced by its solvation was hindered by low hydrophobicity of aromatic backbones and low hydrophilic character of the sulfonic groups in SPEEK. Using DFT methods, Sakai et al. [46] and Doan et al. [47] studied the protonated and deprotonated forms of perfluorosulfonic molecules modeling Nafion membrane. It was demonstrated that the deprotonated sulfonic group is more stable than the protonated state by 3.72 kcal/mol (16 kJ/mol). Using LPNO-CEPA/1 method, we found a similar value of 4.17 kcal/mol (17.44 kJ/mol) for Nafion with three water molecules. Sakai et al. [48] analyzed the energetic stability of membrane models of Nafion and SPEEK at low hydration levels (1–3 molecules of water) in terms of the activation energy (ΔEa) and the enthalpy difference (ΔH) before and after deprotonation of the sulfonic group. It was pointed out that the activation energy of sulfonic groups in the perfluorosulfonic membrane is very low, while ΔEa and ΔH for the aromatic SPEEK membrane are higher than for the perfluorinated Nafion one. Therefore, the deprotonation step was indicated as a non-favorable in the case of SPEEK. Consequently, it can be calculated that the presence of the benzene ring in the side chains of the aromatic membranes decreases the acidity of their functional groups [9]. However, the dissociation of the functional group does not depend solely on the side chain structure but also on the polarity of the absorbed solvent. This fact results from the observation that the ion-pair of Nafion membrane dissociates, when the membrane is solvated in water, while it does not dissociate in methanol.
Figure 6 depicts the binding energy per one solvent molecule of water and methanol as a function of the solvation level. In Nafion, the binding energy of both water and methanol is slightly higher than for the aromatic membranes IonClad and M3. These computations help also to explain the experimental results, indicating that the Nafion membrane absorbs more methanol and water than the aromatic membrane IonClad. As it can be seen in Fig. 6, for the Nafion and IonClad membranes, the binding energy of water (E b /X) decreases with increasing hydration level. In the case of membrane M3, where the side chains are terminated with the carboxyl groups, E b /X rises at low hydration level X = 1–3, but the further increase of the hydration (X = 4–10) leads to a decrease in the interactions of water with the carboxyl groups. For all three studied membranes, the binding energy of methanol continuously goes down with increasing solvation.

Binding energy per solvent molecule vs. number of absorbed solvent molecules in Nafion, IonClad, and M3 membranes in Li+ form
A comparison of the interaction energy of water and methanol with the investigated membranes shows that at solvation level X = 1–8, in the perfluorinated membrane Nafion, water molecules are more weakly bound to the functional groups than methanol molecules (Fig. 6). At higher solvation X = 9–10, the binding energies of water and methanol are practically the same. In the aromatic membrane IonClad, the first methanol molecule is more strongly bound to the functional group than the first water molecule. Subsequently, at X = 2–6, the binding energies of water and methanol have very similar values, whereas, at higher solvation level X = 7–10, water is more tightly linked to IonClad than methanol. Very interesting results are observed for the virtual membrane M3. In this case, the carboxyl group interacts significantly more strongly with the first absorbed methanol molecule than with the first water molecule (Fig. 6). At X = 2–7, the binding energies of methanol and water possess very comparable values; however at higher solvation level X = 8–10, water is more strongly tied to the membrane than methanol.
The experimental results reported by Chaabane et al. [32, 33] showed that membrane CM2, having hydrocarbon side chains, contains less water molecules per sulfonic group (X = 6.03 mol H2O/mol SO3 −) than membrane Nafion 117 (X = 8.80 mol H2O/mol SO3 −) with the perfluorinated side chains. Furthermore, Nafion membrane absorbs much more methanol than CM2 membrane. These results correlate well with the binding energies calculated within this work: water and methanol interactions with the hydrocarbon side chain of the IonClad membrane are weaker than with perfluorinated side chain of the Nafion membrane.
Nafion in Li+, H+, and Na+ forms in water
This part of the paper presents a comparison of the structural features of the Nafion membrane in three ionic forms H+ [26], Na+ [26], and Li+ investigated at various hydration level (X = 0–10).
Table 3 presents the atom and ionic radii of H/H3O+, Li/Li+, and Na/Na+ as well as their hydration numbers in aqueous solutions [49–57] and in Nafion membrane determined in the present study. The computed geometries of the membranes hydrated with ten water molecules are demonstrated in Fig. 7. The functional groups in all investigated ionic forms are found in the dissociated state, but the spatial arrangement of the water molecules in the membrane significantly differs and depends on the counter-ion nature (Fig. 7). We found that amount of the water molecules in the first hydration shell of the counter-ions increases with rising atomic radius of the counter-ion in accordance with the sequence H+ < Li+ < Na+ (Table 3). The hydration numbers of the counter-ions obtained in the simulation for Nafion membranes are consistent with the values measured for the ions in aqueous solutions. Table 4 demonstrates the distances to the closest water molecules from the counter-ions and the sulfonic groups in Nafion membrane hydrated by ten water molecules. The water molecules are located significantly nearer to the proton than to the alkali ions. But in proton form of Nafion, the distance between sulfonic group and the water molecules is slightly higher than in Li+ and Na+ forms. As seen from the Table 4, in all studied ionic forms of the Nafion membrane, the distances between the water molecules and the counter-ions are shorter than the distances determined for the counter-ions in aqueous solutions. In H+ form of Nafion, the water molecules are located around 1.57 Å from the proton, while in the H+(H2O)9 cluster the distance is 2.54 Å, as computed by Bankura et al. [52].

The geometry of different ionic forms of membrane Nafion-H, Nafion-Li, and Nafion-Na hydrated with ten water molecules. In Nafion-H, the proton is marked by the dashed circle
Figure 8 depicts the closest distances between the fixed group and counter-ions (–SO3 −…Cat+) as a function of the hydration level. In the dry Nafion, the distance –SO3 −…Cat+ increases with the atomic radius of the counter-ion in the following sequence H+ < Li+ < Na+. In the proton form, the distance –SO3 −…H+ extremely increases, when the membrane absorbs three and five water molecules. At X = 3, the proton form of the functional group dissociates. At X = 5, the proton jumps to the second layer of the water molecule surrounding the sulfonic group. At hydration level X > 5, the proton marginally continues to move away from the sulfonic group with increasing hydration level. In Li+ and Na+ forms of the Nafion membrane, the distances –SO3 −…Cat+ practically do not change at 0 ≤ X ≤ 6. In these ionic forms, the dissociation of the functional groups occurs, when the functional group is surrounded by seven water molecules. The distances –SO3 −…Li+ and –SO3 −…Na+ reach comparable values, 3.70 and 3.77 Å, respectively. At higher hydration level X > 7, Li+ and Na+ ions are located further away from the sulfonic groups than the protons are. The mean distance –SO3 −…H+ equals 3.46 Å at this hydration range (X > 7). It should be mentioned that prior to the dissociation, the hydrogen is connected to the sulfonic group by a covalent bond, while the alkali metal ions Li+ and Na+ create ionic bonds. After the dissociation, the proton builds the hydronium ion, which has a significantly larger radius than the proton and also than lithium and sodium cations (Table 3). Since the hydronium ion has larger size and lower surface charge density than the studied alkali cations, the hydronium ion is less distanced from the sulfonic group at the higher hydration level (X ≥ 7).

Minimal distance between sulfonic group and counter-ions in Nafion membrane of different ionic forms: H+, Li+, and Na+ vs. number of absorbed water molecules
The water binding energy in Nafion membrane is presented in Fig. 9. The diagram demonstrates the water binding energy per water molecule in Nafion membrane of the three ionic forms investigated, as a function of the hydration level. In general, the binding energy continuously decreases with increasing hydration level (Fig. 9). A pronounced difference in the binding energy is observed at low hydration level 1 ≤ X ≤ 4. At this region of hydration, the water interactions with the membrane are the strongest for the Li+ form and the weakest for the H+ form of the membrane. In the whole investigated range of the hydration level (0 ≤ X ≤ 10), the binding energy per water molecules is stronger for Nafion containing the alkali ions as the counter-ions, because the alkali ions possess quite large surface charge density, which strengthens the electrostatic attraction of the water molecules. The static ab initio calculations are consistent with experimental observations demonstrating that the water diffusion coefficient is higher in H+ form Nafion than in Na+ or Li+ forms [58–60].

Binding energy per water molecule in Nafion membrane in H+, Li+, and Na+ form as a function of the hydration level
Conclusions
The work discusses the interactions of water and methanol molecules with different cationic polymeric membranes as well as the role of the nature of the functional groups and membrane polymeric matrices on these interactions. Using ab initio methods, three cation-exchange membrane models of Nafion, IonClad, and M3 were investigated in Li+ ionic form, in contact with water and methanol molecules, at various solvation level (X = 0–10). The studied membranes possess the perfluorinated backbone, but different side chains terminated with the various functional groups of distinct ionic strength. The simulation of the membranes with lithium as counter-ion showed that in the Nafion membrane, the dissociation of the ion-pairs occurs at the hydration level of seven water molecules. In the aromatic membranes IonClad and M3, no dissociation of the functional groups is observed for the whole investigated range of the hydration level. The presence of the benzene ring in the side chains of the aromatic membranes decreases the acidity of their functional groups. The solvation of all three membranes by methanol does not lead to any dissociation of the functional groups. It indicates the strong role of the polarity of the absorbed solvent on the dissociation phenomenon.
It can be assumed that the ability of the functional group to dissociate becomes more pronounced with the following:
-
1)
growing electron density of the polymer side chains, especially when the electronegative group is directly connected to the functional group
-
2)
increasing polarity of the absorbed solvent
-
3)
increasing acidity of the fixed group
The binding energy of the solvent molecules with the membrane generally decreases with increasing solvation level. In the Nafion membrane, the binding energy of water and methanol is slightly higher than in the aromatic membranes IonClad and M3.
The obtained results for Li+-Nafion in water are compared with earlier published simulation for H+ and Na+ forms. In the dry state of Nafion, the distance between the counter-ion and fixed group increases with growing atom radius of the counter-ion as H+ < Li+ < Na+ and then increases with rising hydration level. For high hydration levels X ≥ 7, this distance practically does not depend on the counter-ion nature and is equal to around 3.67 Å. In the Nafion-H+ membrane, the functional groups dissociate at hydration level of X = 3, while in the Na+ and Li+ forms, the dissociation proceeds at X = 7. In the perfluorinated sulfonate membrane, the binding energy per water molecule increases with rising surface charge density of the counter-ion. Moreover, the water molecules are located closer to the counter-ions in the Nafion membrane than in aqueous solutions.
References
Strathmann H (2004) Ion-exchange membrane separation processes, vol 9. Membrane Science and Technology Elsevier Ltd doi:10.1016/S0927-5193(04)80031-7
Zaidi J, Matsuura T (2009) Polymer membranes for fuel cells. Springer US. doi:10.1007/978-0-387-73532-0
Peighambardoust SJ, Rowshanzamir S, Amjadi M (2010) Review of the proton exchange membranes for fuel cell applications. Int J Hydrogen Energ 35(17):9349–9384. doi:10.1016/j.ijhydene.2010.05.017
Pellegrin M-L, Greiner AD, Aguinaldo J, Diamond J, Gluck S, Burbano MS, Arabi S, Wert J, McCandless R, Padhye LP (2012) Membrane processes. Water Environ Res 84(10):1114–1216
Cabasso I, Liu Z-Z (1985) The permselectivity of ion-exchange membranes for non-electrolyte liquid mixtures. J Membrane Sci 24(1):101–119. doi:10.1016/S0376-7388(00)83212-3
Cabasso I, Liu Z-Z, Makenzie T (1986) The permselectivity of ion-exchange membranes for non-electrolyte liquid mixtures. II. The effect of counterions (separation of alcohol/water mixtures with Nafion membranes). J Membrane Sci 28(2):109–122. doi:10.1016/S0376-7388(00)82204-8
Godino MP, Barragán VM, Villaluenga JPG, Ruiz-Bauzá C, Seoane B (2006) Water and methanol transport in Nafion membranes with different cationic forms: 1. Alkali monovalent cations. J Power Sources 160(1):181–186. doi:10.1016/j.jpowsour.2006.02.006
Zhao Q, Carro N, Ryu HY, Benziger J (2012) Sorption and transport of methanol and ethanol in H+-Nafion. Polymer 53(6):1267–1276. doi:10.1016/j.polymer.2012.01.050
Kujawski W, Staniszewski M, Nguyen TQ (2007) Transport parameters of alcohol vapors through ion-exchange membranes. Separ Sci Technol 57(3):476–482. doi:10.1016/j.seppur.2006.08.027
Kujawski W, Nguyen QT, Neel J (1992) Infrared investigations of sulfonated ionomer membranes. I. Water–alcohol compositions and counterions effects. J Appl Polym Sci 44(6):951–958. doi:10.1002/app.1992.070440603
Hamann CH, Theile V, Koter S (1993) Transport properties of cation-exchange membranes in aqueous and methanolic solutions. Diffusion and osmosis. J Membrane Sci 78(1–2):147–153. doi:10.1016/0376-7388(93)85256-V
Koter S (1994) Transport properties of perfluorinated cation-exchanged membranes in aqueous and methanol solutions of NaCl. Polish J Chem 68:2019–2029
Villaluenga JPG, Barragán VM, Izquierdo-Gil MA, Godino MP, Seoane B, Ruiz-Bauzá C (2008) Comparative study of liquid uptake and permeation characteristics of sulfonated cation-exchange membranes in water and methanol. J Membrane Sci 323(2):421–427. doi:10.1016/j.memsci.2008.06.049
Nandan D, Mohan H, Iyer RM (1992) Methanol and water uptake, densities, equivalental volumes and thicknesses of several uni- and divalent ionic perfluorosulphonate exchange membranes (Nafion-117) and their methanol-water fractionation behaviour at 298 K. J Membrane Sci 71(1–2):69–80. doi:10.1016/0376-7388(92)85007-6
Struis RPWJ, Stucki S, Wiedorn M (1996) A membrane reactor for methanol synthesis. J Membrane Sci 113(1):93–100. doi:10.1016/0376-7388(95)00222-7
A. Elliiott. J, Hanna S, M. S. Elliott A, Cooley GE (1999) Atomistic simulation and molecular dynamics of model systems for perfluorinated ionomer membranes. Phys Chem Chem Phys 1(20):4855–4863. doi:10.1039/A905267D
Khalatur PG, Talitskikh SK, Khokhlov AR (2002) Structural organization of water-containing Nafion: the integral equation theory. Macromol Theor Sim 11(5):566–586. doi:10.1002/1521-3919(20020601)11:5<566::AID-MATS566>3.0.CO;2-0
Yamamoto S, S-a H (2003) A computer simulation study of the mesoscopic structure of the polyelectrolyte membrane Nafion. Polym J 35(6):519–527. doi:10.1295/polymj.35.519
Urata S, Irisawa J, Takada A, Shinoda W, Tsuzuki S, Mikami M (2005) Molecular dynamics simulation of swollen membrane of perfluorinated ionomer. J Phys Chem B 109(9):4269–4278. doi:10.1021/jp046434o
Devanathan R, Venkatnathan A, Dupuis M (2007) Atomistic simulation of Nafion membrane: I. Effect of hydration on membrane nanostructure. J Phys Chem B 111(28):8069–8079. doi:10.1021/jp0726992
Devanathan R, Venkatnathan A, Dupuis M (2007) Atomistic simulation of Nafion membrane. 2. Dynamics of water molecules and hydronium ions. J Phys Chem B 111(45):13006–13013. doi:10.1021/jp0761057
Moilanen DE, Spry DB, Fayer MD (2008) Water dynamics and proton transfer in Nafion fuel cell membranes. Langmuir 24(8):3690–3698. doi:10.1021/la703358a
Wu D, Paddison SJ, Elliott JA (2008) A comparative study of the hydrated morphologies of perfluorosulfonic acid fuel cell membranes with mesoscopic simulations. Energ Environ Sci 1(2):284–293. doi:10.1039/B809600G
Dorenbos G, Suga Y (2009) Simulation of equivalent weight dependence of Nafion morphologies and predicted trends regarding water diffusion. J Membrane Sci 330(1–2):5–20. doi:10.1016/j.memsci.2008.11.056
Nechaeva LS, Butyrskaya EK, Shaposhnik VA (2012) The main role of the hydrogen bond in the ion transport processes of cation exchangers. International Ion Exchange Conference Cambridge, UK, pp. pp 1–pp13
Karpenko-Jereb LV, Kelterer A-M, Berezina NP, Pimenov AV (2013) Conductometric and computational study of cationic polymer membranes in H+ and Na+-forms at various hydration levels. J Membrane Sci 444:127–138. doi:10.1016/j.memsci.2013.05.012
Pluhařová E, Marsalek O, Schmidt B, Jungwirth P (2013) Ab initio molecular dynamics approach to a quantitative description of ion pairing in water. J Phys Chem Lett 4(23):4177–4181. doi:10.1021/jz402177q
Vishnyakov A, Neimark AV (2000) Molecular simulation study of Nafion membrane solvation in water and methanol. J Phys Chem B 104(18):4471–4478. doi:10.1021/jp993625w
Abroshan H, Akbarzadeh H, Taherkhani F, Parsafar G (2011) Effect of water–methanol content on the structure of Nafion in the sandwich model and solvent dynamics in nano-channels; a molecular dynamics study. Mol Phys 109(5):709–724. doi:10.1080/00268976.2010.549846
Urata S, Irisawa J, Takada A, Tsuzuki S, Shinoda W, Mikami M (2005) Intermolecular interaction between the pendant chain of perfluorinated ionomer and methanol. J Fluor Chem 126(9–10):1312–1320. doi:10.1016/j.jfluchem.2005.07.004
Urata S, Irisawa J, Takada A, Shinoda W, Tsuzuki S, Mikami M (2005) Molecular dynamics study of the methanol effect on the membrane morphology of perfluorosulfonic ionomers. J Phys Chem B 109(36):17274–17280. doi:10.1021/jp052647h
Chaabane L, Bulvestre G, Larchet C, Nikonenko V, Deslouis C, Takenouti H (2008) The influence of absorbed methanol on the swelling and conductivity properties of cation-exchange membranes: evaluation of nanostructure parameters. J Membrane Sci 323(1):167–175. doi:10.1016/j.memsci.2008.06.044
Chaabane L, Dammak L, Grande D, Larchet C, Huguet P, Nikonenko SV, Nikonenko VV (2011) Swelling and permeability of Nafion®117 in water–methanol solutions: an experimental and modeling investigation. J Membrane Sci 377(1–2):54–64. doi:10.1016/j.memsci.2011.03.037
Komarov PV, Veselov IN, Chu PP, Khalatur PG (2010) Mesoscale simulation of polymer electrolyte membranes based on sulfonated poly(ether ether ketone) and Nafion. Soft Matter 6(16):3939–3956. doi:10.1039/B921369D
Mahajan CV, Ganesan V (2010) Atomistic simulations of structure of solvated sulfonated poly(ether ether ketone) membranes and their comparisons to Nafion: I. Nanophase segregation and hydrophilic domains. J Phys Chem B 114(25):8357–8366. doi:10.1021/jp104078h
Mahajan CV, Ganesan V (2010) Atomistic simulations of structure of solvated sulfonated poly(ether ether ketone) membranes and their comparisons to Nafion: II. Structure and transport properties of water, hydronium ions, and methanol. J Phys Chem B 114(25):8367–8373. doi:10.1021/jp1040794
Tricoli V, Carretta N, Bartolozzi M (2000) A comparative investigation of proton and methanol transport in fluorinated ionomeric membranes. J Electrochem Soc 147(4):1286–1290. doi:10.1149/1.1393351
Computational Chemistry Comparison and Benchmark DataBase. National Institute of Standards and Technology. http://cccbdb.nist.gov/vibscalejust.asp.
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys 7(18):3297–3305. doi:10.1039/B508541A
Neese F, Wennmohs F, Hansen A (2009) Efficient and accurate local approximations to coupled-electron pair approaches: an attempt to revive the pair natural orbital method. J Chem Phys 130(11):114108. doi:10.1063/1.3086717
Neese F (2010) ORCA Version 28, An ab initio, density functional and semiempirical program package. University Bonn
Eigen M, Tamm K (1962) Schallabsorption in elektrolytlösungen als folge chemischer relaxation I. Relaxationstheorie der mehrstufigen dissoziation. Zeitschrift für Elektrochemie, Berichte der Bunsengesellschaft für Physikalische Chemie 66(2):93–107. doi:10.1002/bbpc.19620660205
Pratihar S, Chandra A (2011) A first principles molecular dynamics study of lithium atom solvation in binary liquid mixture of water and ammonia: structural, electronic, and dynamical properties. J Chem Phys 134(2):024519. doi:10.1063/1.3511702
Kabbalee P, Tongraar A, Kerdcharoen T (2015) Preferential solvation and dynamics of Li+ in aqueous ammonia solution: an ONIOM-XS MD simulation study. Chem Phys 446:70–75. doi:10.1016/j.chemphys.2014.11.012
Wu X, Wang X, He G, Benziger J (2011) Differences in water sorption and proton conductivity between Nafion and SPEEK. J Polym Sci B: Polymer Physics 49(20):1437–1445. doi:10.1002/polb.22326
Sakai H, Tokumasu T (2015) Quantum chemical analysis of the deprotonation of sulfonic acid in a hydrocarbon membrane model at low hydration levels. Solid State Ionics 274:94–99. doi:10.1016/j.ssi.2015.03.005
Doan J, Navarro NE, Kumari D, Anderson K, Kingston E, Johnson C, Vong A, Dimakis N, Smotkin ES (2015) Symmetry-based IR group modes as dynamic probes of Nafion ion exchange site structure. Polymer 73:34–41. doi:10.1016/j.polymer.2015.07.017
Sakai H, Tokumasu T (2014) Reaction analysis for deprotonation of the sulfonic group of perfluorosulfonic acid molecules at low hydration levels. J Phys Chem A 118(1):275–282. doi:10.1021/jp409781s
Riedel E, Janiak C (2007) Anorganische chemie. 7. Auflage. de Gruyter, Berlin, ISBN 978–3–11–018903–2
Robinson RA, Stokes RH (1959) Electrolyte solutions. Academic Press, New York
Hellferich F (1962) Ion-exchange. McGraw-Hill Book Company, Inc., New York
Bankura A, Chandra A (2011) A first principles theoretical study of the hydration structure and dynamics of an excess proton in water clusters of varying size and temperature. Chem Phys 387(1–3):92–102. doi:10.1016/j.chemphys.2011.07.008
Azam SS, Hofer TS, Randolf BR, Rode BM (2009) Hydration of sodium(I) and potassium(I) revisited: a comparative QM/MM and QMCF MD simulation study of weakly hydrated ions. J Phys Chem A 113(9):1827–1834. doi:10.1021/jp8093462
Van Geet AL (1972) Hydration number of sodium ions determined by sodium magnetic resonance. J Am Chem Soc 94(16):5583–5587. doi:10.1021/ja00771a009
Mancinelli R, Botti A, Bruni F, Ricci MA, Soper AK (2007) Hydration of sodium, potassium, and chloride ions in solution and the concept of structure maker/breaker. J Phys Chem B 111(48):13570–13577. doi:10.1021/jp075913v
Yamaguchi T, Ohzono H, Yamagami M, Yamanaka K, Yoshida WH (2010) Ion hydration in aqueous solutions of lithium chloride, nickel chloride, and caesium chloride in ambient to supercritical water. J Mol Liq 153:2–8. doi:10.1016/j.molliq.2009.10.012
Mähler J, Persson I (2012) A study of the hydration of the alkali metal ions in aqueous solution. Inorg Chem 51(1):425–438. doi:10.1021/ic2018693
Legras M, Hirata Y, Nguyen QT, Langevin D, Métayer M (2002) Sorption and diffusion behaviors of water in Nation 117 membranes with different counter ions. Desalination 147(1–3):351–357. doi:10.1016/S0011-9164(02)00608-2
Volkov VI, Volkov EV, Timofeev SV, Sanginov EA, Pavlov AA, Safronova EY, Stenina IA, Yaroslavtsev AB (2010) Water self-diffusion and ionic conductivity in perfluorinated sulfocationic membranes MF-4SK. Russ J Inorg Chem 55(3):315–317. doi:10.1134/S0036023610030010
Volkov VI, Pavlov AA, Sanginov EA (2011) Ionic transport mechanism in cation-exchange membranes studied by NMR technique. Solid State Ionics 188(1):124–128. doi:10.1016/j.ssi.2010.09.046
Acknowledgments
This study was supported by ÖAD, WTZ project PL 08/2012 “Experimental and computational studies of transport mechanism of organic solvents through ion-exchange membranes,” and Lifelong Learning Program Erasmus.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Karpenko-Jereb, L., Rynkowska, E., Kujawski, W. et al. Ab initio study of cationic polymeric membranes in water and methanol. Ionics 22, 357–367 (2016). https://doi.org/10.1007/s11581-015-1551-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-015-1551-7