
Abstract
Phylogenetic analyses of sequences of the nuc rDNA small subunit (18S), internal transcribed spacer (ITS1-5.8S-ITS2 = ITS), and large subunit (28S) region (= 18S-ITS-28S), as well as sequences of this region concatenated with sequences of the largest subunit of RNA polymerase II (RPB1) gene, proved that the species originally described as Acaulospora polonica (phylum Glomeromycota) represents a new genus and a new family of the ancient order Archaeosporales, here introduced into the Glomeromycota under the names Polonospora and Polonosporaceae, respectively. The phylogenetic analyses and BLASTn queries also indicated that the Polonosporaceae with P. polonica comb. nov. still contains several morphologically undescribed taxa at the ranks of genus and species, which have a worldwide distribution.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Among 58 species so far described in the genus Acaulospora (see Online Resource 1 for species authors), family Acaulosporaceae, is A. polonica, accommodated in this genus because forming spores laterally directly on the neck of a sporiferous saccule (Gerdemann and Trappe 1974; Błaszkowski 1988; 2012), as similarly described for A. laevis, the type species of Acaulospora. However, several morphological characters of A. polonica spores did not fit those of most Acaulospora species. All typical Acaulospora species produce spores with a colored spore wall (spore wall 1), forming the spore surface, and two colorless inner walls (spore walls 2 and 3). Spore wall 1 consists of two to three layers, and its laminate layer in many species is ornamented. Spore wall 2 usually contains two tightly adherent thin (< 1 µm thick), smooth, flexible to semi-flexible layers, and occasionally is one-layered. Spore wall 3 consists of two layers, of which layer 1 is thin and ornamented with fine granules, rarely smooth. Layer 2 in most species is thin when mounted in water but becomes plastic, strongly thickening in lactic acid-based mountants, e.g., in polyvinyl alcohol/lactic acid/glycerol (PVLG, Omar et al. 1979), and stains dark in Melzer’s reagent. Only in few species, this layer remains thin in PVLG and stains faintly or does not stain in Melzer’s.
The subcellular spore structure of A. polonica also consists of three walls, but spore wall 1 remains hyaline to white through its entire life cycle and is smooth (Błaszkowski 1988, 2012). However, the fundamental differences between A. polonica and typical Acaulospora species reside in the composition, as well as in the phenotypic and histochemical properties of spore wall 3. In A. polonica, spore wall 3 is also two-layered, but the outer layer 1 is relatively thick, coriaceous sensu Walker (1986), and smooth. Spore wall 3 layer 2 is flexible to semi-flexible and does not thicken in PVLG. In addition, none of spore wall 3 layers 1 and 2 stains in Melzer’s reagent.
Of members of the Glomeromycota-producing spores laterally on the neck of a sporiferous saccule, named acaulosporoid spores (Sieverding and Oehl 2006; Oehl et al. 2011a), the features of spore wall 3 of A. polonica most resemble those of the innermost spore wall 2 of all species of the genera Ambispora and Archaeospora, for example, Am. leptoticha and Ar. trappei, originally described as A. gerdemannii and A. trappei, respectively (Nicolson and Schenck 1979; Morton and Redecker 2001; Sieverding and Oehl 2006; Bills and Morton 2015).
Thus, A. polonica spores have a spore wall 2 with phenotypic features similar to spore wall 2 of typical Acaulospora species and a spore wall 3, whose characters do not fit those of spore wall 3 of other Acaulospora species but share characters of spore wall 2 of acaulosporoid spores of Ambispora and Archaeospora species. Another feature linking A. polonica, typical Acaulospora species, and species of Archaeospora but separating these species from Ambispora species is the place where acaulosporoid spores are formed. Spores of Acaulospora species and Ar. trappei arise directly on the neck of a sporiferous saccule, whereas spores of Ambispora species develop at the tip of a short branch of the neck. Moreover, Ambispora species also produce glomoid spores at tips of cylindric or funnel-shaped sporogenous hyphae, as Glomus macrocarpum and Archaeospora species also form glomoid and entrophosporoid spores (Morton and Redecker 2001; Oehl et al. 2011a). The latter arise inside the neck of a sporiferous saccule, as in a species originally described as G. infrequens (Hall 1977) and later renamed Entrophospora infrequens (Ames and Schneider 1979), as well as in some species described in Entrophospora and then transferred to Acaulospora (Kaonongbua et al. 2010). All specimens of A. polonica found by us so far had acaulosporoid spores only (Błaszkowski 2012). Finally, species of Ambispora and Archaeospora are distinguished by their mycorrhizal structures, which stain faintly, much lighter than those of typical Acaulospora species, or do not stain at all in Trypan blue (Morton and Redecker 2001).
Currently, species of Ambispora and Archaeospora are classified in the families Ambisporaceae and Archaeosporaceae, respectively, both belonging to the order Archaeosporales, that represent ancient members of the Glomeromycota (Morton and Redecker 2001) and molecularly are strongly divergent from the generic Acaulospora clade (Oehl et al. 2011b; Online Resource 8).
Despite possessing the unique morphological characters discussed above, the true placement in the Glomeromycota of the species currently included into Ambispora and Archaeospora was possible only based on phylogenetic analyses of their molecular sequences (Morton and Redecker 2001; Oehl et al. 2011a). Also, the morphological divergences of A. polonica from typical Acaulospora species, as well as the morphological similarities and differences of A. polonica to species of Ambispora and Archaeospora, suggest that A. polonica does not belong to Acaulospora but should represent an undescribed clade located among basal clades of the Glomeromycota. Unfortunately, the determination of the placement of this clade based on morphology of A. polonica spores only has not been possible using current morphological approaches.
Acaulospora polonica has so far been found physically very rarely in the field and has not been grown in culture. Only recently, we found A. polonica abundantly sporulating in pot trap cultures. Therefore, the aims of the studies discussed here were (i) to grow A. polonica in single-species cultures, (ii) to check whether the morphological and histochemical properties of its spores were correctly characterized by Błaszkowski (1988, 2012), (iii) to characterize mycorrhizal structures of this species, and (iv) to determine whether this species forms spore morphotypes other than acaulosporoid, as well as (v) to perform relevant molecular phylogenetic analyses to confirm our hypothesis that this species belongs to an undescribed clade located among basal clades of the Glomeromycota and to determine its position within this phylum.
Materials and methods
Origin of study material
Spores of A. polonica were originally extracted from a trap pot culture inoculated with field-collected rhizosphere soil and root fragments of Rosa rugosa inhabiting maritime dunes of the Baltic Sea. The spores were used to establish single-species pot cultures. The plant used for the trap and single-species cultures was Plantago lanceolata. The field inoculum was collected near the village of Kuźnica (54° 44′ 11′′ N 18° 34′ 47′′ E) located on the Hel Peninsula in northern Poland by J. Błaszkowski 21 Aug 2017.
Establishment and growth of trap and single-species cultures
Methods used to establish trap and single-species cultures, growing conditions, and methods of spore extraction and staining of mycorrhizal structures were as those described previously in Błaszkowski et al. (2012). The only exception was the source of the supplemental lighting. We used LEDDY Retro Fit tube lamps located ca. 40 cm above the cultures. The photosynthetic photon flux density measured at the level of the upper surface of the growing substrate was ca. 40 µmol m2 s−1. The exposure time was 12 h a day. Ten to 20 spores of uniform morphology stored in water for ca. 2 weeks at 4 °C in a refrigerator were used to establish single-species cultures.
Microscopy and nomenclature
Morphological features of spores and the phenotypic and histochemical characters of spore wall layers of A. polonica were characterized based on at least 100 spores mounted in water, lactic acid, PVLG, and a mixture of PVLG and Melzer’s reagent (1:1, v/v). The preparation of spores for study and photography was as those described previously (Błaszkowski 2012; Błaszkowski et al. 2012). Types of spore wall layers are those defined by Błaszkowski (2012) and Walker (1983). Color names are from Kornerup and Wanscher (1983). Nomenclature of fungi and the authors of fungal names are from the Index Fungorum website http://www.indexfungorum.org/AuthorsOfFungalNames.htm. The term glomerospores was used for spores produced by AMF, as proposed by Goto and Maia (2006).
Voucher specimens of the proposed new taxon [spores permanently mounted in PVLG and a mixture of PVLG and Melzer’s reagent (1:1, v/v) on slides] are deposited at Z + ZT (ETH Zurich, Switzerland: ZT Myc 64,926 and 64,927) and in the Laboratory of Plant Protection, Department of Shaping of Environment (LPPDSE), West Pomeranian University of Technology in Szczecin, Poland (holotype, isotypes).
DNA extraction, PCR conditions, cloning, and DNA sequencing
Genomic DNA of A. polonica was extracted from eight samples, each consisting of 5‒10 spores crushed with the end of a preparation needle on sterile microscope slides observed under a dissecting microscope. Details of the treatment of the spores prior to polymerase chain reaction (PCR), the conditions, and primers used in the PCR to obtain 18S-ITS-28S sequences of the fungus were as those described in Błaszkowski et al. (2014, 2015b), Krüger et al. (2009), and Symanczik et al. (2014). RPB1 sequences of P. polonica were obtained by amplification with primer RPB1-4F1 in combination with RPB1-5R, as described in Błaszkowski et al. (2021).
RPB1 sequences of Ar. schenckii, Innospora majewskii, and Paraglomus laccatum were obtained by amplification with primers designed by Stockinger et al. (2014) following the recommended conditions. We used the same DNA, from which 18S-ITS-28S sequences had been obtained (Błaszkowski et al. 2017). The first PCR was performed with primers RPB1-Ac and RPB1-DR2160r, while the second PCR with RPB1-Ac and RPB1-DR1210r + RPB1-DR1210r_Aca_div.
Cloning and sequencing of the PCR products to obtain both types of sequences were as those described by Błaszkowski et al. (2015a). The sequences were deposited in GenBank with the accession numbers MZ359654‒MZ359660 and MZ362263‒MZ362271.
Sequence alignment and phylogenetic analyses
BLASTn’ searches, using 18S-ITS-28S sequences of A. polonica, showed that closest relatives of the species are uncultured fungi of the Archaeosporales. To determine the placement of A. polonica among morphologically and molecularly characterized members of the Archaeosporales, as well as among the uncultured fungi indicated by BLASTn, two alignments were assembled: 18S-ITS-28S and 18S-ITS-28S + RPB1. The 18S-ITS-28S alignment consisted of 60 sequences of the 18S-ITS-28S nuc rDNA region or 28S gene that characterized five species of Archaeospora and four species of Ambispora, A. polonica, and Geosiphon pyriformis (Geosiphonaceae), as well as 20 sequences of uncultured members of the Archaeosporales identified in eleven countries of the world. The outgroup was twelve sequences of members of the Paraglomerales: four species of the genus Paraglomus, Innospora majewskii (Paraglomeraceae), and Pervetustus simplex (Pervetustaceae). In the 18S-ITS-28S + RPB1 alignment, containing all sequences of the 18S-ITS-28S alignment, 15 RPB1 sequences were concatenated with 18S-ITS-28S sequences of eight species of the Archaeosporales and Paraglomerales, all so far provided with sequences of this gene, except for Ar. trappei (reasons for this exception are exposed in the “Discussion” section). The RPB1 sequences covered part of the fourth and fifth exon of the gene and the intron in between.
Before concatenation, each sequence set was aligned separately with MAFFT 7, using E-INS-i as iterative refinement method (http://mafft.cbrc.jp/alignment/server/). Indels were coded for the 18S-ITS-28S alignment as binary characters by means of FastGap 1.2 (Borchsenius 2009), with the possibility to code missing data to be recognized by the phylogenetic inference programs. Sequences from the two alignments were then manually concatenated to produce the 18S-ITS-28S + RPB1 alignment. The alignments are shown in Online Resources 2 and 3.
The sequences of uncultured members of the Archaeosporales were selected as follows: about 450 sequences were downloaded from a BLASTn search using sequences of A. polonica as queries. The sequences were ranging from 99 to 80% of identity with the query. After alignment with MAFFT 7, a shared portion of the 28S gene (ca 550 bp), spanning approximately between the primer sites 28G1 and LSUBr (da Silva et al. 2006; Krüger et al. 2009), was used for clustering the sequences in OTUs in Mothur v.1.44.3 (Schloss et al. 2009), using 0.04 as distance cutoff. Pilot analysis on the same 28S portion from species in the Archaeosporales detected the cutoff value = 0.04 as the best choice to cluster at species level and cutoff value = 0.1 to cluster at genus level. Singletons were not considered for further analysis, resulting in twenty OTUs for 364 sequences. Full-length sequences representative of each OTUs were then used in phylogenetic analysis. A summary of the OTUs composition and the geographical origin of the sequences is shown in Online Resources 4 and 5.
Identity values of the 18S-ITS-28S and RPB1 sequences of A. polonica were calculated separately using BioEdit (Hall 1999). With the same program, we calculated the percentage sequence divergence of this species from sequences of its closest relatives (Fig. 1, Online Resource 6). All comparisons were performed on sequences of the same length.

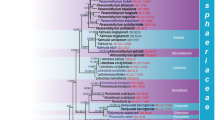
A 50% majority rule consensus phylogram inferred from a Bayesian inference analysis of 18S-ITS-28S + RPB1 sequences of Polonospora polonica, 10 species of the Archaeosporales, 20 OTUs representative sequences from environmental studies (the number of sequences clustered in each OTU is shown between brackets), and six species of the Paraglomerales serving as outgroup. Families in the Archaeosporales, including the new Polonosporaceae, are highlighted by colored boxes. The three distinct clades (POL1‒3) in the Polonosporaceae are separated by red dashed lines. The Bayesian posterior probabilities ≥ 0.90 and ML bootstrap values ≥ 50% are shown near the branches, respectively. Bar indicates 0.1 expected change per site per branch
The phylogenetic position of A. polonica among sequenced species and other potential members of the Archaeosporales was reconstructed based on Bayesian inference (BI) and maximum likelihood (ML) phylogenetic analyses of the alignments, performed via CIPRES Science Gateway 3.3 (Miller et al. 2010). GTR + G + I was chosen as substitution model for the DNA partitions for both BI and ML analysis (Abadi et al. 2019). For the indel partition, F81 model was chosen, as suggested by MrBayes manual. Four Markov chains were run over one million generations in MrBayes 3.2 (Ronquist et al. 2012), sampling every 1000 generations, with a burn-in at 3000 sampled trees. The ML phylogenetic tree inference, using a maximum likelihood/1000 rapid bootstrapping run, was computed with RAxML 8.2.12 (Stamatakis 2014). To improve the accuracy of phylogenetic reconstruction (Lanfear et al. 2012; Nagy et al. 2012), in both BI and ML analyses, the 18S-ITS-28S alignment was divided into six partitions: 18S, ITS1, 5.8S, ITS2, 28S, and the binary (indel) character set. The same partitioning scheme was used in the 18S-ITS-28S + RPB1 alignment with five additional partitions: for the two exons, separate partitions were applied for the first two and for the third codon positions, while a single partition was applied to the intron. In the analysis of the resulting trees, we assumed that clades were supported when BI posterior probability and ML bootstrap support values were ≥ 0.95 and ≥ 70%, respectively. In addition, the 18S-ITS-28S and 18S-ITS-28S + RPB1 trees were compared based on three measures: (i) the number of species clades supported with BI ≥ 0.95 and ML ≥ 70%, (ii) mean supports of nodes with BI ≥ 0.95 and ML ≥ 70%, and (iii) the amount of resolution of each tree. The mean supports of nodes were the sums of BI ≥ 0.95 and ML ≥ 70% supports divided by the number of nodes with BI ≥ 0.95 and ML ≥ 70% present in each tree. The amount of resolution is the number of nodes with BI ≥ 0.95 and ML ≥ 70% divided by the number of all nodes. The phylogenetic trees obtained in the analyses were visualized and rooted in Archaeopteryx.js (https://sites.google.com/site/cmzmasek/home/software/archaeopteryx-js).
Results
General data and phylogeny
In this study, 92 sequences of the 18S-ITS-28S rDNA region or 28S rDNA gene and 15 sequences of the RPB1 gene were analyzed. Of these, 16 were new (7 18S-ITS-28S and 9 RPB1). The 18S-ITS-28S and 28S sequences represented 17, and those of RPB1 eight species of the Archaeosporales ingroup, including the species originally described as A. polonica, and the Paraglomerales outgroup. These sequences were part of two alignments (18S-ITS-28S and 18S-ITS-28S + RPB1) that were analyzed using BI and ML algorithms.
Data about the numbers of base pairs as well as variable and parsimony-informative sites of the 18S-ITS-28S and 18S-ITS-28S + RPB1 alignments are presented in Table 1. The identity values of the six 18S-ITS-28S and three RPB1 sequences of A. polonica were 96.9‒98.9% and 99.3%, respectively.
The topologies of the trees generated in BI and ML analyses of the 18S-ITS-28S and 18S-ITS-28S + RPB1 alignments were identical for both the Archaeosporales and Paraglomerales clades (Fig. 1, Online Resource 6). Also, the measures we used to compare these trees were very similar (Online Resource 7). Because it is widely recommended to reconstruct fungal phylogenies from multiple unlinked loci (Matheny 2005; Miadlikowska et al. 2014; Stadler and Weber 2021), here were discuss the phylogeny of A. polonica mainly based on trees generated in analyses of the 18S-ITS-28S + RPB1 alignment (Fig. 1).
Sequences of A. polonica clustered in a new clade at the rank of family in a sister position to the Archaeosporaceae clade, but this association was weakly supported in the BI analysis only (Fig. 1, Online Resource 6). The new family clade consisted of three subclades (named POL1, 2, and 3) at the rank of genus. POL1 and POL2 formed a sister relationship with good support only in the BI analysis. POL1 consisted of two sister clades, of which one contained sequences of A. polonica and sequences obtained in environmental studies conducted in China, Georgia, Japan, Portugal, and the USA (Michigan and Massachusetts). The sister clade to A. polonica contained only environmental sequences. These were obtained from studies performed in China, Japan, and Portugal, and clustered in three OTUs representing potentially new species. POL2, sister to the generic clade with A. polonica, hosted the highest number of environmental sequences, clustering in 12 OTUs representing several potentially new species. POL2 was also the most geographically widespread; its available environmental sequences originated from Australia, China, Europe, and South and North America (see Online Resource 5 for details). The third generic clade contained environmental sequences from China, Czech Republic, and Norway. The new family clade obtained full supports in both BI (= 1.0) and ML (= 100%) analyses. Also, both analyses strongly supported the generic clades POL1 and POL2. Instead, the generic clade POL3 was supported only in the BI analysis of the 18S-ITS-28S alignment. The species clade with A. polonica obtained strong supports in both BI (= 1.0) and ML (= 90.0%) analyses.
Of the five species of the Archaeosporaceae clade, the closest relative of A. polonica was Ar. ecuadoriana. The 18S-ITS-28S sequence divergence between these species was 24.5‒28.0%.
Considering the results of the phylogenetic analyses and comparisons of sequences described above, we transferred A. polonica to the Archaeosporales and accommodated it in two newly erected taxa, Polonosporaceae fam. nov. and Polonospora gen. nov., as P. polonica comb. nov. In addition, we presented an updated morphological description of P. polonica and its possible distribution.
Taxonomy
Erection of a new family, genus, and combination
Polonosporaceae Błaszk., Niezgoda, B.T. Goto, Magurno, fam. nov.
MycoBank: MB840255.
Type: Polonospora Błaszk., Niezgoda, B.T. Goto, Magurno.
Etymology: Latin, Polonosporaceae, Polono (= from Poland) and sporaceae (= forming spores), referring to Poland, in which spores of the new family were originally found.
Diagnosis: Differs from the Archaeosporaceae and Ambisporaceae in the number of spore walls (three-walled vs. two-walled) and the nucleotide composition of sequences of the 18S-ITS-28S nuc rDNA region and the RPB1 gene.
Description: Forming hypogeous single acaulosporoid glomerospores (= spores) directly on the neck of a sporiferous saccule (Fig. 2a, b). Spores hyaline to white (1A1), usually globose to subglobose, 80–115 μm diam, with three spore walls (spore walls 1–3; Fig. 2a–h). Spore wall 1 consisting of a short-lived, evanescent, thin layer, continuous with the wall of the sporiferous saccule, and a permanent, laminate, thicker layer (Fig. 2c–h). Spore wall 2 composed of one permanent, flexible to semi-flexible layer (Fig. 2c–h). Spore wall three permanent, coriaceous, 1.8–3.6-μm thick, composed of two tightly adherent layers (Fig. 2c–h). Only spore wall 1 layer 2 sometimes stains reddish white (7A2) in Melzer’s reagent.

Polonospora polonica. a, b. Intact spores (s) with the neck of a sporiferous saccule (nss; remnants). c‒h. Spore wall 1 (sw1) layers 1 and 2, spore wall 2 (sw2), and spore wall 3 (sw3) layers 1 and 2; soil debris (sd) covering the upper surface of sw1l1 and cicatrix (c) are indicated in c. a‒f. Spores in PVLG. g, h. Spores in PVLG + Melzer’s reagent. a‒h. Differential interference microscopy. Scale bars: a, b = 20 μm, c‒h = 10 μm
Distribution and habitat: Worldwide, with records of physical specimens and environmental sequences originating from Australia, Czech Republic, China, Georgia, Japan, Norway, Poland, Portugal, and North and South Americas (Online Resource 5). Associated with roots of plants growing in dunes, forests, grasslands, hydrocarbon-polluted sites, tropical forested and cultivated soils, and wetland ecosystems.
Polonospora Błaszk., Niezgoda, B.T. Goto, Magurno, gen. nov.
MycoBank: MB840256.
Type: Polonospora polonica (Błaszk.) Błaszk., Niezgoda, B.T. Goto, Magurno.
Etymology: As that for the Polonosporaceae (see above).
Diagnosis: As that of Polonosporaceae (see above).
Distribution and habitat: Worldwide, with records of physical specimens and environmental sequences originating from China, Georgia, Japan, Lithuania, Poland, Portugal, and the USA (Online Resource 5). Associated with roots of plants growing in gardens, dunes, forests, and highly contaminated sites (Gai et al. 2006; Błaszkowski 2012).
Specimens examined: 95–136, 3804–3821, LPPDSE.
Polonospora polonica (Błaszk.) Błaszk., Niezgoda, B.T. Goto, Magurno, comb. nov. Figure 2a–h.
MycoBank: MB840257.
Basionym: Acaulospora polonica Błaszk., Karstenia 27, 38. 1988.
Description: Glomerospores (= spores) formed singly in soil, arise laterally from the neck of a sporiferous saccule continuous with an extraradical mycorrhizal hypha (Fig. 2a, b). Spores hyaline to white (1A1), glistening, globose to subglobose, (80–)94(–115) μm diam, with three spore walls (Fig. 2a–h). Spore wall 1 with two layers (Fig. 2c–h). Layer 1 evanescent, hyaline, up to 1.5-μm thick, continuous with the wall of the sporiferous saccule, usually highly deteriorated or completely sloughed in mature spores. Layer 2 permanent, laminate, smooth, hyaline to white (1A1), (3.3–)4.7(–5.5) μm thick. Spore wall 2 consists of one, permanent, flexible to semi-flexible, (0.8–)1.1(–1.3) μm thick, hyaline layer (Fig. 2c–h). Spore wall 3 composed of two permanent, hyaline, smooth layers (Fig. 2c–h). Layer 1 semi-flexible, (1.3–)1.8(–2.3) μm thick. Layer 2 flexible to semi-flexible, 0.5–1.3-μm thick, usually tightly adherent to the lower surface of layer 1 and difficult to see. In Melzer’s reagent, only spore wall 1 layer 2 sometimes stains reddish white (7A2). Germination orb not found. Sporiferous saccule hyaline, globose to subglobose, 60–90 μm diam; neck 40–70 μm long, tapering from 10.0–17.5 μm diam at the saccule to 8.0–12.5 μm diam at the point of spore attachment. Saccule wall a hyaline, smooth, 0.5–1.0-μm thick layer. Saccule collapsing at maturity and usually detached in mature spores. Cicatrix a slightly raised collar when seen in cross view, circular, 6–9 μm diam, when observed in plain view.
Mycorrhizal association: In the field, produced spores in rhizosphere of, e.g., Ammophila arenaria, Chamaecyparis lawsoniana, Corynephorus canescens, Juncus balticus, Juniperus communis, R. canina, and Thuja occidentalis (Błaszkowski 1988, 1993a, b, 1994, unpubl. data). Abundantly sporulated in trap cultures inoculated with rhizosphere soil and root fragments of R. rugosa, whose host plant was P. lanceolata. Many attempts at growing P. polonica in single-species cultures failed.
Distribution and habitat: Worldwide, with records of physical specimens and environmental sequences originating from China, Georgia, Japan, Lithuania, Poland, Portugal, and the USA (Online Resource 5). Associated with roots of plants growing in gardens, dunes, forests, and highly contaminated sites (Gai et al. 2006; Błaszkowski 2012).
Specimens examined: Holotype. POLAND. Hel, ca 200 m from the Baltic Sea, under Thuja occidentalis, 21.08.1985, Błaszkowski J., 95, LPPDSE; 96–136 (isotypes), LPPDSE; Kuźnica, under R. rugosa, 21.08.2017, Błaszkowski J., 3810–3821, LPPDSE. LITHUANIA. Kuronian Spit, Parnidos dune, under A. arenaria, 15.09.2013, Błaszkowski J., 3804–3809, LPPDSE.
Discussion
The results of molecular phylogenetic analyses and comparisons of sequences discussed above confirmed our hypothesis that the species originally described as A. polonica (Błaszkowski 1988) does not belong to Acaulospora but represents a new genus in a new family of the Archaeosporales, here introduced into the Glomeromycota under the names Polonospora and Polonosporaceae, respectively. Moreover, our analyses indicated that (i) the Polonosporaceae consists of three groups at the rank of genus: Polonospora, typified by P. polonica comb. nov, and AMF known from molecular environmental studies only (Fig. 1, Online Resources 4, 5) and (ii) the Polonosporaceae with P. polonica and the yet undescribed taxa have a worldwide distribution (Online Resource 5). Unfortunately, our attempts at growing P. polonica in single-species cultures failed. Therefore, the characters of mycorrhizal structures of this species and its potential ability to produce spore morphotypes other than acaulosporoid remain unknown.
Our POL1, POL2, and POL3 correspond to the Arch3, Arch2, and Arch1 clades, respectively, of an undescribed family in the Archaeosporales recognized by Kolaříková et al. (2021) based on analyses of environmental sequences. The Arch1 clade did not receive full support in our analysis, but with the present data, it is not possible to establish if it could represent more than one genus. Notably, considering the full-length sequences of the three OTUs in the clade, one is highly divergent (by 13.0–15.5%) from the other two.
Our analyses indicated a moderate sister relationship of the Polonosporaceae to the Archaeosporaceae, whereas the Kolaříková et al.’s (2021) informal family was a sister to a clade formed by the Ambisporaceae and Geosiphonaceae. This could result from the differences in the phylogenetic approaches. In Kolaříková et al. (2021), only sequences of the unpartitioned 18S-ITS-28S nuc rDNA locus were employed in the BI and ML analyses, while in the present analysis, a partitioning scheme was applied to the alignment containing 18S-ITS-28S sequences concatenated with those of the unlinked protein-coding RPB1 gene. Furthermore, the sequence set used in the present study consisted of sequences of the Archaeosporales and Paraglomerales only (the latter used as outgroup). The sequence set analyzed by Kolaříková et al. (2021) additionally contained sequences of the Diversisporales and Glomerales. Finally, twenty OTUs, representing 364 environmental sequences, and six sequences belonging to P. polonica were used to represent the new family Polonosporaceae, while in Kolaříková et al. (2021), only six OTUs and four sequences from another study (Melo et al. 2018) were used. The genetic diversity of sequence data, the resolution power of their components, the method of analyzing, and the taxonomic composition of the analyzed set may affect the reliability of fungal phylogenies (Matheny 2005; Redecker et al. 2013; Miadlikowska et al. 2014).
We did not concatenate any 18S-ITS-28S sequence of Ar. trappei with the sole available RPB1 sequence of this species (HG315988) obtained from the Ar. trappei INVAM CR401B isolate (Stockinger et al. 2014) because in public data bases, there is no 18S-ITS-28S sequence obtained from this isolate. Schüßler and Walker (2019) demonstrated that the phylogenetic position of Ar. trappei is unknown. In our 18S-ITS-28S and 18S-ITS-28S + RPB1 trees, sequences ascribed to Ar. trappei were also accommodated in three clades separated by Ar. europaea and Ar. schenckii (Fig. 1, Online Resource 6). No living cultures of ex-type material of Ar. trappei are available (Schüßler and Walker 2019). Thus, to unambiguously determine the phylogenetic position of Ar. trappei and other members of this genus within the Archaeosporaceae, an epitype of this species has to be designated based on spores obtained from material collected from the area, where Ar. trappei was originally found (Ames and Linderman 1976).
Apart from Ar. trappei, the Glomeromycota contains many other species, whose natural phylogeny is uncertain or unknown (Schüßler and Walker 2010; Błaszkowski 2012; Redecker et al. 2013; Kolaříková et al. 2021; pers. observ.). One of them is A. gedanensis, whose morphological features (Błaszkowski 1988, 2012) suggest belonging to an undescribed taxon related to members of the Ambisporaceae. As P. polonica, A. gedanensis was also originally found in soils of the Hel Peninsula.
Data availability
Datasets generated during and/or analyzed during the current study are available from the corresponding author upon request.
Code availability
Not applicable
References
Abadi S, Azouri D, Pupko T, Mayrose I (2019) Model selection may not be a mandatory step for phylogeny reconstruction. Nat Commun. https://doi.org/10.1038/s41467-019-08822-w, www.nature.com/naturecommunications
Ames RN, Linderman RG (1976) Acaulospora trappei sp. nov. Mycotaxon 3:565–569
Ames RN, Schneider RW (1979) Entrophospora, a new genus in the Endogonaceae. Mycotaxon 8:347–352
Bills RB, Morton JB (2015) A combination of morphology and 28S rRNA gene sequences provide grouping and ranking criteria to merge eight into three Ambispora species (Ambisporaceae, Glomeromycota). Mycorrhiza 25:485–498. https://doi.org/10.1007/s00572-015-0626-7
Błaszkowski J (1988) Four new species of the Endogonaceae (Zygomycotina) from Poland. Karstenia 27:37–42. https://doi.org/10.29203/ka.1987.252
Błaszkowski J (1993a) Comparative studies of the occurrence of arbuscular fungi and mycorrhizae (Glomales) in cultivated and uncultivated soils of Poland. Acta Mycol 28:93–140. https://doi.org/10.5586/am.1993.013
Błaszkowski J (1993b) The occurrence of arbuscular fungi and mycorrhizae (Glomales) in plant communities of maritime dunes and shores of Poland. Bull Pol Ac Sci 41:377–392
Błaszkowski J (1994) Arbuscular fungi and mycorrhizae (Glomales) of the Hel Peninsula, Poland. Mycorrhiza 5:71–88. https://doi.org/10.1007/BF00204022
Błaszkowski J (2012) Glomeromycota. Szafer Institute of Botany, Polish Academy of Sciences, Kraków, p 303
Błaszkowski J, Kovács GM, Gáspár BK, Balázs TK, Buscot F, Ryszka P (2012) The arbuscular mycorrhizal Paraglomus majewskii sp. nov. represents a distinct basal lineage in Glomeromycota. Mycologia 104:148–156. https://doi.org/10.3852/10-430
Błaszkowski J, Chwat G, Góralska A, Ryszka P, Orfanoudakis M (2014) Septoglomus jasnowskae and Septoglomus turnauae, two new species of arbuscular mycorrhizal fungi (Glomeromycota). Mycol Prog 13:985. https://doi.org/10.1007/s11557-014-0985-z
Błaszkowski J, Chwat G, Góralska A, Bobrowska-Chwat A (2015a) Glomus tetrastratosum, a new species of arbuscular mycorrhizal fungi (Glomeromycota). Mycoscience 56:280–286. https://doi.org/10.1016/j.myc.2014.08.003
Błaszkowski J, Chwat G, Góralska A, Ryszka P, Kovács GM (2015b) Two new genera, Dominikia and Kamienskia, and D. disticha sp. nov. in Glomeromycota. Nova Hedwigia 100:225–238. https://doi.org/10.1127/nova_hedwigia/2014/0216
Błaszkowski J, Jobim K, Niezgoda P, Meller E, Malinowski M, Milczarski P, Sz Zubek, Magurno F, Casieri L, Bierza W, Błaszkowski T, Crossay T, Goto BT (2021) New glomeromycotan taxa, Dominikia glomerocarpica sp. nov. and Epigeocarpum crypticum gen. nov. et sp. nov. from Brazil, and Silvaspora gen. nov. from New Caledonia. Front Microbiol 12:655910. https://doi.org/10.3389/fmicb.2021.655910
Błaszkowski J, Kozłowska A, Crossay T, Symanczik S, Al-Yahya’ei MN (2017) A new family, Pervetustaceae with a new genus, Pervetustus, and P. simplex sp. nov. (Paraglomerales), and a new genus, Innospora with I. majewskii comb. nov. (Paraglomeraceae) in the Glomeromycotina. Nova Hedwigia 105:397–410. https://doi.org/10.1127/nova_hedwigia/2017/0419
Borchsenius F (2009) FastGap 1.2. Department of Biosciences, Aarhus University, Denmark. Published online at http://www.aubot.dk/FastGap_home.htm
Gai JP, Christie P, Feng G, Li XL (2006) Twenty years of research on biodiversity and distribution of arbuscular mycorrhizal fungi in China: a review. Mycorrhiza 16:229–239. https://doi.org/10.1007/s00572-005-0023-8
Gerdemann JW, Trappe JM (1974) The Endogonaceae in the Pacific Northwest. Mycologia Memoir 5:1–76
Goto BT, Maia LC (2006) Glomerospores: a new denomination for the spores of Glomeromycota, a group molecularly distinct from the Zygomycota. Mycotaxon 96:129–132
Hall IR (1977) Species and mycorrhizal infections of New Zealand Endogonaceae. Trans Br Mycol Soc 68:341–356. https://doi.org/10.1016/S0007-1536(77)80186-1
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Kaonongbua W, Morton JB, Bever JD (2010) Taxonomic revision transferring species in Kuklospora to Acaulospora (Glomeromycota) and a description of Acaulospora colliculosa sp. nov. from field collected spores. Mycologia 102:1497–1509. https://doi.org/10.3852/10-011
Kolaříková Z, Slavíková R, Krüger C, Krüger M, Kohout P (2021) PacBio sequencing of Glomeromycota rDNA: a novel amplicon covering all widely used ribosomal barcoding regions and its applicability in taxonomy and ecology of arbuscular mycorrhizal fungi. New Phytol. Accepted Author Manuscript. https://doi.org/10.1111/nph.17372
Kornerup A, Wanscher JH (1983) Methuen handbook of colour, 3rd edn. Eyre Methuen, London, p 252
Krüger M, Stockinger H, Krüger C, Schüßler A (2009) DNA-based level detection of Glomeromycota: one PCR primer set for all arbuscular mycorrhizal fungi. New Phytol 183:212–223. https://doi.org/10.1111/j.1469-8137.2009.02835.x
Lanfear R, Calcott B, Ho SYW, Guindon S (2012) PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29:1695–1701. https://doi.org/10.1093/molbev/mss020
Matheny PB (2005) Improving phylogenetic inference of mushrooms with RPB1 and RPB2 nucleotide sequences (Inocybe; Agaricales). Mol Phylogenet Evol 35:1–20. https://doi.org/10.1016/j.ympev.2004.11.014
Melo CD, Luna S, Krüger C, Walker C, Mendonça D, Fonseca HMAC, Jaizme-Vega M, Machado AC (2018) Communities of arbuscular mycorrhizal fungi under Picconia azorica in native forests of Azores. Symbiosis 74:43–54. https://doi.org/10.1007/s13199-017-0487-2
Miadlikowska J, Kauff F, Högnabba et al (2014) A multigene phylogenetic synthesis for the class Lecanoromycetes (Ascomycota): 1307 fungi representing 1139 infrageneric taxa, 317 genera and 66 families. Mol Phylogenet Evol 79:132–168. https://doi.org/10.1016/j.ympev.2014.04.003
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES science gateway for inference of large phylogenetic trees. Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, pp 1–8. https://doi.org/10.1109/GCE.2010.5676129
Morton JB, Redecker D (2001) Two families of Glomales, Archaeosporaceae and Paraglomaceae, with two new genera Archaeospora and Paraglomus, based on concordant molecular and morphological characters. Mycologia 93:181–195. https://doi.org/10.2307/3761615
Nagy LG, Kocsube S, Csana Z, Kovacs GM, Petkovits T, Lgyi CV, Papp T (2012) Re-mind the gap! insertion – deletion data reveal neglected phylogenetic potential of the nuclear ribosomal internal transcribed spacer (ITS) of fungi. PLoS ONE 7:e49794. https://doi.org/10.1371/journal.pone.0049794
Nicolson TH, Schenck NC (1979) Endogonaceous mycorrhizal endophytes in Florida. Mycologia 71:178–198. https://doi.org/10.2307/3759231
Oehl F, da Silva GA, Goto BT, Sieverding E (2011a) Glomeromycota: three new genera and glomoid species reorganized. Mycotaxon 116:75–120. https://doi.org/10.5248/116.75
Oehl F, da Silva GA, Sánchez-Castro I, Goto BT, Maia LC, Vieira HEE, Barea J-M, Sieverding E, Palenzuela J (2011b) Revision of Glomeromycetes with entrophosporoid and glomoid spore formation with three new genera. Mycotaxon 117:297–316. https://doi.org/10.5248/117.297
Omar MB, Bollan L, Heather WA (1979) A permanent mounting medium for fungi. Bull Brit Mycol Soc 13:31–32
Redecker D, Schüßler A, Stockinger H, Stürmer SL, Morton JB, Walker C (2013) An evidence-based consensus for the classification of arbuscular mycorrhizal fungi (Glomeromycota). Mycorrhiza 23:515–531. https://doi.org/10.1007/s00572-013-0486-y
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542. https://doi.org/10.1093/sysbio/sys029
Schloss PD, Westcott SL, Ryabin et al (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. https://doi.org/10.1128/AEM.01541-09
Schüßler A, Walker C (2010) The Glomeromycota. A species list with new families and new genera. Edinburgh, Royal Botanic Garden Edinburgh, p 56
Schüßler A, Walker C (2019) Archaeospora ecuadoriana sp. nov. from a mountainous biodiversity hotspot area in Ecuador, and transfer of Palaeospora spainiae to Archaeospora, as A. spainiae comb. nov. Mycorrhiza 29:435–443. https://doi.org/10.1007/s00572-019-00913-2
Sieverding E, Oehl F (2006) Revision of Entrophospora and description of Kuklospora and Intraspora, two new genera in the arbuscular mycorrhizal Glomeromycetes. J Appl Bot Food Qual 80:69–81
da Silva GA, Lumini E, Maia L, Bonfante P, Bianciotto V (2006) Phylogenetic analysis of Glomeromycota by partial LSU rDNA sequences. Mycorrhiza 16:183–189. https://doi.org/10.1007/s00572-005-0030-9
Stadler M, Weber E (2021) New developments in mycological taxonomy and nomenclature and news about the future development of Mycological Progress. Mycol Prog 20:223–225. https://doi.org/10.1007/s11557-021-01680-1
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Stockinger H, Peyret-Guzzon M, Koegel S, Bouffaud M-L, Redecker D (2014) The Largest Subunit of RNA Polymerase II as a New Marker Gene to Study Assemblages of Arbuscular Mycorrhizal Fungi in the Field. PLoS ONE 9:e107783. https://doi.org/10.1371/journal.pone.0107783
Symanczik S, Błaszkowski J, Chwat G, Boller T, Wiemken A, Al-Yahya’ei MN (2014) Three new species of arbuscular mycorrhizal fungi discovered at one location in a desert of Oman: Diversispora omaniana, Septoglomus nakheelum and Rhizophagus arabicus. Mycologia 106:243–259. https://doi.org/10.3852/106.2.243
Walker C (1983) Taxonomic concepts in the Endogonaceae: spore wall characteristics in species descriptions. Mycotaxon 18:443–455
Walker C (1986) Taxonomic concepts in the Endogonaceae. II. A fifth morphological wall type in endogonaceous spores. Mycotaxon 25:95–99
Acknowledgements
Part of this work was supported by Polish National Centre of Science, grant no. 2020/37/N/NZ9/00509, as well as by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) that provided research grants to BT Goto (proc. 311945/2019-8). We also thank Universidade Federal do Rio Grande do Norte for covering the costs of B.T. Goto’s stay as collaborative research in West Pomeranian University of Technology in Szczecin in the period from December 2019 to January 2020. The research was funded, in part, by the Institute of Botany at the Jagiellonian University, project no. N18/ DBS/000002.
Funding
To Piotr Niezgoda by Polish National Centre of Science, grant no. 2020/37/N/NZ9/00509, to Bruno Tomio Goto by Conselho Nacional de Desenvolvimento Científico e Tecnológico, proc. 311945/2019–8), to Szymon Zubek by the Institute of Botany at the Jagiellonian University, project no. N18/DBS/000002.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Janusz Błaszkowski, Bruno Tomio Goto, Leonardo Casieri, Franco Magurno, Monika Malicka, Edward Meller, Paweł Milczarski, Piotr Niezgoda, and Szymon Zubek. The first draft of the manuscript was written by Janusz Błaszkowski, and all authors commented on previous versions of the manuscript. Conceptualisation: Janusz Błaszkowski, Bruno Tomio Goto, Leonardo Casieri, Franco Magurno, Sylwia Uszok; methodology: Janusz Błaszkowski, Bruno Tomio Goto, Franco Magurno, Piotr Niezgoda; formal analysis and investigation: Janusz Błaszkowski, Bruno Tomio Goto, Franco Magurno, Monika Malicka, Edward Meller, Paweł Milczarski, Piotr Niezgoda, Sylwia Uszok, and Szymon Zubek; writing original draft preparation: Janusz Błaszkowski, Bruno Tomio Goto, Franco Magurno; writing—review and editing: Janusz Błaszkowski, Bruno Tomio Goto, Franco Magurno, Monika Malicka, Edward Meller, Paweł Milczarski, Sylwia Uszok, Piotr Niezgoda, and Szymon Zubek; funding acquisition: Bruno Tomio Goto, Piotr Niezgoda, Szymon Zubek; resources: Janusz Błaszkowski, Franco Magurno, Piotr Niezgoda; Supervision: Janusz Błaszkowski. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Section editor: Marco Thines
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Błaszkowski, J., Niezgoda, P., Meller, E. et al. New taxa in Glomeromycota: Polonosporaceae fam. nov., Polonospora gen. nov., and P. polonica comb. nov.. Mycol Progress 20, 941–951 (2021). https://doi.org/10.1007/s11557-021-01726-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11557-021-01726-4